





Abstract
Gene expression varies widely in natural populations, yet the proximate and ultimate causes of this variation are poorly known. Understanding how variation in gene expression affects abiotic stress tolerance, fitness, and adaptation is central to the field of evolutionary genetics. We tested the hypothesis that genes with natural genetic variation in their expression responses to abiotic stress are likely to be involved in local adaptation to climate in Arabidopsis thaliana. Specifically, we compared genes with consistent expression responses to environmental stress (expression stress responsive, “eSR”) to genes with genetically variable responses to abiotic stress (expression genotype-by-environment interaction, “eGEI”). We found that on average genes that exhibited eGEI in response to drought or cold had greater polymorphism in promoter regions and stronger associations with climate than those of eSR genes or genomic controls. We also found that transcription factor binding sites known to respond to environmental stressors, especially abscisic acid responsive elements, showed significantly higher polymorphism in drought eGEI genes in comparison to eSR genes. By contrast, eSR genes tended to exhibit relatively greater pairwise haplotype sharing, lower promoter diversity, and fewer nonsynonymous polymorphisms, suggesting purifying selection or selective sweeps. Our results indicate that cis-regulatory evolution and genetic variation in stress responsive gene expression may be important mechanisms of local adaptation to climatic selective gradients.
Introduction
Across the geographic range of a species, populations typically vary in both genome sequence and gene expression. A central goal of evolutionary genetics is to determine the evolutionary drivers of sequence and expression polymorphism and how these affect organismal traits and fitness. Additionally, variants underlying expression polymorphism can be used to generate hypotheses about gene function for molecular study (Hughes et al. 2000; Jansen and Nap 2001; Rockman 2008; Kesari et al. 2012). Natural variation over the geographic range of a species is maintained partly by local adaptation, where directional selection favors different alleles under different environmental conditions (Kawecki and Ebert 2004; Hereford 2009). Local adaptation requires environment-dependent variation in fitness, which is represented quantitatively as a genotype-by-environment interaction, that is, fitness in a given location is dependent on the combination of allelic state and environmental conditions (Hancock et al. 2011; Ågren et al. 2013; Des Marais et al. 2013).
Arabidopsis thaliana (Brassicaceae; hereafter Arabidopsis) represents a unique opportunity to investigate ecological drivers of natural variation from molecular to continental scales (Mitchell-Olds 2001; Tonsor et al. 2005). Arabidopsis exhibits diverse transcriptional, physiological, and fitness responses to abiotic stress (Chaves et al. 2003; Hannah et al. 2006; Yamaguchi-Shinozaki and Shinozaki 2006; Korves et al. 2007; Des Marais et al. 2012; Richards et al. 2012). Common gardens and reciprocal transplants have revealed strong genotype-by-environment effects on fitness (Korves et al. 2007; Fournier-Level et al. 2011) and local adaptation (Ågren and Schemske 2012; Ågren et al. 2013). Climate varies extensively across the range of Arabidopsis (Hoffmann 2002) and temperature and precipitation gradients likely play a major role in local adaptation and natural variation (Fournier-Level et al. 2011; Hancock et al. 2011; Ågren and Schemske 2012; Lasky et al. 2012). Climate adaptation in Arabidopsis likely involves life history and phenological variation that mediates cold and drought responses (McKay et al. 2003; Korves et al. 2007; Lasky et al. 2012; Picó 2012). Recent studies of Arabidopsis have found strong associations between nonsynonymous genetic variation and local climates (Hancock et al. 2011; Lasky et al. 2012) in addition to genotype-by-climate interaction effects on fitness in common gardens (Fournier-Level et al. 2011; Hancock et al. 2011; Ågren and Schemske 2012). However, little is known about the proximal mechanisms that link polymorphisms to fitness variation along climatic gradients.
Abiotic stress responsive gene expression (e.g., drought responsive) is thought to play a central role in stress tolerance and can be an important mechanism of adaptive evolution (King and Wilson 1975; Ferea et al. 1999; Hamblin et al. 2002; Rockman et al. 2003; Lopez-Maury et al. 2008; Jones et al. 2012). Stress responsive gene expression is often conserved among populations, species, families, and kingdoms (Hannah et al. 2006; Des Marais et al. 2012; Rengel et al. 2012). These transcripts may be thought of as consistent expression stress responsive (eSR) genes and likely represent adaptive plastic responses to commonly encountered environmental challenges. As such, they are expected to experience strong positive or purifying selection.
In contrast, the expression response of many genes to stressful conditions depends strongly on natural genotypic variation (Des Marais et al. 2013). This naturally segregating variation in stress response, or expression genotype-by-environment interaction (eGEI), may underlie genotype-by-environment interaction effects on fitness and thus be a mechanism of local adaptation (Gibson and Weir 2005; Whitehead and Crawford 2006; Des Marais et al. 2012). For example, plasticity may increase fitness in variable environments, hence it is expected that gene expression plasticity will be more common in heterogeneous, unpredictable environments (Moran 1992; Alpert and Simms 2002; Sultan and Spencer 2002) whereas fixed phenotypes are favored in less variable environments (Levins 1968; Futuyma and Moreno 1988). However, there exist few direct empirical links between genome-wide expression variation, sequence variation, selective gradients, and fitness (Hannah et al. 2006; Hoekstra and Coyne 2007; Larsen et al. 2007). Genetic variation for expression plasticity can be caused by polymorphisms in cis-binding sites (transcription factor binding sites near a given gene) or trans-regulatory factors (transcription factors acting on distant genes) (Wray et al. 2003; Wray 2007; Wagner and Lynch 2008; Wittkopp and Kalay 2012), both of which may underlie phenotypic evolution (Riechmann et al. 2000; Yvert et al. 2003; Shapiro et al. 2004, 2006; Wittkopp et al. 2008). However, there is little known about the general genome-wide importance of cis-regulatory evolution in response to abiotic stressors. Here, we focused on testing for signatures of selection and adaptation in sequence variation near genes (i.e., cis-regulatory elements) with expressional response to abiotic stress.
Genome scans for loci exhibiting evidence of selection, loci associated with environmental gradients (Fournier-Level et al. 2011; Hancock et al. 2011; Jones et al. 2012; Lasky et al. 2012), and expression responses to abiotic stressors (Hannah et al. 2006; Des Marais et al. 2012) are complementary approaches to understanding environmental drivers of natural variation. Here, we bridge these approaches in a novel effort to elucidate the evolutionary processes shaping expression plasticity. To accomplish this goal, we analyze large data sets on natural variation in genomic sequence, climate of origin, field fitness, and expression response to abiotic stress. We use whole genome expression profiling of diverse genotypes to classify responsive transcripts into two categories, eSR and eGEI. We then test whether these categories tend to harbor different levels of genetic variation indicative of selection, using multiple lines of evidence: selection statistics, associations with climatic gradients, associations with fitness, and sequence diversity. Finally, we ask whether eSR and eGEI genes exhibit overall differences in the frequency and polymorphism of major stress response transcription factor binding motifs.
Results
Diverse Expression Responses to Abiotic Stress
We analyzed single nucleotide polymorphisms (SNPs) and natural variation in gene expression to test for evidence of local adaptation to climate. In two published experiments, 9 or 17 natural accessions were exposed to extended periods of cold (14 days at 4 °C) or drought (7 days drying to 40% of soil extractable water remaining), respectively, and gene expression, compared with controls, was quantified with Affymetrix ATH1 Genome arrays (Hannah et al. 2006; Des Marais et al. 2012). We studied two classes of genes: 1) Those exhibiting common (i.e., shared among genotypes) expression stress responses (eSR genes) and 2) those exhibiting genotype-by-environment interaction effects on expression (eGEI genes; fig. 1). Genes were assigned to expression classes based on two-way analyses of variance (ANOVAs), resulting in 9,305 drought eSR genes, 1,473 drought eGEI genes, 10,060 cold eSR genes, and 2,149 cold eGEI genes (out of 21,428 nuclear genes studied; supplementary figs. S1 and Supplementary Data, Supplementary Material online). Drought and cold eSR genes showed significant overlap (5,576 genes, χ2 test, P < 10−16). For the smaller class of eGEI genes, drought and cold showed near-significant overlap (168 genes, χ2 test, P = 0.07). We likely identified only a subset of eSR and eGEI genes because experiments were conducted across a limited range of environments and genotypes, and on a limited number of replicate individuals.
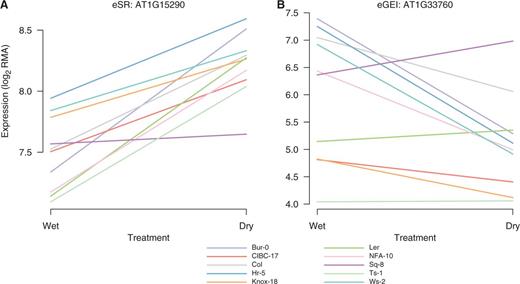
Representative eSR (A) and eGEI (B) expression responses to drought (Des Marais et al. 2012). (A) AT1G15290, a tetratricopeptide repeat-like superfamily protein, has a significant eSR (but not eGEI) effect in response to drought. The AT1G15290 promoter is characterized by an uncommonly high and invariant number of ABREs across 80 accessions. Additionally, AT1G15290 contains a relatively high number of SNPs with high PHS. (B) AT1G33760, a member of the DREB subfamily A-4 of the ERF/AP2 transcription factor family, has a significant eGEI effect in response to drought and is near (<1 kb distant) SNPs with outlier associations with survival in the United Kingdom and growing season precipitation. Additionally, ABRE counts in the AT1G33760 promoter showed relatively high variation across 80 accessions. The ten early-flowering accessions from the original experiment (Des Marais et al. 2012) are shown.
Population Genomic Signatures Suggest Selection on eSR and eGEI Genes
To evaluate evidence for selection, we asked whether eSR and eGEI genes were enriched for SNPs with statistical signatures of selection. We used four statistics calculated for 214,051 SNPs among 1,307 worldwide accessions, the first two of which were reported previously (Horton et al. 2012) and the other two of which we calculated in this study. First, we used pairwise haplotype sharing (PHS) as evidence for loci that are current or recent targets of selective sweeps (Toomajian et al. 2006). Second, we used composite likelihood ratio (CLR) tests to detect sequences that have swept to high frequency (Horton et al. 2012). Third, we calculated SNP minor allele frequency (MAF). High MAF suggests that polymorphism is maintained by selection whereas low MAF suggests the action of purifying selection or selective sweeps (Tajima 1989). Fourth, we calculated Fst among Eurasian accessions; high Fst is expected for loci involved in local adaptation, although here calculating Fst requires arbitrary designations of populations. We considered gene sets with a high frequency of extreme SNP selection statistics (in the 0.05 tail) as “enriched” (Segrè et al. 2010). We generated null expectations for gene set enrichment by permuting gene classifications as eSR or eGEI among genes on the microarray. In reporting enrichment test results, we refer to these permuted null distributions as “genomic controls.”
We found that both cold and drought eSR genes were significantly enriched with SNPs having higher PHS than that of genomic controls (permutation test, cold: z = 2.38, P = 0.0258, drought: z = 2.57, P = 0.0198), whereas eGEI genes showed no significant association with PHS (results summarized in table 1; see supplementary table S1, Supplementary Material online, for more detail). No gene set showed enrichment for high CLR (supplementary table S1, Supplementary Material online). Both cold and drought eSR genes were depleted of SNPs with high MAF compared with genomic controls (cold: z = −4.18, P < 0.0002, drought: z = −2.43, P = 0.0168) and compared with eGEI genes (cold: z = −3.10, P = 0.0022, drought: z = −2.62, P = 0.0106; supplementary table S1, Supplementary Material online). By contrast, drought eGEI genes had a significantly high proportion of SNPs with elevated MAF (z = 1.96, P = 0.0434) and cold eGEI genes had nonsignificantly elevated MAF (z = 1.66, P = 0.1002) compared with genomic controls. Drought eGEI genes were significantly enriched with high Fst compared with genomic controls (z = 2.46, P = 0.0142) and compared with eSR genes (z = −2.30, P = 0.0258), but other gene sets were not significantly enriched for Fst (supplementary table S1, Supplementary Material online). The enrichment of eSR genes with PHS and low MAF may indicate that many eSR genes have experienced partial or ongoing sweeps. By contrast, the relative lack of sweeps in eGEI genes, high MAF of eGEI genes, and the high Fst of drought eGEI genes is consistent with the hypothesis that variants at these genes are involved in local adaptation.
Summary of Statistical Results for Comparisons with Null Expectations from Genomic Controls (i.e., permutations of gene categories).
| SNP or Gene Statistic | Sig. Result for Cold eSR Genes | Sig. Result for Drought eSR Genes | Sig. Result for Cold eGEI Genes | Sig. Result for Drought eGEI Genes |
|---|---|---|---|---|
| PHS | + | + | o | o |
| CLR | o | o | o | o |
| MAF | − | − | o | + |
| Fst | o | o | o | + |
| Climate assn., early-flowering accessions | −/o | o | +/o | +/o |
| Climate assn., late-flowering accessions | −/o | o | o | +/o |
| Survival | −/o | −/o | o | o |
| Silique N | −/o | o | o | o |
| Promoter θ | − | − | o | + |
| Ka/Ks | − | − | − | o |
| Singleton proportion, nonsynonymous SNPs | + | + | o | o |
| Mean frequency ABREs | o | + | + | o |
| Mean frequency DRE/CBFs | o | + | + | o |
| Variance in frequency of ABREs | − | − | o | + |
| Variance in frequency of DRE/CBFs | o | o | − | o |
| SNP or Gene Statistic | Sig. Result for Cold eSR Genes | Sig. Result for Drought eSR Genes | Sig. Result for Cold eGEI Genes | Sig. Result for Drought eGEI Genes |
|---|---|---|---|---|
| PHS | + | + | o | o |
| CLR | o | o | o | o |
| MAF | − | − | o | + |
| Fst | o | o | o | + |
| Climate assn., early-flowering accessions | −/o | o | +/o | +/o |
| Climate assn., late-flowering accessions | −/o | o | o | +/o |
| Survival | −/o | −/o | o | o |
| Silique N | −/o | o | o | o |
| Promoter θ | − | − | o | + |
| Ka/Ks | − | − | − | o |
| Singleton proportion, nonsynonymous SNPs | + | + | o | o |
| Mean frequency ABREs | o | + | + | o |
| Mean frequency DRE/CBFs | o | + | + | o |
| Variance in frequency of ABREs | − | − | o | + |
| Variance in frequency of DRE/CBFs | o | o | − | o |
Note.—Results are separated by statistic (e.g., PHS) and gene set tested (cold and drought eSR and eGEI genes). “+” indicates significantly high or strong statistics in the gene set (enrichment); “−” indicates significantly low or weak statistics in the gene set (depletion); “o” indicates nonsignificant results; “+/o” or “−/o” indicates multiple tests in the category and mixed results.
Summary of Statistical Results for Comparisons with Null Expectations from Genomic Controls (i.e., permutations of gene categories).
| SNP or Gene Statistic | Sig. Result for Cold eSR Genes | Sig. Result for Drought eSR Genes | Sig. Result for Cold eGEI Genes | Sig. Result for Drought eGEI Genes |
|---|---|---|---|---|
| PHS | + | + | o | o |
| CLR | o | o | o | o |
| MAF | − | − | o | + |
| Fst | o | o | o | + |
| Climate assn., early-flowering accessions | −/o | o | +/o | +/o |
| Climate assn., late-flowering accessions | −/o | o | o | +/o |
| Survival | −/o | −/o | o | o |
| Silique N | −/o | o | o | o |
| Promoter θ | − | − | o | + |
| Ka/Ks | − | − | − | o |
| Singleton proportion, nonsynonymous SNPs | + | + | o | o |
| Mean frequency ABREs | o | + | + | o |
| Mean frequency DRE/CBFs | o | + | + | o |
| Variance in frequency of ABREs | − | − | o | + |
| Variance in frequency of DRE/CBFs | o | o | − | o |
| SNP or Gene Statistic | Sig. Result for Cold eSR Genes | Sig. Result for Drought eSR Genes | Sig. Result for Cold eGEI Genes | Sig. Result for Drought eGEI Genes |
|---|---|---|---|---|
| PHS | + | + | o | o |
| CLR | o | o | o | o |
| MAF | − | − | o | + |
| Fst | o | o | o | + |
| Climate assn., early-flowering accessions | −/o | o | +/o | +/o |
| Climate assn., late-flowering accessions | −/o | o | o | +/o |
| Survival | −/o | −/o | o | o |
| Silique N | −/o | o | o | o |
| Promoter θ | − | − | o | + |
| Ka/Ks | − | − | − | o |
| Singleton proportion, nonsynonymous SNPs | + | + | o | o |
| Mean frequency ABREs | o | + | + | o |
| Mean frequency DRE/CBFs | o | + | + | o |
| Variance in frequency of ABREs | − | − | o | + |
| Variance in frequency of DRE/CBFs | o | o | − | o |
Note.—Results are separated by statistic (e.g., PHS) and gene set tested (cold and drought eSR and eGEI genes). “+” indicates significantly high or strong statistics in the gene set (enrichment); “−” indicates significantly low or weak statistics in the gene set (depletion); “o” indicates nonsignificant results; “+/o” or “−/o” indicates multiple tests in the category and mixed results.
eGEI Genes, But Not eSR Genes, Are Frequently Enriched with SNPs Associated with Climate
We were interested in determining whether climatic gradients shape genetic variation in expression response. We conducted a new genome-wide association study (GWAS) with climate, controlling for population structure (Kang et al. 2008), using the same 214,051 SNPs as above, restricted to approximately 1,000 accessions from the native range of Arabidopsis (Hoffmann 2002; Anastasio et al. 2011). We used permutations to test whether outlier SNP-climate associations (5% lower tail of P values) were more or less frequent among SNPs linked to eSR and eGEI genes (supplementary fig. S3, Supplementary Material online). We tested for enrichment with SNP associations to 11 moisture and cold-related climate variables (Lasky et al. 2012).
Plant adaptations to abiotic stress are often associated with genetic variation in flowering time and life history (McKay et al. 2003; Donohue 2005; Korves et al. 2007). Arabidopsis individuals can be classified as rapid cycling, and early-flowering, or as late-flowering (and likely winter annual). The majority of accessions in published data are early-flowering, which occupy a relatively wider range of climate conditions (Banta et al. 2012; Lasky et al. 2012). Previously, we found that early- and late-flowering accessions showed evidence of distinct patterns of local adaptation to climate (Lasky et al. 2012). As a result, we followed Des Marais et al. (2012) and analyzed climate enrichment for early-flowering and late-flowering accessions separately (supplementary fig. S4, Supplementary Material online, for results on combined accessions).
Climate enrichments were strongly divergent between eSR and eGEI genes. Compared with genomic controls, eGEI genes in early-flowering accessions were significantly enriched for associations with four climate variables, and eSR genes were significantly depleted in associations with five climate variables (figs. 2 and 3). In direct comparison, eGEI genes had significantly more climate-associated SNPs than those of eSR genes along 9 of 11 studied climatic gradients. eGEI were most enriched for associations with minimum growing season temperatures compared with eSR genes (z = −3.024, P = 0.0024; supplementary table S2, Supplementary Material online). Late-flowering drought eGEI genes were significantly enriched with associations to coefficient of variation (CV) of monthly precipitation compared with genomic controls (z = 2.33, P =0.0182). Late-flowering eGEI genes also typically had higher climate enrichment than that of eSR genes (figs. 2 and 3).
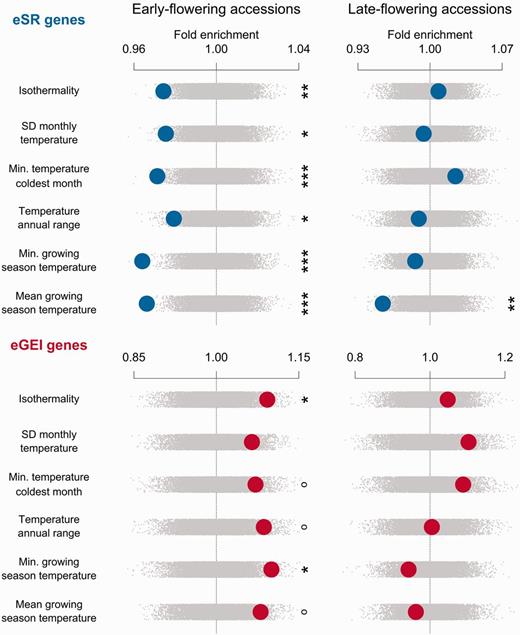
Enrichment of gene sets with associations to cold-related climate variables for early- and late-flowering accessions. Observed enrichments are calculated as a z score using the distribution from null permutations. A high z score indicates a gene set has greater outlier climate-SNP associations compared with the genome-wide expectation, whereas a low z indicates few outlier climate-SNP associations. Enrichments are shown with large circles with eSR genes in the top rows and eGEI genes in the bottom rows. Null permutations are shown as small gray dots (°P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.005).
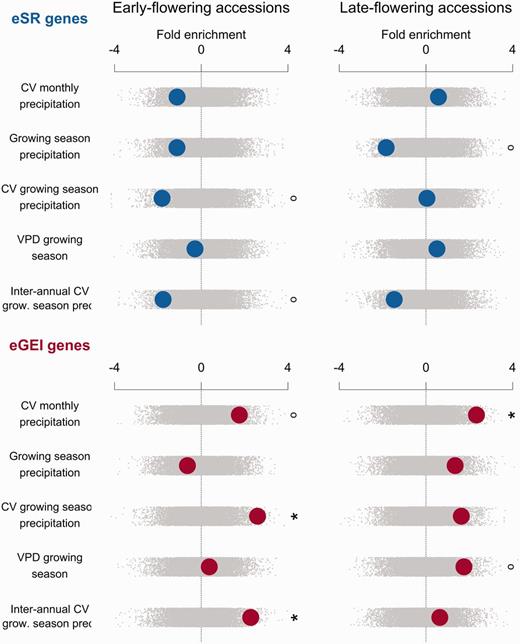
Enrichment of gene sets with associations to drought-related climate variables for early- and late-flowering accessions. Observed enrichments are calculated as a z score using the distribution from null permutations. A high z score indicates a gene set has greater outlier climate-SNP associations compared with the genome-wide expectation, whereas a low z indicates few outlier climate-SNP associations. Enrichments are shown with large circles with eSR genes in the top rows and eGEI genes in the bottom rows. Null permutations are shown as small gray dots (°P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.005).
eSR Genes Have Weak SNP Associations with Fitness in Field Common Gardens
Genes involved in local adaptation may exhibit associations between allelic variation and fitness in common garden field experiments (Fournier-Level et al. 2011; Ågren et al. 2013). Thus, we conducted a new association study of two fitness components—published silique number and survival data from four European field sites of diverse climates (Wilczek et al. 2009; Fournier-Level et al. 2011).
Cold and drought eSR genes almost always had fewer associations with fecundity and survival than those of genomic controls, across all field sites (supplementary table S3 and Supplementary Data, Supplementary Material online). Compared with genomic controls, cold eSR genes were significantly depleted in survival associations in the United Kingdom (permutation test: z = −2.92, P = 0.0046) and Spain (z = −3.67, P = 0.0008) as well as silique number in Finland (z = −2.19, P = 0.0338), whereas drought eSR genes were significantly depleted in survival associations in the United Kingdom (z = −2.08, P = 0.0360). Additionally, eSR genes had significantly fewer survival associations in Spain than those of eGEI genes (z = −2.36, P = 0.0202). The few fitness associations for eSR genes combined with high PHS suggest that purifying or widespread positive selection on eSR genes precludes their association with variation in fitness. eGEI genes showed greater (though nonsignificant) enrichment with fitness associations relative to eSR and genomic controls in 10 of 16 combinations of fitness component, abiotic stressor, and site (supplementary table S3, Supplementary Material online). The higher frequency of fitness associations for eGEI compared with eSR genes may be due to fitness effects of plasticity at eGEI loci. However, some eGEI fitness enrichments were weaker than genomic controls, possibly because many eGEI genes have weak fitness effects or because fitness associations did not identify causal loci.
eSR and eGEI Genes Differ in the Frequency of Promoter Polymorphisms
If eGEI genes are linked to cis-regulatory variants associated with climate, we predict that their proximal promoters exhibit elevated polymorphism. However, if eGEI effects were solely driven by trans-regulatory variants we would not expect elevated promoter polymorphism. eSR genes may be subject to stronger positive or purifying selection, such that we expect low polymorphism in their promoter and coding regions. We thus tested the hypothesis that sequence diversity of eSR and eGEI genes shows signatures of differing selection regimes. Among 80 resequenced accessions (Cao et al. 2011), segregating nucleotide diversity in the proximal (−1,000 bp) promoters of eSR genes was significantly less than genomic controls (drought: z = −3.56, P = 0.0066, cold: z = −3.30, P = 0.0329). This low diversity suggests the action of recent selective sweeps or strong purifying selection on eSR promoters (fig. 4). By contrast, the promoters of eGEI genes exhibited high nucleotide diversity compared with genomic controls (drought: z = 2.190, P = 0.0248, cold: z = 1.58, P = 0.1181) and compared with eSR genes (drought: z = −3.58, P = 0.0042, cold: z = −3.32, P = 0.0043). We next looked at the ratio of nonsynonymous to synonymous polymorphism (Ka/Ks) in coding sequences and found that in both eSR (drought: z = −15.76, P < 0.0002, cold: z = −16.35, P < 0.0002) and eGEI genes (drought: z = −0.74, P = 0.4651, cold: z = −4.76, P < 0.0002) this ratio was lower than genomic controls (supplementary table S4, Supplementary Material online). We also tested the proportion of nonsynonymous variants comprised of singletons, as purifying selection is expected to limit the frequency of nonsynonymous variants. We found that nonsynonymous polymorphisms in eSR genes were significantly more likely to be singletons compared with genomic controls (drought: z = 3.87, P < 0.0002, cold: z = 4.26, P < 0.0002), whereas eGEI genes did not significantly differ from genomic controls in the proportion of singletons (drought: z = −0.12, P = 0.9090, cold: z = −1.03, P = 0.3064).
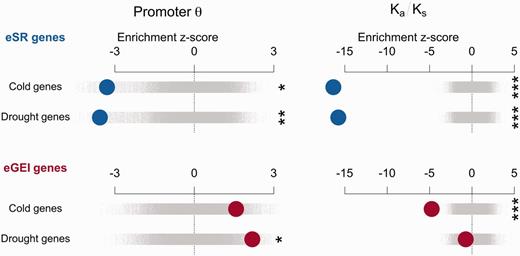
The enrichment of candidate gene sets with promoter diversity or ratio of nonsynonymous/synonymous polymorphisms in 80 resequenced accessions. Observed enrichments are calculated as a z score using the distribution from null permutations of gene sets. Enrichments are shown with large circles with eSR genes in the top rows and eGEI genes in the bottom rows. Null permutations are shown as small gray points (*P < 0.05, **P < 0.01, ***P < 0.005).
Together, these two results suggest that both eSR and eGEI genes are under purifying selection comparable to or stronger than genomic controls. By contrast, the excess promoter polymorphism in eGEI genes suggests that the cis-regulatory sequences of these two categories of genes are under different selective pressures. This polymorphism is potentially maintained by divergent selection across sites or reduced selective constraints on promoters of eGEI genes. Our finding that the coding sequences of eGEI genes have low Ka/Ks ratios and nonsynonymous singleton polymorphisms at a rate similar to the genome-wide average indicates that eGEI genes experience some degree of purifying selection whereas their promoters are more evolutionarily labile.
eGEI Genes Have Elevated Polymorphism of Abscisic Acid-Responsive Cis-Regulatory Motifs
We studied whether the high versus low polymorphism in the promoters of eGEI and eSR genes, respectively, would be reflected in known transcription factor binding sites involved in abiotic stress responses. The number of such binding sites in a promoter can affect the level of stress-responsive gene expression (Narusaka et al. 2003). Thus, we quantified the number of two major types of stress responsive motifs in promoters across 80 resequenced accessions (Cao et al. 2011). Abscisic acid-responsive elements (ABRE) are bound by multiple ABRE binding protein transcription factors and are characterized by a conserved ACGT core motif (Fujita et al. 2005; Zhang et al. 2005; Maruyama et al. 2012; Fujita et al. 2013). Dehydration responsive elements or C-repeat binding factors (DRE/CBF) are bound by dehydration and cold responsive element binding proteins and are characterized by a common GCCGAC motif (Baker et al. 1994; Zhang et al. 2005; Geisler et al. 2006; Lim et al. 2007).
The mean frequency of ABRE and DRE/CBF motifs was generally higher in eSR and eGEI genes than in genomic controls, suggesting that these motifs were involved in expression responses of these genes (fig. 5 and supplementary tables S6 and Supplementary Data, Supplementary Material online). We also analyzed the variance in motif counts among accessions. eSR genes showed significantly low variance in ABRE counts among accessions compared with genomic controls (drought: z = −7.08, P < 0.0002, cold: z = −3.38, P = 0.0164) and compared with eGEI genes (drought: z = −4.48, P = 0.0060, cold: z = −2.93, P < 0.0002; fig. 5 and supplementary tables S8 and Supplementary Data, Supplementary Material online), suggesting that ABREs in eSR genes are under purifying selection. By contrast, ABRE counts were more variable among accessions than expected for drought eGEI genes (z = 2.16, P = 0.0336), suggesting that variability in ABREs is a potential mechanism for eGEI effects. DRE/CBFs showed less than expected variance for eSR and eGEI genes, significantly so for cold eGEI genes (read in both directions, z = −2.21, P = 0.0270).
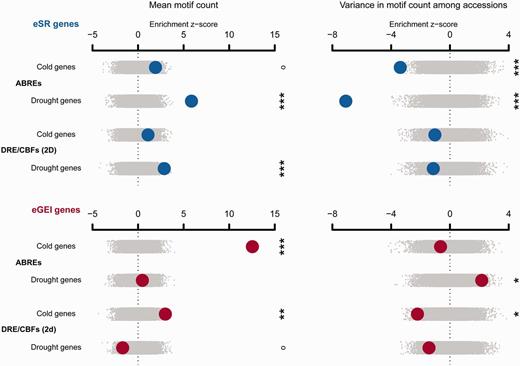
The enrichment of candidate gene sets with mean motif frequency or variance in motif frequency among 80 resequenced accessions. Observed enrichments are calculated as a z score using the distribution from null permutations. Enrichments are shown with large circles with eSR genes in the top rows and eGEI genes in the bottom rows. Null permutations are shown as small gray dots (°P < 0.1, *P < 0.05, **P < 0.01, ***P < 0.005).
Discussion
Extensive empirical work detailing the molecular mechanisms of transcriptional regulation has led to many hypotheses about their role in adaptive evolution (Wray et al. 2003). However, progress has been limited by the challenge of genome-wide expression profiling across diverse genotypes and environments. The general empirical significance of phenotypic plasticity in local adaptation to environment is also not well known, despite many theoretical studies of plasticity and adaptation (Price et al. 2003; Ghalambor et al. 2007). Researchers previously have characterized genes with eSR and eGEI responses to abiotic stress in Arabidopsis (Hannah et al. 2006; Des Marais et al. 2012; Richards et al. 2012). However, the role of expression plasticity in local adaptation to climate by natural populations is poorly known. Furthermore, the general empirical importance of cis-regulatory evolution in abiotic stress response is not well known. Here, we found widespread evidence that eSR and eGEI genes responsive to abiotic stress are subject to distinct selective pressures resulting in divergent patterns of cis-regulatory evolution. Our approach joined the complementary information from sequence polymorphism (Cao et al. 2011; Hancock et al. 2011; Horton et al. 2012) to laboratory stress response (Hannah et al. 2006; Des Marais et al. 2012). We uncovered evidence that conserved expression responses (eSR) tend to be subject to positive selection and that genetic variation in expression plasticity may be involved in local adaptation to climate.
The Role of Gene Expression in Adaptation
Researchers have previously detected patterns suggestive of local adaptation via transcriptomic plasticity, despite considerably smaller sample sizes. For example, Larsen et al. (2007) found many eGEI genes across two flounder populations in different salinity environments, despite low neutral divergence between populations. Additionally, eGEI genes were associated with traits related to fitness (Larsen et al. 2007). In stickleback, Jones et al. (2012) found that intergenic, putative regulatory regions that are highly conserved across species showed high sequence divergence between salt and freshwater populations. These studies and our results suggest that cis-regulatory evolution is a source of variation for local adaptation. Authors have hypothesized that cis-regulatory evolution is a more efficient mechanism of adaptation compared with protein coding sequence changes due to limited pleiotropy and incomplete dominance (Prud’homme et al. 2007; Wray 2007). However, the predominance of cis-regulatory change in adaptive evolution has been debated (Hoekstra and Coyne 2007; Stern and Orgogozo 2008) and its empirical importance has not been well demonstrated, particularly within species (reviewed by Wray et al. 2003; Wray 2007). In this study, genome-wide analysis suggests that cis-regulatory variation affects expression of eGEI genes. Further direct functional assays will be required to confirm the role of such variants in local adaptation to climate. It is likely that evolution of trans-regulatory elements also plays a major role in eGEI effects and local adaptation to climate though our analyses were not designed to test this hypothesis. Future studies might use crossing experiments to map expression quantitative trait loci (eQTL) to generate conclusive evidence for cis-regulation. eQTL mapping can also be used to identify trans-regulatory loci and associated evidence of selection (e.g., Lowry et al. 2013).
Causes of Genetic Variation in Expression Polymorphism in Response to Abiotic Stress
The high variance in ABRE motif frequency for drought eGEI genes suggests a cis-regulatory explanation for many genotypic differences in response to drought. Combined with the observed climate enrichments of eGEI genes, our results suggest that genetic variation in expression response to drought caused by variation in the presence of ABRE motifs may be a mechanism of local adaptation. Although evolution of many DRE/CBF motifs may also be involved in local adaptation to climate, their low variability in the promoters of eGEI genes may indicate that selective gradients do not typically maintain high variability in DRE/CBFs counts. In contrast to eGEI genes and ABREs, our finding that eSR genes have consistently high frequency and low variance in the frequency of motifs suggests that motif frequency for eSR genes is under purifying selection. These motifs likely promote binding by transcription factors resulting in conserved expression responses that consistently increase fitness in the face of cold and drought (Fujita et al. 2005).
The climate enrichments of SNPs near eGEI genes may indicate that genotype-by-environment effects on expression underlie local adaptation to climate. Two factors suggest that local adaptation involving cis-regulatory evolution may be the cause of our results. First, the fact that eGEI genes, by definition, have genetic variation for plasticity suggests that variation in nearby SNPs could be linked to variation in cis-regulatory elements. Second, eGEI genes have expression responses to laboratory stressors that are likely representative of conditions associated with climate gradients in our study. However, some eGEI fitness enrichments were weaker than genomic controls, possibly because individual eGEI genes have very small fitness effects. Alternatively, the relative importance of expression plasticity—compared with other molecular mechanisms—in local adaptation may vary among sites, potentially weakening eGEI association with fitness. Additionally, there may be a mismatch between the scale of adaptation to climate and fitness variation in the field experiments (Wilczek et al. 2009; Fournier-Level et al. 2011), which were conducted across relatively small spatiotemporal scales (i.e., four sites and one year) on fewer than 200 genotypes. Testing whether eGEI effects are involved in local adaptation will require experiments to link specific eGEI variants with genotype-by-environment effects on fitness across sites.
The high polymorphism associated with promoters of eGEI genes may indicate that many are evolving neutrally, and thus not involved in local adaptation. However, several lines of evidence indicate that the coding regions of most eGEI genes are not evolving neutrally. First, Ka/Ks in eGEI genes was lower than (for cold) or equal to (for drought) than genomic controls, suggesting a persistent effect of purifying selection at these loci. Second, eGEI genes were enriched with SNPs with higher MAF than that of genomic controls, consistent with spatially variable selection. Third, eGEI genes were often enriched for climate associations after controlling for population structure. It is important to note that apparent relaxed selection on a locus is not mutually exclusive with a locus being involved in local adaptation. For example, Fournier-Level et al. (2011) found that alleles associated with low fitness typically had more narrow climatic distributions than those of alleles associated with high fitness, suggesting that most loci involved in local adaptation are effectively neutral across large portions of a species range. As with all enrichment analyses, an important caveat is that enrichment of a test statistic in a gene list does not require that all genes in the list be causally linked to the statistic. Rather, enrichment merely suggests that on average genes in the list are more often involved in some process generating the test statistic compared with the genome average.
Phenology, Expression Polymorphism, and Local Adaptation
The observed differences in climate enrichments between flowering time groups may be due to distinct responses to climatic gradients (Lasky et al. 2012). Previous studies on Arabidopsis (Korves et al. 2007; Méndez-Vigo et al. 2011; Banta et al. 2012; Kronholm et al. 2012; Picó 2012; Lovell et al. 2013) and other species (Franks et al. 2007; Lowry 2012; Lowry et al. 2014) are consistent with the hypothesis that phenology mediates local adaptation to climate. We observed more frequent climate associations among eGEI genes for early flowering compared with late-flowering accessions, especially for cold eGEI genes. Our finding appears contrary to the hypothesis that late-flowering accessions exhibit stronger local adaptation because they employ a stress-tolerant life history, compared with early flowering accessions that escape abiotic stress (McKay et al. 2003; Lasky et al. 2012). However, late-flowering accessions may be more likely to experience cold after germination in the autumn, may be more tolerant to freezing (Korves et al. 2007), and thus might exhibit local adaptation via more constitutive and fewer plastic responses.
Causes of Low Polymorphism in Consistent Stress Responsive Genes
The significantly few climate and fitness associations, combined with the low variation in promoters and amino acid sequences of eSR genes, suggest the effects of purifying selection or recent positive selection and conserved responses among accessions. The low frequency of climate associations in eSR genes is consistent with the hypothesis that most eSR genes are involved in essential, highly beneficial and conserved responses to stress (Lowry et al. 2013). Expression is often highly conserved, even across divergent taxa, such as aging-associated changes in Caenorhabditis elegans and Drosophila melanogaster, which are separated by approximately 1 Gy of evolution (McCarroll et al. 2004). These molecular responses may be essential to all genotypes so that the great majority of changes to them are deleterious, resulting in stabilizing selection and a low likelihood of involvement in local adaptation and fitness variation (Blekhman et al. 2008; Hodgins-Davis and Townsend 2009; Lowry et al. 2013). Note that patterns of selection on eSR genes may partly be due to gene functions unrelated to the abiotic factors we studied, such as biotic interactions. Our findings that eSR genes are characterized by low sequence diversity in both promoters and coding sequence and high PHS suggest strong selective constraints or recent selective sweeps. Selective sweeps might have been driven by climate change or range expansion into new climates following the end of the last glacial maximum, as Arabidopsis expanded from refugia (Sharbel et al. 2000). Alternatively, recent bottlenecks during expansion may have generated extended haplotypes with mildly deleterious alleles, followed by ongoing strong purifying selection on eSR genes that acts to eliminate those haplotypes.
Conclusions
Plant responses to their environment involve changes across levels of biological organization. Individual studies typically examine either plasticity in expression of individual genes or continental-scale allele frequency clines. Here, we linked plasticity responses in individual genes to allele frequency clines across Eurasia along gradients in climate heterogeneity. Through multiple dimensions of genomic and environmental variation, we demonstrate that distinct evolutionary processes appear to act on genes with conserved versus genetically variable abiotic stress responses. Our findings suggest that selection on cis-regulatory elements is a major factor influencing phenotypic plasticity in response to abiotic stressors. By contrast, genes with conserved stress responses appear to be subject to strong purifying selection in both promoter and coding sequences. Future research that integrates experimentally measured phenotypic variation with spatial variation in allele frequency and environment is a promising approach to revealing the evolutionary mechanisms affecting natural variation and local adaptation.
Materials and Methods
Data
Expression Data
Each gene expression experiment used the Affymetrix ATH1 Genome array. Estimates of drought-responsive gene expression are from Des Marais et al. (2012). In brief, these experiments exposed ten early-flowering and seven late-flowering natural accessions of Arabidopsis, grown in potting medium, to a gradual 7-day controlled dry-down. On day 7, three replicate plants each from treatment and control environments were harvested and placed in RNALater.
In the second experiment, seven early-flowering and two late-flowering accessions were subjected to cold acclimation (Hannah et al. 2006). Once treatment plants began bolting they were transferred from a 20/18 °C day/night chamber to a constant 4 °C treatment, with a 16-h photoperiod (Hannah et al. 2006). Control plants remained in the 20/18 °C day/night conditions. After 14 days, whole rosettes of ten plants per accession in each treatment were harvested and pooled for RNA extraction (Hannah et al. 2006).
We completed analyses of gene expression data from both experiments using filtered Robust-MultiChip Average expression (RMA) values (Irizarry et al. 2003). CEL files were imported into JMP Genomics and gene expression measures were generated using the RMA function (background corrected, log2 transformed, quantile normalized, median-polish summary) with a custom CDF file constructed from the TAIR version 10 of the Arabidopsis genome (available for download at http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/CDF_download.asp; Dai et al. 2005). Expression measures were then processed by ANOVA. In each case, we fit a fixed effect general linear model including a term for “accession” (i.e., genotype), “treatment” (i.e., environment), and their interaction. In all analyses, we controlled for multiple testing using a positive false-discovery rate of 0.05 (Storey 2003). eSR and eGEI gene sets were defined as all genes having a significant treatment or genotype-by-treatment interaction effect, respectively, at the positive false-discovery rate of 0.05. ANOVA and FDR were previously published for drought experiments (Des Marais et al. 2012) although our analyses of data from Hannah et al. (2006) were new for our present study.
The drought experiment by Des Marais et al. (2012) split accessions into flowering time groups (early- vs. late-flowering) when collecting microarray data. We followed by splitting accession expression data into groups that were early versus late-flowering in the absence of vernalization, using flowering status reported in the original studies (Hannah et al. 2006; Des Marais et al. 2012). We then analyzed expression data and generated eSR and eGEI gene sets for each flowering time category. We also generated gene sets for accessions of both flowering time categories combined. For the cold experiment, we conducted ANOVA and FDR on all accessions to generate gene sets for combined flowering time categories. However, microarray data were collected separately for flowering time groups in the drought experiment (Des Marais et al. 2012), thus block effects could be confounded with flowering time effects in combined statistical analysis of expression. For this reason, we simply combined drought gene sets from each flowering time category to generate drought gene sets for both flowering time categories combined.
Genomic Data
We used two published data sets on genome-wide sequence variation in natural accessions. First, we used data on 1,307 accessions genotyped with a custom Affymetrix SNP chip characterizing 214,051 SNPs (version 3.06; Hancock et al. 2011; Horton et al. 2012; see supplementary table S5, Supplementary Material online, for a list of all accessions). We focused on accessions found in the native Eurasian range (Hoffmann 2002) and eliminated likely contaminants (Anastasio et al. 2011) giving 1,003 total accessions from Eurasia. Sampling occurred across the world but was densest in Northern and Western Europe and sparser in Eastern Europe and Central Asia. Second, we used published resequencing data for 80 natural accessions to characterize SNP diversity and promoter motifs (Cao et al. 2011). Accessions were sequenced using the Illumina Genome Analyzer platform and then were aligned to the Col-0 reference genome.
Climate Data
We compiled and calculated climate data for collection locations of accessions in a previous study (Lasky et al. 2012), where a more detailed description of climate data can be found. Climate data sources were publicly available but varied in spatiotemporal resolution and parameters (Kalnay et al. 1996; New et al. 2002; Hijmans et al. 2005). We used monthly data on temperature, precipitation, and vapor pressure deficit (VPD) to estimate growing season conditions for each accession (Lasky et al. 2012) in addition to several derived variables of hypothesized biological importance (Hijmans et al. 2005) (see supplementary material S1, Supplementary Material online, for more detail).
Fitness Data
We studied two aspects of fitness, silique (fruit) number and survival, using published data from four field sites (Wilczek et al. 2009; Fournier-Level et al. 2011). Sites were located in Norwich, United Kingdom, Oulu, Finland, Valencia, Spain, and Halle, Germany. At the sites in the United Kingdom, Spain, and Germany, 157 accessions were sowed in outdoor plots whereas 68 were sowed in Finland. Wilczek et al. (2009) conducted plantings to match germination of local populations, planting in the autumn in the United Kingdom, Germany, and Spain, planting in the spring in the United Kingdom, and planting in the summer in the United Kingdom and Finland. We tested all eight fitness variables (four sites and two aspects of fitness) for SNP associations (Kang et al. 2008).
Statistical Analyses
Enrichment of Genes with Transcriptional Plasticity
We assessed whether genes having abiotic stress treatment effects (eSR) or accession by treatment effects (eGEI) on expression were linked to SNPs indicating statistical signatures of selection or SNPs associated with climate and fitness. We used a permutation enrichment test based on that of Segrè et al. (2010). The test compares the proportion of candidate genes (eSR or eGEI genes) linked to nearby SNPs with strong associations (lower 0.05 quantile of P values) with the proportion of randomly selected genes having nearby SNPs with strong associations (supplementary fig. S3, Supplementary Material online). The use of a tail test statistic at the SNP level (e.g., Horton et al. 2012) is justified because the central tendency of associations near a given gene is likely heavily influenced by effectively neutral SNPs, even in genes that are under selection.
In enrichment tests, SNPs are typically assigned to genes if they fall within a fixed distance of the gene (e.g., Segrè et al. 2010). However, linkage disequilibrium (LD) varies widely across the genome (Horton et al. 2012). Thus, we considered SNPs linked to specific genes based on local LD calculated across the genome (see supplementary material S1, Supplementary Material online). We created a null distribution using permutation of 10,000 random sets of genes from the microarray (i.e., genomic controls), where random sets had the same number of genes as the observed set. These permutations test the null hypothesis that the microarray candidate genes co-occurred randomly with respect to strong associations (Segrè et al. 2010).
We tested the enrichment of gene sets from drought microarrays with SNP associations to drought-related climate variables and the enrichment of gene sets from cold-acclimation microarrays with SNP associations to cold-related climate variables. We tested the climate enrichments of gene sets within each flowering time group separately and among all accessions. Compared with climate associations, fewer accessions (<200) were used in fitness experiments and resequencing panels and as a result we did not test fitness enrichments and sequence evidence for selection within flowering time groups.
Sequence Signatures of Selection
CLR was previously calculated by Horton et al. (2012) for 1,193 accessions. PHS was previously calculated by Toomajian et al. (2006) for 1,144 accessions. We calculated MAF on the full panel of 1,307 accessions for each SNP (Horton et al. 2012). We calculated Fst on the 1,003 Eurasian accessions that remained after eliminating likely contaminants (Anastasio et al. 2011). Previously Horton et al. (2012) calculated Fst on a large global panel including the more recently introduced populations in North America where the signal of local adaptation might be weaker (Platt et al. 2010). Here, we divided the 1,003 Eurasian accessions into populations based on a 10° × 10° latitude/longitude grid, further subdividing populations from continental Europe from the British Isles. We used the implementation of Weir and Hill (2002) in the R package “dirmult” (Tvedebrink 2010) to estimate Fst. In order to avoid spurious statistics from rare alleles, we restricted PHS, CLR, and Fst analyses to SNPs with MAF of at least 0.1 (Atwell et al. 2010).
GWAS Mixed Model
We used the Efficient Mixed-Model Association (EMMA) linear model of Kang et al. (2008) to test SNP associations with climate and fitness. EMMA includes a kinship random effect (calculated as identity-in-state) to attempt to control for population structure and is more effective at doing so than permutation-based partial Mantel tests (Hancock et al. 2011; Guillot and Rousset 2013; see supplementary material S1 and Supplementary Data, Supplementary Material online, for more detail).
Climate GWAS Stratified by Flowering Time
We followed Des Marais et al. (2012) and our previous study (Lasky et al. 2012), conducting a subset of climate GWAS on putative early-flowering groups. Because flowering time data were only available for 476 of the 1,307 accessions with genomic data, in the previous study we used data from the 476 to predict flowering time variation in the remaining accessions (Lasky et al. 2012). We then used these empirical flowering time data to categorize accessions using a SNP-based model of flowering-time category for accessions lacking data (see supplementary material S1, Supplementary Material online, for more detail).
Polymorphism in Resequenced Genomes
We used 80 whole genome sequences to test for evidence of differing selection on eSR and eGEI genes (Cao et al. 2011). For each gene we calculated nucleotide diversity (θ) from segregating sites in a 1,000-bp promoter (Watterson 1975) and the ratio of nonsynonymous to synonymous substitutions (Ka/Ks). Based on the TAIR10 annotation, we used custom Perl scripts to extract the complete coding sequences and 1,000-bp upstream of predicted transcriptional start site from the 80 resequenced Arabidopsis accessions reported by Cao et al. (2011). Nucleotide diversity in the promoter sequences was estimated as θ. For each 1,000-bp promoter, we estimated nucleotide diversity from segregating sites (S) as θ = S/a1, where a1 = 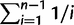 and n is the number of sequences (Watterson 1975). As a measure of the strength of purifying selection in the coding sequences of genes, we estimated the ratio of nonsynonymous to synonymous substitutions using the yn00 module of PAML (Yang 1997). As a second measure of the strength of purifying selection, we estimated the Allele Frequency Spectra of each gene using a modified version of PolyMORPHOrama (Bachtrog and Andolfatto 2006) provided by E. Josephs. We then calculated the proportion of nonsynonymous SNPs in each gene that were represented as singletons. In this context, singleton nonsynonymous SNPs are inferred to be young, slightly deleterious alleles which have not yet been removed from the population by purifying selection. Higher values of the ratio of singleton to total nonsynonymous SNPs are therefore indicative of stronger purifying selection.
and n is the number of sequences (Watterson 1975). As a measure of the strength of purifying selection in the coding sequences of genes, we estimated the ratio of nonsynonymous to synonymous substitutions using the yn00 module of PAML (Yang 1997). As a second measure of the strength of purifying selection, we estimated the Allele Frequency Spectra of each gene using a modified version of PolyMORPHOrama (Bachtrog and Andolfatto 2006) provided by E. Josephs. We then calculated the proportion of nonsynonymous SNPs in each gene that were represented as singletons. In this context, singleton nonsynonymous SNPs are inferred to be young, slightly deleterious alleles which have not yet been removed from the population by purifying selection. Higher values of the ratio of singleton to total nonsynonymous SNPs are therefore indicative of stronger purifying selection.
We tested the hypothesis that eSR genes and eGEI genes had Ka/Ks and promoter θ equal to each other and equal to genome-wide controls using a permutation test similar to that described above for climate and fitness enrichments. Mean Ka/Ks and promoter θ were calculated for each gene as observed test statistics. We then circularly permuted gene classifications as eSR, eGEI or neither, 10,000 times and calculated null test statistics. We then calculated the tail density of the null distribution with a more extreme Ka/Ks or θ than the observed mean and doubled the tail density to get a two-tailed P value.
We investigated whether well-known stress response motifs in promoters were associated with eSR and eGEI genes. We quantified ABRE and DRE/CBF motifs occurring within 1,000-bp upstream (putative promoters). For each gene, we calculated the mean frequency and variance in frequency of a given motif across accessions. We then tested whether eSR and eGEI genes had nonrandom and distinct mean and variance of motifs, using permutations of gene categories as described previously. We aggregated statistics across several motif variants for each core motif using the method of O’Brien (1984), in addition to testing each variant independently (see supplementary material S1, Supplementary Material online).
Acknowledgments
The authors thank Geoffrey Morris, Saunak Sen, and several anonymous reviewers for helpful comments. They also thank the Weijia Xu and the Texas Advanced Computing Center for assistance with computing. This work was supported by the National Science Foundation, IOS-0922457 to T.E.J. and T.H.K, DEB-0618347 to T.E.J., DEB-0618294 to J.H.R., and DEB-0618302 to J.K.M. and United States Department of Agriculture NIFA-AFRI postdoctoral fellowships 2011-67012-30696 to D.B.L. and 2011-67012-30663 to D.L.D.
References
Author notes
Associate editor: Stephen Wright
†Present address: Arnold Arboretum and Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}