





Abstract
Many of the protists thought to represent the deepest branches on the eukaryotic tree are assigned to a loose assemblage called the “excavates.” This includes the mitochondrion-lacking diplomonads and parabasalids (e.g., Giardia and Trichomonas) and the jakobids (e.g., Reclinomonas). We report the first multigene phylogenetic analyses to include a comprehensive sampling of excavate groups (six nuclear-encoded protein-coding genes, nine of the 10 recognized excavate groups). Excavates coalesce into three clades with relatively strong maximum likelihood bootstrap support. Only the phylogenetic position of Malawimonas is uncertain. Diplomonads, parabasalids, and the free-living amitochondriate protist Carpediemonas are closely related to each other. Two other amitochondriate excavates, oxymonads and Trimastix, form the second monophyletic group. The third group is comprised of Euglenozoa (e.g., trypanosomes), Heterolobosea, and jakobids. Unexpectedly, jakobids appear to be specifically related to Heterolobosea. This tree topology calls into question the concept of Discicristata as a supergroup of eukaryotes united by discoidal mitochondrial cristae and makes it implausible that jakobids represent an independent early-diverging eukaryotic lineage. The close jakobids-Heterolobosea-Euglenozoa connection demands complex evolutionary scenarios to explain the transition between the presumed ancestral bacterial-type mitochondrial RNA polymerase found in jakobids and the phage-type protein in other eukaryotic lineages, including Euglenozoa and Heterolobosea.
Introduction
Determining the deep-level structure of the phylogenetic tree of eukaryotes is key to understanding the evolutionary history of complex cells. Of central importance are the various “excavates,” a collection of 10 distinct groups of unicellular eukaryotes united primarily by similarities of cell ultrastructure (Simpson 2003). Early molecular phylogenies of small-subunit ribosomal RNA (SSU rRNA) sequences and elongation factor proteins placed two mitochondrion-lacking (amitochondriate) groups of excavates, Diplomonadida and Parabasala, among the three deepest branches in the eukaryotic tree (Sogin 1989; Sogin et al. 1989; Hashimoto et al. 1994; Yamamoto et al. 1997). This placement catalyzed extensive investigations into the genome organization and cell biology of model diplomonads and parabasalids, in particular, the human parasite Giardia intestinalis (Gillin, Reiner, and McCaffery 1996; McArthur et al. 2000). Some more recent phylogenetic analyses and the recognition of organelles seemingly of mitochondrial origin in both parabasalids and diplomonads have weakened the arguments that these groups represent deep-branching eukaryotes (Embley and Hirt 1998; Roger 1999; Philippe et al. 2000; Baldauf 2003; Tovar et al. 2003; Arisue, Hasegawa, and Hashimoto 2005). However, basal positions for both groups remain “textbook science,” and modified proposals potentially affording them a primitive status continue to be advanced (Chihade et al. 2000; Dyall, Brown, and Johnson 2004).
More recently, a different group of excavates, Jakobida, was found to have the most bacterial-like (primitive) mitochondrial genomes known (Lang et al. 1997; Gray, Burger, and Lang 1999; Gray, Lang, and Burger 2004). Jakobid mitochondrial genomes retain more protein-coding genes than those of other eukaryotes. Most importantly, they encode 2–4 subunits of a bacterial-type RNA polymerase, whereas the mitochondrial RNA polymerase of all other studied eukaryotes is a nonhomologous single subunit “phage-type” enzyme, typically encoded by the nuclear genome. Under the simplest evolutionary scenario, the bacterial-type mitochondrial RNA polymerase was the ancestral form inherited from the endosymbiotic α-proteobacterium that gave rise to mitochondria and was replaced once in eukaryotic history by the phage-type enzyme. If this scenario is correct, this replacement event must have happened after the divergence of jakobids from other eukaryotes, and jakobids must represent one of the earliest diverging eukaryotic lineages.
Determining the evolutionary significance of any particular excavate group requires a resolution of its phylogenetic relationships with other excavates. Despite genomic sequencing of some species that are human pathogens, there has been virtually no molecular data available that can be compared across all excavates. Some recent phylogenetic analyses do include taxa from most or all of the 10 excavate groups but use data from one or, at most, two genes (Archibald, O'Kelly, and Doolittle 2002; Simpson et al. 2002b; Cavalier-Smith 2003; Keeling and Leander 2003; Simpson 2003). Many relationships among excavates remain essentially unresolved. In particular, robust and precise phylogenetic positions for diplomonads, parabasalids, and jakobids have remained elusive (Baldauf et al. 2000; Gray, Lang, and Burger 2004).
We have assembled the first multigene data set of eukaryotes that includes a taxonomically comprehensive representation of excavates. Our detailed analyses cement a close, yet not specific, relationship between diplomonads and parabasalids and demonstrate a specific relationship between jakobids and the supergroup “Discicristata” (Euglenozoa and Heterolobosea), especially Heterolobosea. This position for jakobids requires complex scenarios to explain their primitive-looking mitochondrial RNA polymerases and questions the validity of Discicristata as a natural (monophyletic) “supergroup” of eukaryotes.
Materials and Methods
Material Sources
Trimastix marina was purified by serial dilution from an isolation by J. D. Silberman (University of Arkansas) from William's Lake, Nova Scotia, Canada (44°39′N; 63°34′W) and was maintained on American Type Culture Collection (ATCC) 802 media. The Carpediemonas membranifera isolate examined has been described previously (Simpson and Patterson 1999). Genomic DNA (gDNA) was isolated from both cultures using standard protocols (Clark and Diamond 1991). Rhynchopus sp. (ATCC 50230) was grown and gDNA was extracted as described previously (Simpson, Lukeš, and Roger 2002). Rhynchomonas nasuta gDNA was a kind gift from M. Atkins (Woods Hole Oceanographic Institute, Woods Hole, Mass.). Reclinomonas americana (ATCC 50283) gDNA and Malawimonas jakobiformis gDNA were kind gifts from B. F. Lang (Université de Montreal, Canada). Naegleria gruberi (strain NEG-M) gDNA and Trimastix pyriformis (ATCC 50562) cDNA were kindly provided by Å. Sjögren and J. D. Silberman, respectively.
Gene Discovery
Six slowly evolving nuclear-encoded genes were examined—those for α-tubulin, β-tubulin, elongation factor 1α (EF-1α), elongation factor 2 (EF-2), cytosolic heat shock protein 70 (HSP70), and cytosolic heat shock protein 90 (HSP90). A total of 26 near-complete or complete coding sequences were determined from various excavates. HSP90 and EF-2 sequences from Spironucleus barkhanus and EF-2 from Naegleria gruberi were sequenced from cDNA clones identified in expressed sequence tag (EST) surveys. All other sequences were obtained by degenerate polymerase chain reaction (PCR) from gDNA or cDNA templates using a variety of primer combinations, including several new primers with broad applicability (see Supplementary Material online). PCR amplifications were performed with annealing temperatures of 48–55°C. Amplifications from gDNA templates other than Rhynchopus and Rhynchomonas included 5% w/v acetamide in the reaction cocktail. PCR products were gel-purified and cloned into TA plasmid vectors (TOPO series, Invitrogen, Carlsbad, Calif.) in Escherichia coli. One to five positive clones were partially sequenced, and a single clone of each distinct paralog encountered (usually only one) was selected for complete bidirectional sequencing. New sequences have been deposited in GenBank accession numbers (DQ295211–DQ295236).
For alignment, sequences were translated conceptually to amino acids. Where present, spliceosomal introns were detected by eye and eliminated. Amino acid sequences from the examined genes were aligned by eye with homologues from taxa representing a broad diversity of eukaryotes. Some sequences were obtained from publicly accessible genome or EST projects. Where multiple paralogs of a gene were available, the least divergent sequence was generally used. When deep paralogy was encountered (within animals and plants), preliminary phylogenetic analyses were run to ensure the selection of an othologous set of sequences from within these groups wherever possible. The six examined genes were concatenated. In six cases (Trichomonas, Eimeria, Stylonychia, Tetrahymena, Porphyra, and Monosiga), data from two nominal species were combined as one taxon. Some highly divergent taxa (e.g., microsporidia) and redundant close relatives were excluded, leaving 44 taxa as a broad representation of eukaryotes. Fifteen excavates were retained, representing nine of the 10 excavate groups—the omitted excavate group, retortamonads was excluded solely because of a lack of data and is known with confidence to be specifically related to diplomonads based on SSU rRNA analyses (Silberman et al. 2002) and HSP90 protein trees (A. G. B. Simpson, unpublished data). Ambiguously aligned regions were excluded, leaving a total of 3,142 sites, with every taxon including >75% of the analyzed sequence from 4+ genes and >50% of all examined sites (average 91%) and with “taxonomically isolated” taxa such Naegleria, Reclinomonas, and Malawimonas represented by sequences from all six genes and >80% of examined sites. Species names and included genes are tabulated in the supplementary material (Supplementary Material online), and data sets are available by request to A. G. B. Simpson.
We did not attempt to root the tree using deep eukaryotic paralogs and/or prokaryotic orthologs as outgroups. All possible outgroup sequences would be very distant from the ingroup and have very different patterns of evolutionary rates at sites, a potential source of phylogenetic artefact (Inagaki et al. 2004). As a result, any such rooting of the eukaryotic tree would almost certainly be unreliable (Philippe et al. 2000) and, worse, could bias the estimation of relationships among the ingroup. Our data set would be particularly poorly suited to outgroup analysis as some genes are especially dissimilar to their nearest eukaryotic paralog (e.g., EF-2), while others are extremely distant from the nearest widespread prokaryotic genes (tubulins).
Phylogenetic Analysis Under a “Linked” Model
Initially, we used a standard linked (concatenated) approach, with a single set of branch lengths and a single among-site rate variation (ASRV) distribution imposed across the whole multigene data set. The maximum likelihood (ML) tree was searched for with PROML 3.6b (Felsenstein 2004) using the Jones-Taylor-Thornton (JTT) amino acid substitution matrix and ASRV modeled by a Γ distribution approximated by four equally probable discrete categories (five random addition sequences and global rearrangements were used). To assess the robustness of our tree, a 500 replicate “fast” ML bootstrap analysis was performed using PHYML (Guindon and Gascuel 2003), under the same model but with an eight-category Γ approximation. The bootstrap analysis was repeated with diplomonads excluded (200 replicates). The α parameters and discrete rates governing the Γ distribution were estimated from the data using Tree-Puzzle 5.1 (Schmidt et al. 2002). Although the Whelan and Goldman (WAG) substitution matrix conferred a higher likelihood on the data, the JTT matrix was used because PROML does not support the WAG matrix. Irrespective, we repeated the bootstrap analysis described above using the WAG substitution matrix and 200 replicates and found almost no difference in the support across the tree (not shown).
Phylogenetic Analysis Under an “Unlinked” Model
Previous analyses of multigene data sets indicate that model fit can be significantly improved if separate sets of parameters are allowed for the different genes (Bapteste et al. 2002; Pupko et al. 2002). To accommodate within-taxon rate heterogeneity across genes, a second set of analyses was performed under an unlinked model where different branch lengths (and Γ shapes for ARSV) were allowed for each gene. This model can be examined in the Bayesian analysis program MrBayes 3.14 (Ronquist and Huelsenbeck 2003; Nylander et al. 2004). The WAG substitution matrix was applied, with a four–discrete-category Γ approximation for each data set (“WAG + Γ4 model”). The α parameter values were optimized during the analysis. Several analyses were performed using different random starting trees, with one cold and two heated Markov chain Monte Carlo (MCMC) chains (“temperature” parameter = 0.2), and sampling every 100 generations. Three “long” analyses were run for 2 × 106 generations (with a very conservative 106 generations burn-in) and three “short” analyses for 5 × 105 generations (3 × 105 generations burn-in). The three long runs stabilized in two different regions of tree space, and in all, three different topologies of maximum posterior probability were recovered. Accordingly, all eight trees with a posterior probability >0.001 in any one run were compared to the ML tree from the linked analysis and to other user-defined trees constituting minor rearrangements of likely trees (total 202 trees). The user-defined trees included topologies where excavates were monophyletic, where jakobids were not specifically related to Heterolobosea, or where jakobids were not specifically related to Heterolobosea plus Euglenozoa. For each tree, total log-likelihood (ln L) under the unlinked WAG + Γ4 model was obtained from the sum of ln Ls for each gene calculated separately using Tree-Puzzle. This “unlinked model” conferred much greater likelihood on the data than did the analogous linked model (Δln L = 1460 − 1540, depending on the tree). This difference was highly significant in likelihood ratio tests (P ≪ 10−5). A subset of these trees (65) were compared using “approximately unbiased” (AU) tests of significance (Shimodaira 2002), under the unlinked WAG + Γ4 model. For each tree, site likelihoods for each gene were calculated using Tree-Puzzle 5.2. Using these site likelihoods, AU tests were performed using CONSEL 0.1 (Shimodaira and Hasegawa 2001), with default scaling and replicate values.
Statistical uncertainty of phylogenetic estimates was assessed by ML bootstrapping. One-hundred and two bootstrap replicates were generated with partitioned resampling, such that each gene contributed its original number of sites to each replicate (implemented using SEQBOOT and a perl script: b3boot.pl). Each replicate was examined using MrBayes with the same model and parameters as above except that the MCMC analysis was run for 2 × 105 generations, with 1.5 × 105 generations burn-in (trials showed that >90% of runs stabilized in regions of at least local parameter optimality within this period). For each bootstrap replicate, three independent runs from different random starting trees were performed, and the tree of highest posterior probability from the run with the highest harmonic mean likelihood was selected as an approximation of the ML tree (in other words, a Bayesian analysis was used as an ML estimator for each bootstrapped data set). Even with multiple runs there was probably a larger than normal amount of semirandom phylogenetic error associated with each bootstrap replicate due to incomplete convergence to global optima—thus the bootstrap values for nodes should perhaps be considered somewhat “conservative.” This bootstrap analysis took several processor-months to complete.
Single-Gene Jackknifing
To assess whether a discordant signal from any one gene was having a strong effect on our results, we excluded each of the six genes in turn and ran fast 200 or 500 replicate ML bootstrap analyses under the linked model, as described above (for logistical reasons a parallel unlinked analysis was not performed). As reported below, substantial changes in the bootstrap support for important groups were observed only when one particular gene was excluded—α-tubulin. Consequently, the complete array of linked and unlinked analyses described above was repeated with α-tubulin removed, including AU tests and bootstrap analysis (105 replicates) under the unlinked model.
Additional Taxa
After the main analyses described here were performed, additional data become available from some major taxa not included in our original analysis, notably, chlorarachniophytes (Rhizaria: Cercozoa) and cryptophytes (Harper, Waanders, and Keeling 2005). To test whether this new data affected our inferences, we constructed a new data set containing additional taxa as follows: the basidiomycete fungus Cryptococcus (all genes), the cyanidialean red alga Cyanidioschyzon (all genes), a composite cryptophyte taxon (with all genes except EF-2), the chlorarchniophyte Bigelowiella, the dinoflagellate Heterocapsa, and the raphidophycean stramenopile Heterosigma (the latter three all missing both EF-1α and EF-2). For the new data set, a 200 replicate bootstrap analysis under the linked model was performed, as above. Our original inferences were largely robust to the inclusion of these additional taxa, except that the position of Bigelowiella was unstable (a substantial minority of bootstrap replicates united Bigelowiella and Reclinomonas). Therefore, the linked model bootstrap analysis was repeated with Bigelowiella removed and also with EF-1α and EF-2 excluded, both with and without Bigelowiella. In addition, we compared several plausible trees where Bigelowiella either formed a clade with Reclinomonas or did not under an unlinked model, by likelihood ratios and an AU test, as described above. For logistical reasons we did not repeat the full ML analysis under the unlinked model.
Results
Analysis of the Complete Data Set
The linked and unlinked analyses give very similar optimal trees (fig. 1), representing a broadly reasonable view of eukaryotic phylogeny, as recovered in recent multigene analyses (Baldauf et al. 2000; Bapteste et al. 2002; Lang et al. 2002; Philippe et al. 2004). Animals and choanoflagellates are sister taxa and are strongly united with fungi to form the opisthokonts. Dictyostelium and Entamoeba form a clade, consistent with the proposed Amoebozoa supergroup (Cavalier-Smith 1998). We also recover a very strongly supported relationship between alveolates (ciliates, dinoflagellates, and apicomplexans) and stramenopiles, represented by a diatom and an oomycete. Land plants plus green algae (Viridiplantae) are specifically related to red algae (rhodophytes), including the cryptophyte nucleomorph genome, consistent with a larger “Plantae” clade. However, when cryptophyte nuclear genes were included, these branched as the immediate sister to Viridiplantae, interrupting the monophyly of Plantae (linked model, see Supplementary Material online).
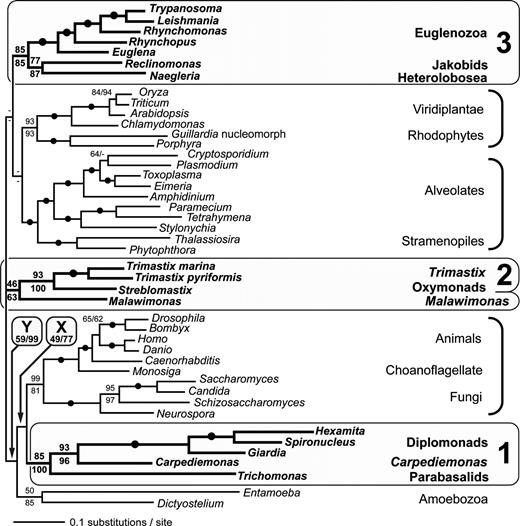
ML phylogenetic tree of eukaryotes inferred from six slowly evolving nuclear-encoded proteins. Best topology under unlinked model shown (i.e., with gene-specific branch lengths). Numbers on branches represent ML bootstrap support values for the unlinked model (upper numbers) and linked model (lower numbers). Filled circles represent bipartitions receiving >95% support with both methods. Dashes represent values <50% not critical to the study. Excavates are identified by gray shading. “1” “2,” and “3” indicate well-supported excavate groups. Note that Malawimonas is uncertainly placed. “X” and “Y” denote better-supported bipartitions that separate Group 1 from other excavates.
Within this background tree, both linked and unlinked analyses place all excavates except Malawimonas in three distinct and strongly supported groups, labeled “1” “2,” and “3” in figure 1.
Excavate Group 1 includes diplomonads, Carpediemonas, and the parabasalid Trichomonas. Diplomonads are most closely related to Carpediemonas, with very strong support, with parabasalids as their sister group. Excavate Group 1 receives strong bootstrap support with both methods (85%, 100%). The group remains strongly supported when diplomonads are excluded (97% bootstrap support with the linked model—tree not shown), so the high support is not due to artificial attraction specifically between parabasalids and the long-branching diplomonads.
Excavate Group 2 unites oxymonads and the two Trimastix spp. Bootstrap support is very strong with both phylogenetic methods employed. This assemblage corresponds to the taxon Preaxostyla (Simpson 2003).
Excavate Group 3 unites the evolutionarily important jakobids (represented by Reclinomonas) with two well-known protist groups—Euglenozoa (which includes the sleeping sickness and Chagas' disease parasites, as well as the model alga Euglena) and Heterolobosea (e.g., Naegleria). Bootstrap support is strong with both methods (85%). With one exception, alternative trees where Excavate Group 3 is not monophyletic confer markedly less likelihood on the data (unlinked model: Δln L > 35) and, where tested, are rejected by AU tests (P < 0.005). The single unrejected tree (Δln L = 25.5; P = 0.174) adds the uncertainly positioned Malawimonas to Excavate Group 3 as the sister of jakobids. Unexpectedly, we recover a specific relationship between Reclinomonas and the heteroloboseid Naegleria, interrupting the Discicristata grouping. This jakobids plus Heterolobosea clade (JH) receives strong bootstrap support (77%/87%), although alternative relationships within Excavate Group 3 are not rejected by AU tests.
The position of Malawimonas is unresolved. In the unlinked analysis, Malawimonas falls as the sister to Excavate Group 2 (fig. 1), while the ML tree from the linked analysis places Malawimonas at the base of Excavate Group 3 (not shown). Both positions receive only weak bootstrap support under either of the evolutionary models.
In all of our analyses, Excavate Groups 2 and 3 plus Malawimonas are separated by one or two internal branches, which always receive weak bootstrap support (≪50%). These bipartitions still show <50% bootstrap support if the taxon Malawimonas is pruned from the bootstrap trees after phylogenetic estimation, indicating that the weak support is not merely due to the uncertain position of Malawimonas. Excavate Group 1, however, always branches within an opisthokont-Amoebozoa clade, as the sister to opisthokonts, and is therefore separated from Excavate Group 2 by two branches, labeled “X” and “Y” in figure 1. These branches receive strong bootstrap support in the linked analysis (X: 77%; Y: 99%). They receive weaker support in the unlinked analysis (X: 49%; Y: 59%), due partly to the more uncertain position of Entamoeba. In fact, all examined trees in which excavates are constrained to be monophyletic are significantly worse explanations of the data under the unlinked model (Δln L > 100) and are rejected by AU tests at low α levels (P < 0.005).
We performed an abbreviated linked analysis including several phylogenetically important taxa that became available after we had begun the computationally intensive linked analysis. The excavate clades described above and their statistical support are essentially unaffected by the inclusion of these new data, except that the bootstrap support for Excavate Group 3 and for its subclade “JH” both decline to 40%–50% (see Supplementary Material online). At issue is the position of the chlorarachniophyte Bigelowiella because (1) a substantial minority of bootstrap replicates (33%) unite Bigelowiella and the jakobid Reclinomonas, and (2) when Bigelowiella is excluded, bootstrap support is reasonably high for both Excavate Group 3 and “heteroloboseids plus jakobids” (78%/80%). Bigelowiella is unusual within the data set because it is taxonomically very isolated (it is the only member of the supergroup Rhizaria that could be included) yet includes data from just four of the six studied genes and 55% of sites. Unexpectedly, excluding the two genes for which there are no Bigelowiella data increases dramatically the bootstrap support for the heteroloboseid-jakobid clade (76%, only marginally lower than that seen when Bigelowiella is excluded from this data set—86%). We suspect that the substantial attraction between Reclinomonas and Bigelowiella particular to the six-gene data set might be an artefact related to the problem of estimating a single branch length across all genes under the linked model. Consistent with this hypothesis, a relationship between Bigelowiella and Reclinomonas is associated with a relatively low likelihood under the unlinked model (39.4 ln L worse than the best plausible tree examined) and is rejected by an AU test under this model (largest P = 0.026).
Single-Gene Jackknifing
In order to examine the contributions of different genes to our tree, we removed every individual gene in turn from the six-gene data set and compared the bootstrap support for important bipartitions. We reasoned that modest reductions in support for a given bipartition would suggest an additive phylogenetic signal from multiple genes. On the other hand, large reductions may indicate that the support for a bipartition is concentrated in a single gene and might result from a gene-specific phylogenetic artefact or nonstandard evolution history (e.g., lateral gene transfer). In general, there are only modest changes in the support for Excavate Groups 1, 2, and 3 and for the grouping of JH, suggesting that the signals for these clades are contributed by multiple genes (table 1, columns 1–4). However, when α-tubulin is excluded, support for the association of Excavate Group 1 with opisthokonts (bipartition X) decreases from 77% to just 16%. Support also falls for the (Group 1, opisthokonts, and Amoebozoa) clade—“bipartition Y” (table 1, columns 5 and 6). This indicates that α-tubulin alone contributes the bulk of the signal placing Excavate Group 1 specifically with opisthokonts.
Bootstrap Support for Important Groups (linked model), When Individual Genes Excluded from the Analysis (single-gene jackknifing)
Excluded | Number of Sites | 1 | 2 | 3 | JH | X | Y |
|---|---|---|---|---|---|---|---|
| None | 3,142 | 100 | 100 | 85 | 87 | 77 | 99 |
| Tub-α | 2,721 | 95 | 99 | 79 | 82 | 16a | 66a |
| Tub-β | 2,717 | 99 | 99 | 71 | 73 | 82 | 94 |
| EF-1α | 2,734 | 100 | 91 | 77 | 81 | 80 | 99 |
| EF-2 | 2,398 | 100 | 100 | 69 | 70 | 69 | 96 |
| HSP70 | 2,584 | 97 | 92 | 67 | 71 | 64 | 96 |
| HSP90 | 2,556 | 100 | 97 | 70 | 84 | 83 | 95 |
Excluded | Number of Sites | 1 | 2 | 3 | JH | X | Y |
|---|---|---|---|---|---|---|---|
| None | 3,142 | 100 | 100 | 85 | 87 | 77 | 99 |
| Tub-α | 2,721 | 95 | 99 | 79 | 82 | 16a | 66a |
| Tub-β | 2,717 | 99 | 99 | 71 | 73 | 82 | 94 |
| EF-1α | 2,734 | 100 | 91 | 77 | 81 | 80 | 99 |
| EF-2 | 2,398 | 100 | 100 | 69 | 70 | 69 | 96 |
| HSP70 | 2,584 | 97 | 92 | 67 | 71 | 64 | 96 |
| HSP90 | 2,556 | 100 | 97 | 70 | 84 | 83 | 95 |
NOTE.—Groups 1, 2, and 3 are major clades of excavates. JH represents the clade of jakobids and Heterolobosea. X and Y unite Excavate Group 1 with opisthokonts, and with opisthokonts and Amoebozoa (see fig. 1).
Note the large reduction in support for X and Y specifically when α-tubulin is omitted.
Bootstrap Support for Important Groups (linked model), When Individual Genes Excluded from the Analysis (single-gene jackknifing)
Excluded | Number of Sites | 1 | 2 | 3 | JH | X | Y |
|---|---|---|---|---|---|---|---|
| None | 3,142 | 100 | 100 | 85 | 87 | 77 | 99 |
| Tub-α | 2,721 | 95 | 99 | 79 | 82 | 16a | 66a |
| Tub-β | 2,717 | 99 | 99 | 71 | 73 | 82 | 94 |
| EF-1α | 2,734 | 100 | 91 | 77 | 81 | 80 | 99 |
| EF-2 | 2,398 | 100 | 100 | 69 | 70 | 69 | 96 |
| HSP70 | 2,584 | 97 | 92 | 67 | 71 | 64 | 96 |
| HSP90 | 2,556 | 100 | 97 | 70 | 84 | 83 | 95 |
Excluded | Number of Sites | 1 | 2 | 3 | JH | X | Y |
|---|---|---|---|---|---|---|---|
| None | 3,142 | 100 | 100 | 85 | 87 | 77 | 99 |
| Tub-α | 2,721 | 95 | 99 | 79 | 82 | 16a | 66a |
| Tub-β | 2,717 | 99 | 99 | 71 | 73 | 82 | 94 |
| EF-1α | 2,734 | 100 | 91 | 77 | 81 | 80 | 99 |
| EF-2 | 2,398 | 100 | 100 | 69 | 70 | 69 | 96 |
| HSP70 | 2,584 | 97 | 92 | 67 | 71 | 64 | 96 |
| HSP90 | 2,556 | 100 | 97 | 70 | 84 | 83 | 95 |
NOTE.—Groups 1, 2, and 3 are major clades of excavates. JH represents the clade of jakobids and Heterolobosea. X and Y unite Excavate Group 1 with opisthokonts, and with opisthokonts and Amoebozoa (see fig. 1).
Note the large reduction in support for X and Y specifically when α-tubulin is omitted.
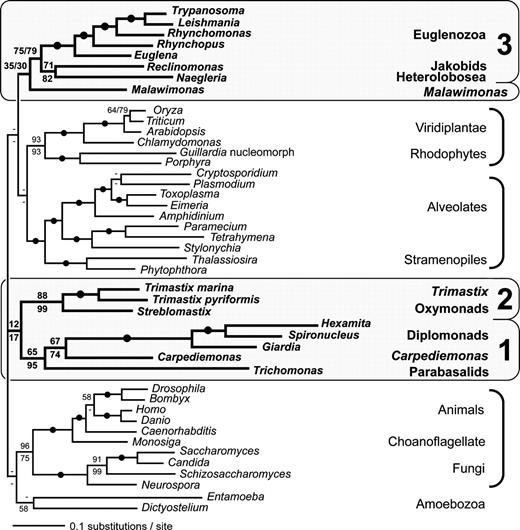
We subsequently repeated the complete ML analysis with α-tubulin omitted (fig. 2). The linked and unlinked ML trees from these analyses are similar to those from the full data set, with one important exception—there are no excavate groups within the opisthokont-Amoebozoa clade. In fact, Excavate Group 1 now branches as the specific sister to Excavate Group 2, albeit with very weak bootstrap support (12/17% or 13/27% if the destabilizing taxon Entamoeba is pruned). After exclusion of α-tubulin, some trees in which excavates are monophyletic are not rejected in AU tests at a 0.05 α level (Δln L = 24, P = 0.141).
Discussion
A Multigene, ML Examination of Excavate Evolution
This study is the first comprehensive multigene analysis of excavate phylogeny. Some previous analyses included a good sampling of excavates but used only one or two molecular markers, usually just SSU rRNA sequences (Simpson et al. 2002b; Cavalier-Smith 2003; Keeling and Leander 2003; Simpson 2003; Nikolaev et al. 2004). It is essential to verify these results by using larger multigene data sets because the phylogenetic estimates from single molecular markers are often poorly resolved (e.g., different analyses of the same gene give markedly different phylogenetic estimates) and, in a worst-case scenario, can be positively misleading. Independent data sets that can verify SSU rRNA analyses are doubly important as eukaryotic SSU rRNAs show considerable length variation in many regions along the sequence. This renders both the alignment itself and the selection of “unambiguously aligned sites” for analysis controversial and potentially influenced by the prior phylogenetic beliefs of the researcher. By contrast, the protein sequences examined here display little length variation, making alignment and site selection trivial concerns. Other recent analyses include data from several-to-many protein-coding genes but include many fewer (2–5) of the 10 excavate groups currently recognized (Baldauf et al. 2000; Bapteste et al. 2002; Lang et al. 2002; Arisue, Hasegawa, and Hashimoto 2005; Harper, Waanders, and Keeling 2005). Such analyses may give misleading pictures of the evolution of excavate eukaryotes, even if the phylogenetic trees reconstructed are topologically correct.
In this analysis, we assess the robustness of our trees using nonparametric bootstrapping. This contrasts with some recent studies of deep eukaryotic phylogeny where Bayesian posterior probabilities are used as the primary measure of robustness when complex (computationally intensive) evolutionary models are employed (Stiller and Hall 2002; Yoon, Hackett, and Bhattacharya 2002; Nikolaev et al. 2004). While they measure different properties, posterior probabilities are routinely much less conservative than bootstrap proportions and are more prone to give strong support for incorrect bipartitions when the evolutionary model is misspecified (Suzuki, Glazko, and Nei 2002; Cummings et al. 2003; Douady et al. 2003; Erixon et al. 2003). Furthermore, there is intrinsic serial correlation in trees and parameters explored during the MCMC analysis, and convergence is difficult to assess. Bayesian analyses can stabilize in locally optimal, rather than globally optimal, regions of parameter space, giving the potential for catastrophic inaccuracy if convergence is not, or cannot be, verified (this possibility is illustrated by intermediate steps in our unlinked analyses, where initially identical long MCMC runs started from different random trees estimated posterior probabilities of 0 and 1 for the same bipartition—data not shown). Bootstrap resamplings are intrinsically independent and, with the number of bootstrap replicates routinely examined in phylogenetic analyses (rarely <50), will not be subject to the same possibility of catastrophe (for a given tree-searching strategy). For all of these reasons, we consider strong bootstrap values as more reliable indication of a well-supported grouping than very high posterior probabilities.
The Evolutionary Position of Jakobids
Our study provides the first robust indication of the evolutionary position of jakobids—they are close relatives of Heterolobosea and Euglenozoa. Previous studies of tubulins and CCTα proteins and some recent analyses of SSU rRNA genes have hinted at this relationship, but the grouping has usually received very weak statistical support (Edgcomb et al. 2001; Archibald, O'Kelly, and Doolittle 2002; Simpson et al. 2002b; Cavalier-Smith 2003, 2004; Nikolaev et al. 2004). In our best estimate, jakobids are actually specifically related to Heterolobosea. This result conflicts with well-sampled SSU rRNA trees, which usually group Heterolobosea and Euglenozoa to the exclusion of jakobids (Cavalier-Smith 2003, 2004; Simpson 2003; Berney, Fahrni, and Pawlowski 2004; Nikolaev et al. 2004). Euglenozoa and Heterolobosea have highly divergent SSU rRNA sequences, and it is plausible that their grouping in SSU rRNA trees could be a long-branch attraction artefact. By contrast, none of Euglenozoa, Heterolobosea, or jakobids are particularly long branches (or otherwise remarkable) in our analysis. Further, multigene studies are required to definitively resolve the exact branching pattern between Euglenozoa, Heterolobosea, and jakobids, and these should incorporate an improved taxon sampling of the latter two groups. In fact, we recover the same basic jakobid-heteroloboseid clade in preliminary multiprotein analyses that include additional jakobid taxa (not shown—A. G. B. Simpson, unpublished data).
Historically, mitochondrial cristae have been the single most important morphological character for deep eukaryote phylogeny (Taylor 1976; Patterson 1994). Heterolobosea and Euglenozoa have unusual “discoidal” mitochondrial cristae. This shared character was central in uniting these two groups as the taxon Discicristata, along with gene phylogenies that did not include jakobids (Keeling and Doolittle 1996; Cavalier-Smith 1998; Baldauf et al. 2000; Baldauf 2003). By contrast, jakobids have tubular or flattened cristae (O'Kelly 1993)—the most common forms in eukaryotes. In light of our results, it is possible that discoidal cristae evolved independently in Heterolobosea and Euglenozoa. Alternatively, because Malawimonas also has discoidal cristae (O'Kelly and Nerad 1999), it is not impossible that discoidal cristae were ancestral for all excavates and thus appeared earlier than the last common ancestor of Euglenozoa and Heterolobosea (even if Euglenozoa and Heterolobosea were found to be sister taxa to the exclusion of jakobids). Either way, on both phylogenetic and morphological grounds, the current widely accepted concept of the supergroup Discicristata is open to dispute and could well be untenable.
Implications for Mitochondrial RNA Polymerase Evolution
The specific relationship between jakobids, Heterolobosea, and Euglenozoa has important implications for proposals that jakobids represent primitive eukaryotes. While jakobids have some bacterial-type RNA polymerase subunits encoded by their mitochondrial genomes (Lang et al. 1997; Gray et al. 2004), both Heterolobosea and Euglenozoa are known to have standard eukaryotic viral-type mitochondrial RNA polymerases encoded by their nuclear genomes (Cermakian et al. 1996; Clement and Koslowsky 2001). The jakobid bacterial-type RNA polymerase can be considered a uniquely primitive character only if the root of the eukaryotic tree lies exactly on the jakobid branch. This rooting position would imply that “Excavate Group 3” cladistically includes all other living eukaryotes. If the placement of jakobids in our ML topology is accurate, it would also imply that Euglenozoa and Heterolobosea are more distantly related than are animals and plants, for example. Because a close relationship between Euglenozoa and Heterolobosea is now widely accepted, this would constitute a major upheaval of the established tree of eukaryotes.
There are several evolutionary scenarios that might account for the distribution of mitochondrial RNA polymerases in eukaryotes without uprooting the entire eukaryotic tree. All of them are complex or invoke apparently rare or dramatic evolutionary events. Firstly, the last common ancestor of eukaryotes may have had both viral- and bacterial-type mitochondrial RNA polymerases, which were then differentially lost in various eukaryotic lineages (Stechmann and Cavalier-Smith 2002). However, if jakobids are deeply nested within other eukaryotes, several independent losses of the bacterial type would have to be inferred, unless some extant eukaryotes still carry both forms (this has yet to be documented). Secondly, the bacterial type alone might be ancestral for living eukaryotes, with the viral-type in Euglenozoa and Heterolobosea being acquired much later by lateral gene transfers from other eukaryotes, or perhaps viruses or plasmids. Again, if jakobids and Heterolobosea are specifically related, two independent transfers (at the very least) would be required. Finally, the viral-type polymerase might be ancestral for all eukaryotes, with the bacterial type representing a more recent lateral transfer from a prokaryote into the mitochondrial genome of an ancestral jakobid. While mitochondria are overwhelmingly viewed as gene donors rather than gene recipients (Adams and Palmer 2003; Burger, Gray, and Lang 2003), the probable transfer of apparently functional genes into mitochondrial genomes has now been documented in land plants, fungi, and cnidarians (Paquin, Laforest, and Lang 1994; Pont-Kingdon et al. 1998; Bergthorsson et al. 2003, 2004; Davis and Wurdack 2004). In some land plants, the transferred gene did not directly supplant an existing mitochondrial gene but instead replaced (or exists in concert with) a gene that has long since been transferred to the nucleus in the host lineage (Bergthorsson et al. 2003). This latter situation is most closely analogous to the scenario by which jakobid mitochondrial RNA polymerase might have been acquired by lateral transfer.
Other Excavate Groups
Our multigene analyses confirm some relationships among other excavates suggested by earlier single-gene analyses. We recovered a strong specific relationship between Trimastix and oxymonads, previously inferred only from SSU rRNA trees (Dacks et al. 2001; Simpson et al. 2002b; Keeling and Leander 2003). We also confirm a close relationship between diplomonads and the obscure free-living amitochondriate organism Carpediemonas (Simpson, MacQuarrie, and Roger 2002; Simpson et al. 2002b). Most interestingly, we recovered a specific relationship between diplomonads, Carpediemonas, and parabasalids with high support. This latter result bridges the gap between two classes of prior phylogenetic studies. (1) Several protein analyses unite diplomonads and parabasalids but do not include any other excavates except Euglenozoa and Heterolobosea and, in one instance, oxymonads (Embley and Hirt 1998; Baldauf et al. 2000; Arisue, Hasegawa, and Hashimoto 2005; Harper, Waanders, and Keeling 2005). (2) Some recent excavate-rich SSU rRNA and tubulin analyses show a specific relationship between parabasalids and the total diplomonad-Carpediemonas clade, usually with weak support (Simpson et al. 2002b; Cavalier-Smith 2003; Keeling and Leander 2003; Simpson 2003). It is also consistent with recent evidence that common ancestors of diplomonads and parabasalids acquired at least two genes by lateral transfer (Henze et al. 2001; Andersson, Sarchfield, and Roger 2005).
Finding relatives of diplomonads and parabasalids has been a long-standing problem. Our six-gene analysis locates Excavate Group 1, including diplomonads and parabasalids, as the specific relatives of opisthokonts. This position is suspicious because it interrupts the association of opisthokonts and Amoebozoa, a grouping for which there is increasing evidence from other analyses and data (Baldauf et al. 2000; Bapteste et al. 2002). However, this placement of Excavate Group 1 is due largely to a “conflicting signal” of uncertain origin from just one gene (α-tubulin). In fact, when α-tubulin is excluded, our best trees place Excavate Groups 1 and 2 together. This basic relationship has been recovered (with extremely weak support) in a small minority of SSU rRNA gene trees (Simpson et al. 2002b; Cavalier-Smith 2003; Berney, Fahrni, and Pawlowski 2004; Cavalier-Smith 2004). Interestingly, all lineages in Excavate Groups 1 and 2 are anaerobes that lack classical aerobic mitochondria, hinting that they may derive from a common ancestor that had already lost aerobic mitochondrial functions (Cavalier-Smith 2003; Simpson and Roger 2004). We refer to this as the “neoarchezoa hypothesis” (Simpson and Roger 2004).
Very recently, Hampl et al. (2005) presented a multigene analysis including several excavate groups—diplomonads and parabasalids (Excavate Group 1), Euglenozoa, and most interestingly, an oxymonad (representing our Excavate Group 2). Their analyses examined just under half as many taxa as our study but included more genes (up to nine total). As in our study, they identified a particularly strong incongruity between α-tubulin and the “majority” phylogenetic signal with respect to the placement of diplomonads and parabasalids. With or without this data, they also recovered a specific relationship between Excavate Groups 1 and 2 but with quite strong ML bootstrap support under a linked model (and posterior probability 1 under an unlinked model).
Excavate Monophyly and Estimating the Eukaryote Tree
It has been argued that all excavates from a monophyletic supergroup of eukaryotes—Excavata—largely on the basis of morphological data (Cavalier-Smith 2002; Simpson et al. 2002a; Simpson 2003). Once the aberrant signal from α-tubulin is excluded, our analysis neither supports nor statistically rejects the monophyly of all excavate groups. This mirrors the results from recent excavate-rich SSU rRNA analyses where certain taxon and alignment combinations yield a monophyletic excavate assemblage with almost no statistical support (Cavalier-Smith 2003; Nikolaev et al. 2004), while other analyses do not recover excavate monophyly but are unable to reject it either (Simpson et al. 2002b; Simpson 2003). If excavates are monophyletic, there seems to be little phylogenetic signal indicating that this remaining in molecular sequences. Considerably more data, perhaps a hundred or more genes, from an appropriate sample of excavates will be required to better examine the excavate monophyly. Unfortunately, most of the best studied excavates (e.g., Giardia, Trichomonas, and trypanosomatids) are among the worst “long branching taxa” in the entire eukaryotic tree. It will be important to ensure that relationships between excavates recovered in phylogenomic multigene analyses are due to authentic historical signal rather than analysis artefact (Sullivan and Swofford 2001). The inclusion of lesser known but shorter-branching excavates in larger multigene analysis could reduce the chance of phylogenetic artifact, either by breaking long branches or by acting as surrogates for related long-branch taxa which could then be excluded from consideration. The latter strategy may have improved phylogenetic accuracy in some single-gene analyses involving excavates (Simpson et al. 2002b; Cavalier-Smith 2003; Nikolaev et al. 2004).
Ultimately, we will need to examine directly the positions of the major excavate groups relative to the root of the eukaryotic tree. Perhaps, the best evidence pinpointing the placement of the eukaryotic root are the phylogenetic distributions of complex molecular characters, namely, fused dihydrofolate reductase and thymidylate synthase (DHFR-TS) genes and a three-enzyme fusion in the pyrimidine biosynthesis pathway (Stechmann and Cavalier-Smith 2002, 2003). Unfortunately, DHFR and TS genes are missing altogether in some critical excavates (e.g., Giardia), while recent evidence indicates that the pyrimidine biosynthesis enzyme fusion has a complex evolutionary history (Arisue, Hasegawa, and Hashimoto 2005), making these data hard to interpret at present, especially with respect to placement of excavates. Analysis where eukaryotes are rooted by outgroups represent a more traditional avenue, however, sophisticated multigene analyses including a few excavate groups are strongly suspected to be affected by analysis artefact (Bapteste et al. 2002; Arisue, Hasegawa, and Hashimoto 2005). Trees of genes universal to eukaryotes almost invariably exhibit a very long internal branch joining the eukaryote clade to other sequences, while the deep internal branches within eukaryotes are relatively short. Under these conditions, analysis artefact can overwhelm historical signal irrespective of the amount of data (Philippe et al. 2000, 2004). For instance, there are often distinctly different patterns of evolutionary rates at sites across a gene (“covarion shifts”) between eukaryotes and other sequences (Inagaki et al. 2004). Evolutionary models currently used for phylogenetic reconstruction do not model covarion shifts, making these a difficult-to-counteract source of artefact. The impact of a covarion shift could be reduced by exclusion of alignment positions that differ substantially in evolutionary rate across a particular pair of subtrees (Inagaki et al. 2004). However, for a reliable estimate of the eukaryotic root, new models that can account for covarion shifts will be indispensable.
Martin Embley, Associate Editor
The authors thank E. Susko (Dalhousie University) for discussions, J. D. Silberman (University of Arkansas) for Trimastix pyriformis cDNA, T. Hashimoto (University of Tsukuba) for sharing data prior to publication, J. Leigh (Dalhousie) for two Python scripts, and C. Blouin (Dalhousie) for supplementary computational resources. A.G.B.S. is supported as a scholar of the Canadian Institute for Advanced Research (CIAR). A.J.R. is supported as a fellow of the CIAR, by a New Investigator Salary Award from the Canadian Institutes for Health Research/Peter Lougheed foundation, and the Alfred P. Sloan foundation. Y.I. is supported by an institutional grant from Nagahama Institute of Bioscience and Technology. The research was supported by Natural Sciences and Engineering Research Council of Canada grant 227085-00 to A.J.R. Computational resources were funded by the “Prokaryotic Genome Evolution and Diversity” Genome Atlantic/Genome Canada large-scale project. Some sequences from Phytothphora sojae, Thalassiosira pseudonana, and Chlamydomonas reinhardtii were derived from genome sequence data from the Joint Genome Institute (Calif.). Some sequences for Eimeria tenella were derived from an in progress genome sequencing project at the Sanger Institute (Cambridge, United Kingdom).
References
Adams, K. L., and J. D. Palmer.
Andersson, J. O., S. W. Sarchfield, and A. J. Roger.
Archibald, J. M., C. J. O'Kelly, and W. F. Doolittle.
Arisue, N., M. Hasegawa, and T. Hashimoto.
Baldauf, S. L., A. J. Roger, I. Wenk-Siefert, and W. F. Doolittle.
Bapteste, E., H. Brinkmann, J. A. Lee et al. (11 co-authors).
Bergthorsson, U., K. L. Adams, B. Thomason, and J. D. Palmer.
Bergthorsson, U., A. O. Richardson, G. J. Young, L. R. Goertzen, and J. D. Palmer.
Berney, C., J. F. Fahrni, and J. Pawlowski.
Burger, G., M. W. Gray, and B. F. Lang.
———.
———.
Cermakian, N., T. M. Ikeda, R. Cedergren, and M. W. Gray.
Chihade, J. W., J. R. Brown, P. R. Schimmel, and L. Ribas de Pouplana.
Clark, C. G., and L. S. Diamond.
Clement, S. L., and D. J. Koslowsky.
Cummings, M. P., S. A. Handley, D. S. Myers, D. L. Reed, A. Rokas, and K. Winka.
Dacks, J. B., J. D. Silberman, A. G. B. Simpson, S. Moruya, T. Kudo, M. Ohkuma, and R. Redfield.
Davis, C. C., and K. J. Wurdack.
Douady, C. J., F. Delsuc, Y. Boucher, W. F. Doolittle, and E. J. P. Douzery.
Dyall, S. D., D. M. Brown, and P. J. Johnson.
Edgcomb, V. P., A. J. Roger, A. G. B. Simpson, D. Kysela, and M. L. Sogin.
Erixon, P., B. Svennblad, T. Britton, and B. Oxelman.
Felsenstein, J.
Gillin, F. D., D. S. Reiner, and J. M. McCaffery.
Gray, M. W., B. F. Lang, and G. Burger.
Guindon, S., and O. Gascuel.
Hampl, V., D. S. Horner, P. Dyal, J. Kulda, J. Flegr, P. Foster, and T. M. Embley.
Harper, J. T., E. Waanders, and P. J. Keeling.
Hashimoto, T., Y. Nakamura, F. Nakamura, T. Shirakura, J. Adachi, N. Goto, K. Okamoto, and M. Hasegawa.
Henze, K., D. S. Horner, S. Suguri, D. V. Moore, L. B. Sanchez, M. Müller, and T. M. Embley.
Inagaki, Y., E. Susko, N. M. Fast, and A. J. Roger.
Keeling, P. J., and W. F. Doolittle.
Keeling, P. J., and B. S. Leander.
Lang, B. F., G. Burger, C. J. O'Kelly, R. Cedergren, G. B. Golding, C. Lemieux, D. Sankoff, M. Turmel, and M. W. Gray.
Lang, B. F., C. J. O'Kelly, T. A. Nerad, M. W. Gray, and G. Burger.
McArthur, A., H. Morrison, J. Nixon et al.
Nikolaev, S. I., C. Berney, J. F. Fahrni, I. Bolivar, S. Polet, A. P. Mylnikov, V. V. Aleshin, N. B. Petrov, and J. Pawlowski.
Nylander, J. A. A., F. Ronquist, J. P. Huelsenbeck, and J. L. Nieves-Aldrey.
O'Kelly, C. J.
O'Kelly, C. J., and T. A. Nerad.
Paquin, B., M.-J. Laforest, and B. F. Lang.
Patterson, D. J.
Philippe, H., P. Lopez, H. Brinkmann, K. Budin, A. Germot, J. Laurent, D. Moreira, M. Müller, and H. Le Guyader.
Philippe, H., E. A. Snell, E. Bapteste, P. Lopez, P. W. H. Holland, and D. Casane.
Pont-Kingdon, G., N. A. Okada, J. L. Macfarlane, C. T. Beagley, C. D. Watkins-Sims, T. Cavalier-Smith, G. D. Clark-Walker, and D. R. Wolstenholme.
Pupko, T., D. Huchon, Y. Cao, N. Okada, and M. Hasegawa.
Ronquist, F., and J. P. Huelsenbeck.
Schmidt, H. A., K. Strimmer, M. Vingron, and A. von Haeseler.
Shimodaira, H.
Shimodaira, H., and M. Hasegawa.
Silberman, J. D., A. G. B. Simpson, J. Kulda, I. Cepicka, V. Hampl, P. J. Johnson, and A. J. Roger.
Simpson, A. G. B.
Simpson, A. G. B., J. Lukeš, and A. J. Roger.
Simpson, A. G. B., E. K. MacQuarrie, and A. J. Roger.
Simpson, A. G. B., and D. J. Patterson.
Simpson, A. G. B., R. Radek, J. B. Dacks, and C. J. O'Kelly.
Simpson, A. G. B., and A. J. Roger.
Simpson, A. G. B., A. J. Roger, J. D. Silberman, D. Leipe, V. P. Edgcomb, L. S. Jermiin, D. J. Patterson, and M. L. Sogin.
Sogin, M. L.
Sogin, M. L., J. H. Gunderson, H. J. Elwood, R. A. Alonso, and D. A. Peattie.
Stechmann, A., and T. Cavalier-Smith.
Stiller, J. W., and B. D. Hall.
Sullivan, J., and D. L. Swofford.
Suzuki, Y., G. V. Glazko, and M. Nei.
Tovar, J., G. Leon-Avila, L. B. Sanchez, R. Sutak, J. Tachezy, M. Van Der Giezen, M. Hernandez, M. Muller, and J. M. Lucocq.
Yamamoto, A., T. Hashimoto, E. Asaga, M. Hasegawa, and N. Goto.
Author notes
*Canadian Institute for Advanced Research, Program in Evolutionary Biology, Dalhousie University, Halifax, Nova Scotia, Canada; †Department of Biology, Dalhousie University, Halifax, Nova Scotia, Canada; ‡Center for Computational Sciences and Institute of Biological Sciences, University of Tsukuba, Tsukuba, Ibaraki, Japan; and §Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, Nova Scotia, Canada

{kind=link}
{kind=link}