





Abstract
Accumulating evidence indicates that estrogen regulates diverse but interdependent signaling pathways via estrogen receptor (ER)-dependent and -independent mechanisms. However, molecular relationship between these pathways for gene regulation under the direction of estrogen remains unknown. To address this possibility, our uterine analysis of Wnt/β-catenin downstream effectors revealed that lymphoid enhancer factor 1 (Lef-1) and T cell factor 3 (Tcf-3) are up-regulated temporally by 17β-estradiol (E2) in an ER-independent manner. Lef-1 is abundantly up-regulated early (within 2 h), whereas Tcf-3 is predominantly induced after 6 h, and both are sustained through 24 h. Interestingly, activated Lef-1/Tcf-3 molecularly interacted with ERα in a time-dependent manner, suggesting they possess a cross talk in the uterus by E2. Moreover, dual immunofluorescence studies confirm their colocalization in uterine epithelial cells after E2. Most importantly, using chromatin immunoprecipitation followed by PCR analyses, we provide evidence for an interesting possibility that ERα and Tcf-3/Lef-1 complex occupies at certain DNA regions of estrogen-responsive endogenous gene promoters in the mouse uterus. By selective perturbation of activated Lef-1/Tcf-3 or ERα signaling events, we provide in this study novel evidence that cooperative interactions, by these two different classes of transcription factors at the level of chromatin, direct uterine regulation of estrogen-responsive genes. Collectively, these studies support a mechanism that integration of a nonclassically induced β-catenin/Lef-1/Tcf-3 signaling with ERα is necessary for estrogen-dependent endogenous gene regulation in uterine biology.
ESTROGENS PRIMARILY REGULATE growth and differentiation, as well as variety of other functions in different target tissues (1–3). The uterus is a primary target of estrogen for various functions during the reproductive cycle and pregnancy (4). Estrogenic effects in uterus are known to be biphasic: early (phase I) responses that occur within 6 h, whereas the late (phase II) responses occur between 18–30 h. In mice, estrogen induces uterine epithelial cell proliferation and is essential for the maintenance of normal epithelial morphogenesis, cytodifferentiation, and secretory activity. The biological effect of estrogen is classically known to be mediated through estrogen receptor (ER)α and ERβ, members of the superfamily of nuclear receptors that can function as ligand-inducible transcription factors (5, 6). However, a large body of evidence, primarily based on both pharmacological and genetic studies, strongly suggested that estrogen can have effects without involving nuclear ERs in the uterus (7–13). In this mechanism, we have previously showed that one of the downstream pathways is the rapid onset of canonical Wnt signaling by estrogen in the mouse uterus (9, 10).
The most extensively studied, canonical Wnt-signaling pathway involves regulation of β-catenin. The activation of Wnt signaling stabilizes intracellular β-catenin by antagonizing kinase activity of glycogen synthase kinase 3β. In the absence of Wnt activation, glycogen synthase kinase 3β forms a multimolecular complex via axin (a bridging molecule), adenomatous polyposis coli, and β-catenin, resulting in β-catenin phosphorylation and subsequently its degradation by ubiquitination pathway. The active intracellular β-catenin is considered to be the stabilized dephosphorylated form (14–16), which translocates to the nucleus and forms a complex with downstream effectors such as the lymphoid enhancer factor (Lef)/T cell factor (Tcf) family of transcription factors to stimulate transcription of Wnt target genes (17, 18). The members of this family consist of four members: Lef-1, Tcf-1(Tcf-7), Tcf-3 (Tcf-7l1), and Tcf-4 (Tcf-7l2). They all recognize a nucleotide sequence motif: 5′-CTTTGWW-3′ (where W indicates A or T) for gene transcription (19–21). In general, Tcf/Lef family members possess an identical domain, high mobility group, for DNA binding, and a conserved N-terminal sequence that is required for β-catenin binding. Consistent with the developmental roles of Wnt signaling, tissue-specific distribution of expression of this Tcf/Lef family members has been shown to be regulated during pregnancy primarily in the embryo, as well as in the uterus (22–24). However, their roles in uterine estrogen signaling remain poorly understood.
In the mouse, it is well recognized that uterine estrogen signaling for late growth response is critically dependent on ERα (25). The manifestation of this signaling is believed to be the result of ERα-dependent gene transactivation via interaction with nuclear transcription protein factors on the promoter of target genes. In this regard, we previously showed that Wnt/β-catenin signaling critically controls estrogen-dependent late signaling in mice (10). In the same study, it was speculated that nonclassical Wnt/β-catenin effectors probably mediate a cross talk with ERα, to achieve a combinatorial function to drive DNA transcription. Indeed, our present study shows for the first time that Lef-1 and Tcf-3 genes are regulated by estrogen in the mouse uterus, in an early responsive manner without involving classical ERs. Furthermore, we show that early β-catenin activation correlates with the regulation of expression and interaction of Lef-1/Tcf-3 together with ERα under the direction of estrogen.Most importantly, studies further provide evidence that activated Lef-1/Tcf-3 undergoes cooperative interactions with ERα for gene regulation both at the protein-protein, as well as protein-DNA levels. Selective perturbation of either Wnt/β-catenin or ERα signaling events detects specific regions on certain estrogen-responsive gene promoter at which two classes of transcription factors cooperate with each other to regulate its expression in the mouse uterus. Overall, our studies provide novel evidence to support that a nonclassically induced β-catenin/Lef-1/Tcf-3 signaling potentially controls estrogen-sensitive endogenous gene promoters via cooperative interactions with ERα.
RESULTS
Estrogen Regulates Uterine Lef-1 and Tcf-3 mRNAs without Involving ERα
We have previously shown that 17β-estradiol (E2) activates the early canonical Wnt/β-catenin signaling pathway in the mouse uterus (10). Because activation of the Wnt/β-catenin pathway utilizes downstream effectors Tcf/Lef family of transcription factors to transactivate Wnt signaling target genes, we sought to examine whether the Tcf/Lef family of genes is controlled by E2 in the mouse uterus. Adult ovariectomized mice were given an injection of oil (a vehicle) or E2 (100 ng/mouse) to analyze uterine expression at indicated times (Fig. 1). As shown in Fig. 1A, our initial comparative RT-PCR analysis in wild-type mice revealed that E2 was capable of inducing Ltf gene expression at all time points (Fig. 1A), a well-characterized estrogen-responsive gene in mouse uterus (7, 8), suggesting the tissues were indeed estrogen responsive. In the case of Lef-1, E2 caused rapid up-regulation of Lef-1 mRNAs within 1 h (∼4 fold), followed by a decline from 6 h through 24 h, but remained high (∼2-fold) as compared with the basal level (Fig. 1A). In contrast, Tcf-3 mRNAs were also rapidly up-regulated by E2 within 1 h (∼2-fold) but maintained a gradual increase until 12 h (7 fold); thereafter there was a slight decline at 24 h (Fig. 1A). In the case of Tcf-4, the expression of mRNAs was detected in the uterus, at constitutively higher levels without showing any regulation by E2 (data not shown). In addition, the levels of Tcf-1 mRNAs were below the detection limit (data not shown).
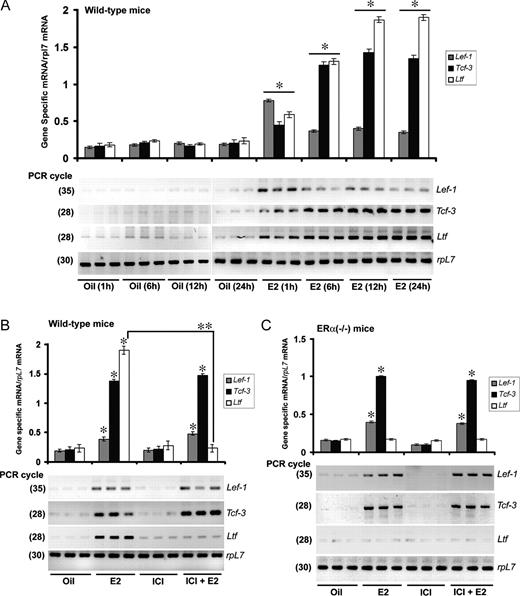
E2 Regulates Uterine Expression of Lef-1 and Tcf-3 mRNAs in Wild-Type and ERα(−/−) Mice, and This Expression Is Unresponsive to ICI A, Temporal effects of E2 in wild-type mice. Adult ovariectomized wild-type mice were given a single injection of E2 (100 ng/mouse) or oil (as vehicle control) and killed at indicated times. In this representative figure, independent preparations of total RNAs from three different mice were analyzed by comparative RT-PCR at indicated PCR cycle numbers to achieve linear amplification for genes of interest. Amplified DNA bands were visualized by ethidium bromide staining. B, Effects of ICI on E2-dependent regulation in wild-type mice. Adult ovariectomized wild-type mice were given an injection of oil, E2 (100 ng/mouse), ICI (500 μg/mouse), or ICI 30 min before an injection of E2 and killed 12 h after the last injection. Comparative RT-PCR analyses using three independent total RNA samples were as described in panel A. C, Effects of ICI on E2-dependent regulation in ERα(−/−) mice. Adult ovariectomized ERα(−/−) mice were given an injection of oil, E2 (100 ng/mouse), ICI (500 μg/mouse), or ICI 30 min before an injection of E2 and killed 12 h after the last injection. Comparative RT-PCR analyses using three independent total RNA samples were followed as described in Fig. 1A. All experiments in panels A–C were repeated at least three times. In the bar plots, the abundance of mRNAs for each gene expression was quantitated by analysis of band intensities using densitometric scanning and was corrected against rpL7. *, Values are statistically different (P < 0.05, Student’s t test) from the corresponding control groups. **, Statistically different between the compared groups (P < 0.05, Student’s t test).
Next we wanted to examine whether the above estrogen-regulated responses are dependent on ERs. To address this question, ovariectomized wild-type or ERα(−/−) mice were given an injection of ICI 182,780 (ICI), E2, ICI in conjunction with E2, or vehicle (oil) and analyzed 12 h after the last treatment, as detailed in Materials and Methods. Previous studies have established that E2 regulates Ltf gene expression in an ER-dependent manner in the uterus (7, 8). Consistently, we observed that the E2-regulated inductive response for Ltf mRNAs in wild-type mice was indeed antagonized by ICI, whereas ICI alone was not effective (Fig. 1B). Furthermore, an E2-dependent Ltf gene response was undetected in ERα(−/−) mice (Fig. 1C). Overall, these results suggest that ICI was indeed capable of antagonization of an ER-dependent uterine gene expression. In contrast, our analysis of expression for Lef-1 and Tcf-3 mRNAs revealed that ICI was unable to oppose E2-dependent gene responses in both wild-type and ERα(−/−) mice (Fig. 1, B and C), whereas ICI was not responsive by itself. Collectively, these results suggest that estrogen temporally regulates Lef-1 and Tcf-3 mRNAs without involving ER.
Estrogen Prompts Lef-1 and Tcf-3 Proteins Temporally in Conjunction with Activation of β-Catenin in the Mouse Uterus
We next asked whether this estrogen-regulated response, as observed in RNA level, was reflected in protein level. Ovariectomized wild-type mice were injected with E2 (100 ng/mouse) and analyzed at 1, 2, 6, and 24 h. Oil-injected mice at 24 h were used as a vehicle control. As shown in Fig. 2, our Western blot analysis clearly demonstrated that Lef-1 was dramatically up-regulated (∼4.5-fold) by E2 at 2 h, then sharply declined by 6 h, but again increased (∼2-fold) at 24 h. In contrast, Tcf-3 showed up-regulation (∼3 fold) by E2 after 6 h and then was maintained high until 24 h. Because activated β-catenin essentially utilizes Tcf/Lef for gene transactivation function, we also analyzed the activated and total β-catenin in uterine extracts after E2 treatment. Our analysis revealed that E2-dependent activation of β-catenin was strongly induced by 2 h and maintained through 24 h, whereas the total pool of β-catenin had also somewhat increased during this time frame. These results are consistent with our previous observations (10). Because we suspected that canonical Wnt pathway activation probably mediates a cross talk with ERα to maintain estrogen signaling in the mouse uterus (10), we wanted to examine further the status of ERα levels under above similar conditions. Our results clearly revealed that ERα is abundantly expressed in the uterus by E2. Overall, results suggest that the status of canonical activation of β-catenin is closely followed by the up-regulation of Lef-1 and Tcf-3 levels by E2 in the mouse uterus.
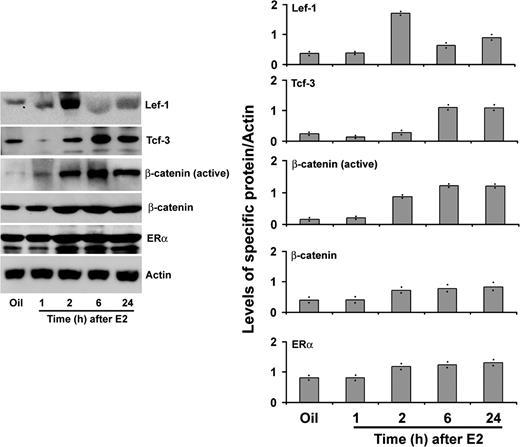
Temporal Analysis of E2-Dependent Regulation of Lef-1, Tcf-3, Activated/Total β-Catenin, and ERα Proteins in Uterine Tissue Extracts of Wild-Type Mice Adult ovariectomized mice were given a single injection of E2 (100 ng/mouse) and killed at indicated times. Mice injected with oil were killed after 24 h and served as control. Whole uterine tissue extracts were analyzed by Western blotting using primary antibodies against Lef-1, Tcf-3, β-catenin (active), β-catenin, ERα, and actin. Relative changes in protein levels were measured by densitometric analysis of band intensities followed by correction with that of β-actin. Values with range of responses from two different set of experiments are shown as dots in the bar plots.
E2 Influences Molecular Association between ERα and β-Catenin-Stimulated Lef-1/Tcf-3 in a Time-Specific Manner in the Mouse Uterus
It is well established that canonical Wnt signaling activation leads to molecular association between β-catenin and Tcf/Lef proteins for their gene transcription function; we thus wanted to examine whether estrogen-stimulated β-catenin is capable of interaction with the inducible Lef-1/Tcf-3 proteins in the mouse uterus. Moreover, we wanted to examine whether this activated Lef-1/Tcf-3 can have a cross talk with ERα via protein-protein interaction. To investigate these possibilities, coimmunoprecipitation studies were carried out in oil- or E2-treated uterine tissue extracts, using β-catenin or ERα antibodies, followed by Western blotting for detection of associated proteins, β-catenin, Lef-1, Tcf-3 or ERα, present in the complex. As shown in Fig. 3A, the interaction between β-catenin and Lef-1 was indeed detected at both 2- and 24-h time points by E2. In contrast, Tcf-3 was interacted with β-catenin primarily at 24 h by E2 (Fig. 3A). Similar results were obtained after immunoprecipitation with Lef-1 or Tcf-3 (data not shown). Furthermore, analysis of β-catenin pulled-down protein complex also detected ERα association after E2 treatment (Fig. 3A). Moreover, ERα-pulled down assays also detected Lef-1 at both time points, whereas Tcf-3 was detected primarily at the latter time point (Fig. 3B). Interestingly, these interactions also appeared to contain β-catenin (Fig. 3B), suggesting that Lef-1/Tcf-3-mediated interaction with ERα depends on activation by β-catenin. Additionally, immunoprecipitation using preimmune serum did not detect any specific bands for β-catenin, Lef-1, Tcf-3, and ERα by Western blotting (data not shown). Collectively, these results suggest that estrogen-directed β-catenin activation leads to molecular association between Lef-1/Tcf-3 and ERα in a multimeric complex in timely fashion in the mouse uterus.
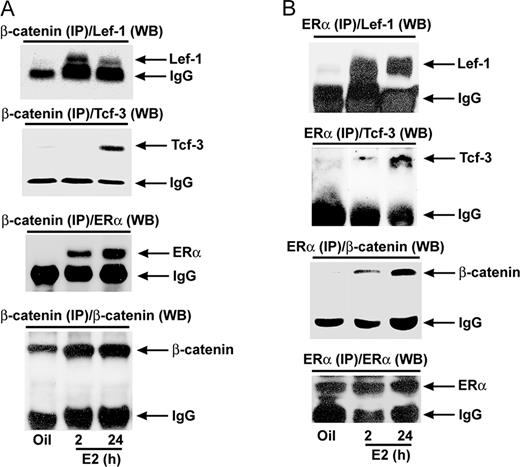
Analysis of E2-Dependent Protein-Protein Interaction between Wnt/β-Catenin-Activated Tcf-3/Lef-1 and ERα in the Mouse Uterus Adult ovariectomized wild-type mice were given a single injection of E2 (100 ng/mouse) and killed at indicated times. Mice injected with oil were killed after 24 h and served as control. Whole uterine tissue extracts were immunoprecipitated with β-catenin (A)- or ERα (B)-specific antibodies followed by Western blotting for Lef-1, Tcf-3, β-catenin, and ERα. Arrows denote position of detected protein bands. The intense band detected at approximately 55 kDa in all panels represents heavy chain subunit of IgG (this is shown as internal loading control). In control experiments, imunoprecipitation using normal serum did not detect any specific bands by Western blotting (data not shown). These experiments were repeated at least three times with similar results. IP, Immunoprecipitation; WB, Western blot.
ERα Colocalizes with Lef-1 and Tcf-3 in the Uterine Luminal and Glandular Epithelial Cells by E2
Previously, we demonstrated that estrogen-driven activated β-catenin is exclusively localized in uterine luminal and glandular epithelial cell nuclei in wild-type mice (10). Consistent with our previous report, our dual immunofluorescence studies for ERα and Lef-1/Tcf-3 revealed that there is an intense nuclear colocalization of these proteins primarily in the uterine luminal and glandular epithelial cells in wild-type mice after treatment of E2 for 24 h, although a weak subepithelial stromal expression of Lef-1 or Tcf-3 was also detected in a colocalization manner with ERα (Fig. 4). However, it is to be noted that our analyses of expression of these proteins after oil treatments also showed a colocalization pattern of much lesser intensity in similar cell types (Fig. 4).
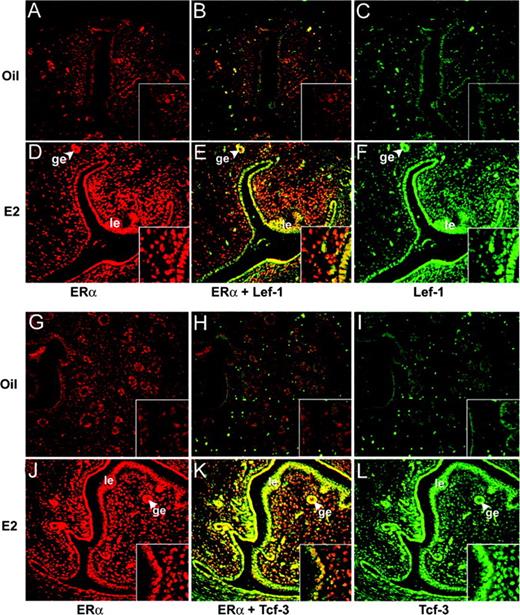
Dual Immunofluorescence Staining for Lef-1 (A–F) or Tcf-3 (G–L) in Conjunction with ERα in the Uterus by Oil (A–C and G–I) or E2 (D–F and J–L) Ovariectomized wild-type mice were treated with oil or E2 (100 ng/mouse) and killed after 24 h. Formalin-fixed paraffin-embedded uterine sections (6 μm) were incubated with primary antibodies for ERα and Tcf-3/Lef-1 followed by incubation of secondary antibodies tagged with Texas red (red) and FITC (green), respectively. Superimposed (merge) images showing yellow-colored cells represent expression of both. No immunostaining was noted when sections were incubated with preimmune serum instead of primary antibody (data not shown). Pictures (A–L) were taken at ×20, and the insets were at ×40. le, Luminal epithelium; ge, glandular epithelium. These experiments were repeated three times with three to four mice in each group, and similar results were obtained.
Chromatin Immunoprecipitation (ChIP) Analysis Identifies Differential Recruitments of ERα and Lef-1/Tcf-3 at the Level of Estrogen-Sensitive Uterine Gene Promoters in Mice
Previous studies established that estrogen-dependent uterine stimulation of c-Myc and Cdkn1a genes occurs early (26–28), whereas that of Pgr, Ltf, Ccnd1 and Mmp7 genes are influenced during the late phase (8, 29, 30). Furthermore, it has been well documented that estrogenic regulation of c-Myc, Cdkn1a, Pgr, Ltf, and Ccnd1 are mediated via ER (8, 11, 28, 31–33). Interestingly, studies also indicate that c-Myc, Cdkn1a, Ccnd1, and Mmp7 are regulated via Wnt-responsive element (WRE) (34–37). However, endogenous regulation of these uterine gene promoters via specific responsive sites has been poorly understood. Therefore, as described in Materials and Methods, a computer-based analyses for at least 10-kb regions of the promoters were undertaken to identify the putative sites, which were confirmed by the ChIP-PCR. In this regard, it is to be noted that our analyses have included previously reported consensus sites for the presence of estrogen-responsive ERα occupancy through estrogen-responsive element (ERE) (at position A, Fig. 5) for Ltf and Pgr (11), and through activator protein 1 (AP1) (at position A, Fig. 5) for Ccnd1 (11). A Tcf/Lef-responsive region (at position A, Fig. 5) for mouse Mmp7, as previously reported (38), was also included in our analysis. Different proximal regions (A, B, and C), containing ERE, SP1 (Sp1 transcription factor binding) or WRE sites in the promoters for c-Myc and Cdkn1a, and regions for Ccnd1, Pgr, Ltf, and Mmp7 (B and C) were designed based on the computer. It is interesting to note that there is a distinct region at position B (Fig. 5) for Ltf, Ccnd1, Cdkn1a, and Mmp7 gene promoters, containing two consensus sites (either for WRE and ERE/AP1/SP1) in close proximity that were not separable by our ChIP-PCR analysis.
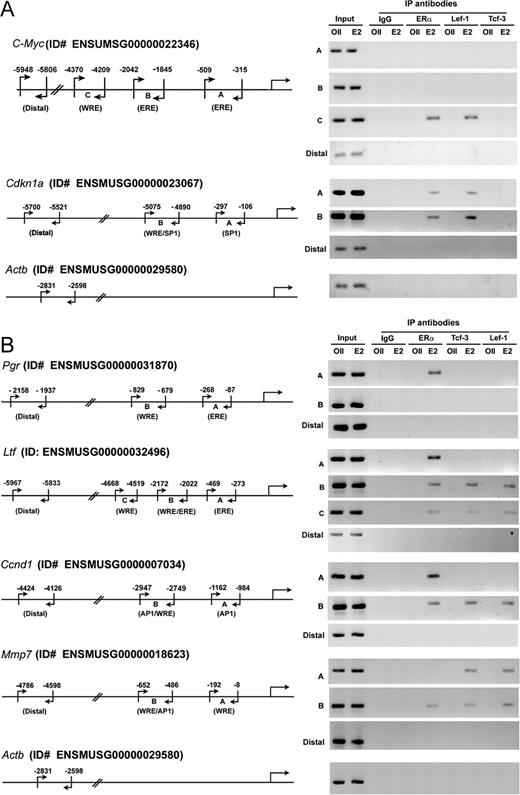
Analysis of Estrogen-Dependent Recruitments of ERα and Lef-1/Tcf-3 at Specific Sites of c-Myc, Cdkn1a, Pgr, Ltf, Ccnd1, and Mmp-7 Gene Promoters Analysis of recruitments for ERα and Lef-1/Tcf-3 on endogenous uterine gene promoters for c-Myc and Cdkn1a at 2 h (A), and for Pgr, Ltf, Ccnd1, and Mmp7 at 24 h (B) after E2 or oil treatments. Adult ovariectomized wild-type mice were given a single injection of E2 (100 ng/mouse) or oil (as vehicle control) and killed at indicated times. ChIP analyses were performed using ERα, lef-1, or Tcf-3 antibodies, or normal serum IgG (as control), followed by PCR. The specific regions as depicted in this figure were analyzed by a set of primers as described in Materials and Methods. The presence of the promoter-specific DNA before immunoprecipitation was confirmed by PCR (input). The details for PCR amplification and the number of cycles parameter were the same as described in Materials and Methods. Analysis of Actb gene promoter was used in parallel (as a control) to compare the status of transcription factors recruitment on an estrogen-insensitive gene promoter. The amplified product sizes (bp) were for c-Myc: 170 (A), 199 (B), 162 (C), and 159 (distal); Cdkn1a: 192 (A), 186 (B), and 180 (distal); Pgr: 191 (A), 151(B), and 222 (distal); Ltf: 199 (A), 151 (B), 150 (C), and 249 (distal); Ccnd1: 179 (A), 199 (B), and 300 (distal); Mmp7: 185 (A), 167 (B), and 167 (distal); Actb: 234. ID#, Identification number; IP, immunoprecipitation.
Because estrogen-dependent activation, as well as interaction of Lef-1/Tcf-3 toward ERα occurred differentially with times, we wanted to examine whether these transcription factors provide evidence for specific or differential recruitments on selected uterine promoters for early genes c-Myc and Cdkn1a (Fig. 5A) or for late genes Pgr, Ltf, Ccnd1 and Mmp7 (Fig. 5B) after E2 treatment of mice. Consistent with above predominant expression and activation of Lef-1, but not Tcf-3 by E2 at 2 h, our chromatin recruitment analysis revealed that Lef-1, but not Tcf-3, was indeed detected at promoter regions that contain WRE sites for c-Myc and Cdkn1a genes (Fig. 5A). In contrast, binding of Lef-1 or Tcf-3 was noted to the promoters for late responsive genes Ltf, Ccnd1, and Mmp7 by E2 after 24 h, whereas Pgr was unable to show any such interactions (Fig. 5B). These results are again consistent with the status of expression and activation of Lef-1 and Tcf-3 at these time points by E2. To our surprise, Lef-1 occupancy was noted at position A (which does not contain a recognizable WRE, but an SP1 site) for Cdkn1a gene promoter by E2 after 2 h (Fig. 5A).
In the case of ChIP studies for ERα, as expected, our analysis detected its occupancy at promoter regions, which contain designated ERE/AP1/SP1 sites for Cdkn1a, Pgr, Ltf, Ccnd1, and Mmp7 genes by E2 (Fig. 5, A and B). However, the c-Myc gene was unable to recruit ERα, despite the presence of two detected EREs (at positions A and B) (Fig. 5A). In contrast, to our surprise, the recruitment of ERα was noted by E2 at position C for c-Myc and Ltf promoters, that do not possess an ERα-recognizable cis-acting element, but WRE site (Fig. 5, A and B).
From the above analyses, note that both ERα and Lef-1/Tcf-3 were occupied at a common region that contains a single recognizable site, e.g. c-Myc (WRE at position C), Cdkn1a (SP1 at position A), and Ltf (WRE at position C) (Fig. 5, A and B). In contrast, both factors were able to recruit at a distinct region that contains dual recognizable sites, e.g. at position B for Cdkn1a (WRE and SP1), Ltf (WRE and ERE), Ccnd1 (WRE and AP1), and Mmp7 (WRE and AP1) (Fig. 5, A and B).
The above detected interactions by E2 were specific, because the oil treatment did not reveal such results (Fig. 5, A and B). Moreover, the detected bands were specific, because immunoprecipitation with normal serum (IgG) or analyses of distal regions for the above estrogen-sensitive gene promoters, as well as an estrogen-insensitive gene promoter (Actb) did not reveal any such results (Fig. 5, A and B). In addition, detection of bands in both oil- or E2-treated groups before immunoprecipitation (input) indicates that the presence of a gene-specific promoter was not a limiting factor (Fig. 5, A and B).
Overall, our results indicate that estrogen directs primarily Lef-1 recruitment on early-responsive gene promoters, whereas the same directs recruitment of both Lef-1 and Tcf-3 in an indistinguishable manner on late-responsive gene promoters. Moreover, our results show that DNA-specific interactions of ERα and Lef-1/Tcf-3 appeared to be mediated either individually or in combination on distinct regions of estrogen-sensitive endogenous promoters.
Blockage of Wnt/β-Catenin or ERα-Mediated Signaling Pathway Revealed Cooperative Interactions by ERα and Lef-1/Tcf-3 at the level of Endogenous Promoters for Estrogenic Regulation of Mouse Uterine Genes
We have shown previously that adenovirus-driven overexpression of secreted frizzled related protein-2 (SFRP-2), an antagonist of Wnt signaling, abrogated E2-induced canonical activation of β-catenin pathway in the mouse uterus (10). We have undertaken a similar approach, to examine the above E2-dependent recruitments of Lef-1/Tcf-3, as well as the recruitment of their interacted ERα on both early and late gene promoters. As previously demonstrated (10), we have observed that E2-induced uterine activation of β-catenin was suppressed by the SFRP-2 overexpressing virus rAdSFRP-2(S) (data not shown). Because above studies provided evidence for the presence of E2-dependent recruitments of Lef-1/Tcf-3 and ERα on certain regions of early and late gene promoters, we next analyzed their effects after perturbation of Wnt signaling (Figs. 6A and 7A). Consistent with the above results, recruitments for Lef-1 (Fig. 6A) on c-Myc and Cdkn1a, and for Tcf-3 (Fig. 7A) and Lef-1 (data not shown) on Ltf, Ccnd1, and Mmp7 were revealed by control virus expressing green fluorescent protein (GFP) (rAdGFP) in the presence of E2. In contrast, the inclusion of rAdSFRP-2(S) virus in conjunction with E2 caused total elimination of recruitments for Lef-1 on early genes (Fig. 6A) or for Tcf-3 (Fig. 7A) and Lef-1(data not shown) on late genes Ltf, Ccnd1, and Mmp7, suggesting the nature of recruitments for these transcription factors was indeed dependent on β-catenin-mediated activation. Interestingly, to our surprise, we also noted that the above loss of recruitments for Tcf-3/Lef-1 also severely affected ERα occupancy at distinct regions of four genes (e.g. c-Myc at position C; Cdkn1a at position A; Ltf at positions B and C; and Mmp7 at position B) (Figs. 6A and 7A), suggesting these gene-specific promoters possess regulation via cooperative interactions between Lef-1/Tcf-3 and ERα under the direction of E2 in the uterus. In this regard, it is important to note that the loss of Tcf-3 (Fig. 7A) or Lef-1 (Fig. 6A) did not cause concomitant elimination of ERα recruitments for cyclin D1 and Cdkn1a at position B, respectively, suggesting that this ERα is independently interacted without involving Lef-1/Tcf-3. Furthermore, amplification of bands before ChIP (input) reveals that the presence of DNA-specific regions was not a limiting factor for interactions (Figs. 6A and 7A).
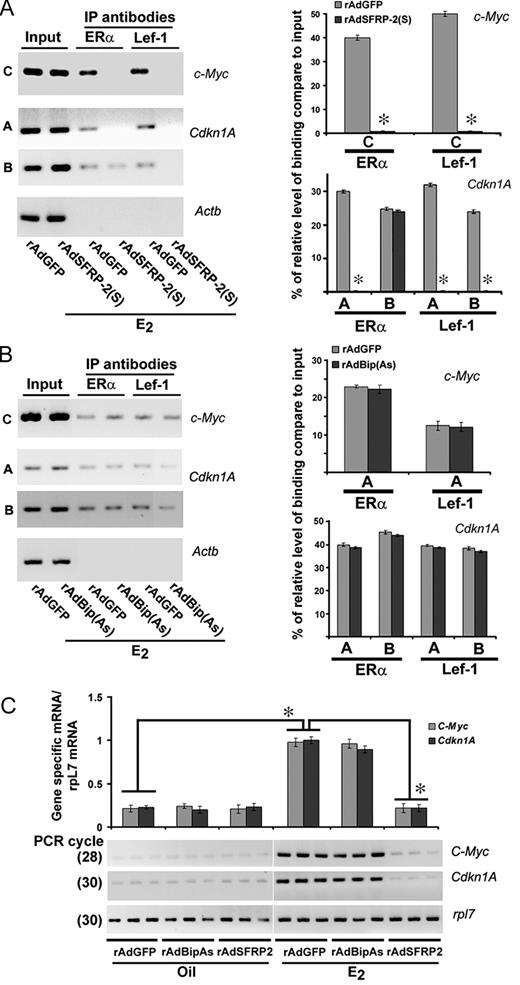
Selected Perturbation of Wnt/β-Catenin or ERα Signaling Axis Determines a correlation for DNA-Specific Recruitments of Lef-1/Tcf-3 and/or ERα with the Alteration of Estrogenic Early Gene Expression in the Mouse Uterus A, Analysis of recruitment of ERα and Lef-1 on c-Myc, Cdkn1A, and Actb promoters. ChIP analysis was performed using ERα and lef-1-specific antibodies in the uteri of mice, after administration of rAdSFRP2(S) or rAdGFP (as control), followed by E2 (100 ng/mouse) for 2 h. B, Analysis of recruitment of ERα and Lef-1 on c-Myc, Cdkn1A, and Actb promoters. ChIP analysis was performed using ERα and lef-1-specific antibodies in the uteri of mice, after administration of rAdBip(As) or rAdGFP (as control), followed by E2 (100 ng/mouse) for 2 h. Representative figures for recruitments of individual transcription factors on different gene promoters are shown (A and B). The amplified product sizes for different regions of the promoters were the same as described in Fig. 5. Three independent analyses were performed to compare the results, as shown in the bar plot (A and B). The quantitation of relative levels for binding of transcription factors on different gene promoters is represented as percent of input. *, Values are statistically different (P < 0.05, Student’s t test) from the corresponding control groups. C, RT-PCR analysis of c-Myc and Cdkn1A gene expression after adenovirus administration. Uterine tissues were collected after rAdBipAs, rAdSFRP-2(s), or rAdGFP (as control) administration, followed by oil or E2 (100 ng/mouse) for 2 h. Ribosomal protein L-7 (rpl-7) was used as a constitutive gene control. Comparative RT-PCR analyses using three independent total RNA samples were followed as described in Fig. 1A. These experiments were repeated at least three times. In the bar plots, the abundance of mRNAs for each gene expression was quantitated by analysis of band intensities using densitometric scanning and was corrected against rpL7. *, Values are statistically different (P < 0.05, Student’s t test) from the corresponding control groups. IP, Immunoprecipitation.
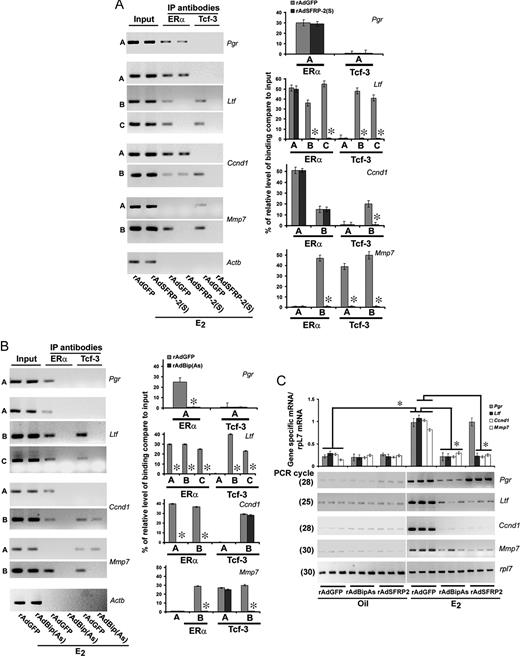
Selected Perturbation of Wnt/β-Catenin or ERα Signaling Axis Determines a Correlation for DNA-Specific Recruitments of Lef-1/Tcf-3 and/or ERα with the Alteration of Estrogenic Late Gene Expression in the Mouse Uterus A, Analysis of recruitment of ERα and Lef-1 on Pgr, Ltf, Ccnd1, Mmp7, and Actb promoters. ChIP analysis was performed using ERα and lef-1-specific antibodies in the uteri of mice, after administration of rAdSFRP2(S) or rAdGFP (as control), followed by E2 (100 ng/mouse) for 24 h. B, Analysis of recruitment of ERα and Lef-1 on c-Myc, Cdkn1A, and Actb promoters. ChIP analysis was performed using ERα and lef-1-specific antibodies in the uteri of mice, after administration of rAdBip(As) or rAdGFP (as control), followed by E2 (100 ng/mouse) for 24 h. Representative figures for recruitments of individual transcription factors on different gene promoters are shown (A and B). The amplified product sizes for different regions of the promoters were the same as described in Fig. 5. Three independent analyses were performed to compare the results, as shown in bar plot (A and B). The quantitation of relative levels for binding of transcription factors on different gene promoters is represented as percent of input. *, Values are statistically different (P < 0.05, Student’s t test) from the corresponding control groups. C, RT-PCR analysis of c-Myc and Cdkn1A gene expression after adenovirus administration. Uterine tissues were collected after rAdBipAs, rAdSFRP-2(s), or rAdGFP (as control) administration, followed by oil or E2 (100 ng/mouse) for 24 h. Ribosomal protein L-7 (rpl-7) was used as a constitutive gene control. Comparative RT-PCR analyses using three independent total RNA samples were as described in Fig. 1A. These experiments were repeated at least three times. In the bar plots, the abundance of mRNAs for each gene expression was quantitated by analysis of band intensities using densitometric scanning and was corrected against rpL7. *, Values are statistically different (P < 0.05, Student’s t test) from the corresponding control groups. IP, Immunoprecipitation.
Previously we also established that adenovirus-driven suppression of Bip, an endoplasmic reticulum protein, specifically interferes with ERα-mediated gene activation functions primarily for late-responsive genes by E2 in the mouse uterus (11). Here, we also examined whether this similar approach is effective to alter ERα and/or Lef-1/Tcf-3 recruitments on the above gene promoters. In our initial studies, consistent with our previous report (11), we observed that rAdBipAs was indeed able to suppress E2-dependent uterine Bip expression (data not shown). Furthermore, as shown in Fig. 7B, we noted that suppression of Bip indeed affected the recruitment of ERα on late-responsive genes Pgr, Ltf, and Ccnd1 as reported previously (11), as well as Mmp7 by E2. Interestingly, the loss of ERα recruitment leads to complete elimination of Tcf-3 (Fig. 7B) or Lef-1 (data not shown) occupancy at distinct regions for Ltf at positions B and C, and for Mmp7 at position B. Consistent with the above, these results again suggest a cooperative interaction mediated by Tcf-3/Lef-1 and ERα at the level of Ltf and Mmp7 gene promoters in the presence of E2. It should be noted here again that loss of ERα did not cause elimination of Tcf-3 (Fig. 7B) or Lef-1 (data not shown) for cyclin D1 at position B, suggesting that ERα is independently recruited by E2. Moreover, detection of bands before ChIP (input) reveals the presence of DNA-specific regions, suggesting the above loss was not due to a limitation of the presence of DNA fragments. Furthermore, consistent with our previous observations (11), suppression of Bip via rAdBipAs was not antagonistic for ERα recruitments on early genes in the presence of E2 (Fig. 6B). In the similar context, E2-dependent Lef-1 recruitment was also not affected (Fig. 6B).
To gain better insights about the signal-specific recruitments of above transcription factors on gene-specific promoters, we next analyzed estrogen-responsive gene expression after the perturbation of individual signaling in the uteri of mice in the presence of oil or E2. Inhibition of Wnt signaling [via rAdSFRP-2(S)] causes repression of E2-dependent expression for early genes c-Myc and Cdkn1a (Fig. 6C) and for late genes Ltf, Ccnd1, and Mmp7, when compared with the control virus (rAdGFP)-injected group in the presence or absence of E2 (Fig. 7C). However, E2-dependent Pgr expression did not show such alteration. In contrast, Bip-mediated inactivation of ERα [via rAdBip(As)] caused attenuation of expression for late genes (Pgr, Ltf, Ccnd1, and Mmp7) (Fig. 7C), but not the early genes (c-Myc and Cdkn1a) (Fig. 6C), when compared against the control virus (rAdGFP)-injected groups in the presence or absence of E2.
Collectively, the above results suggest that two classes of transcription factors Lef-1/Tcf-3 and ERα can have a cross talk for regulation of expression at the level of uterine gene promoters for c-myc, Cdkn1a, Ltf, and Mmp7 by E2. However, cyclin D1 did show the impact of individual signaling for gene expression, but was unable to show any cooperative interactions at the level of its gene promoter. Moreover, we were able to define DNA-responsive regions for estrogen-directed signal-specific interactions of ERα and Lef-1/Tcf-3 on distinct sets of promoters in the mouse uterus.
DISCUSSION
Our recent concepts regarding canonical Wnt signaling suggest that functional activation of β-catenin-mediated pathway may interact with multiple nuclear signal transducers to coordinate tissue or cell-specific functions (10, 39–44). Consistent with this current notion, we demonstrated here, for the first time, that protein-protein interaction exists between activated Tcf-3/Lef-1 and ERα for estrogen signaling in the mouse uterus. More specifically, using an adenovirus-directed approach for selective abrogation of individual signaling events induced by activated Tcf/Lef or ERα, we were able to establish the presence of a synergistic control via these two classes of factors at the level of chromatin that directs endogenous regulation of estrogen-sensitive genes. This remarkable finding of ERα-independent early β-catenin/Lef/Tcf expression contributing to ERα-dependent gene-regulatory events in the uterus adds new insights to our understanding of uterine biology.
In the present study, we observed that of four Tcf/Lef families of genes, only Lef-1 and Tcf-3 were up-regulated by E2 both at mRNA and protein levels (Figs. 1A and 2). In this regard, expression of both genes at mRNA levels appeared to be induced rapidly within 1 h, although they were differentially induced with time at protein levels. However, Lef-1 is primarily regulated early (by 2 h), as opposed to Tcf-3, which was undetected until 6 h but during the late times after E2, both proteins appeared to be detected with a predominant expression for Tcf-3. These results suggest that Lef-1 is a primary downstream effector for estrogen-dependent Wnt/β-catenin signaling during the early phase, whereas Lef-1/Tcf-3 primarily directs late signaling. This speculation is consistent with our temporal analysis of β-catenin activation in the mouse uterus by E2 (Fig. 2). The discrepancy with respect to the time of accumulation of these protein factors, as compared with their mRNAs, under the direction of E2 appears to suggest that they may possess differential control at the level of translational and/or degradation efficiency in the uterus.
We have previously demonstrated that several Wnt-signaling genes, including β-catenin, SFRP-2, Wnt4, Wnt5a, and Fz were regulated early by estrogen in an ER-independent manner in the mouse uterus (2, 10). Consistent with our previous observations, in this study we also noted that the regulation of Lef-1 and Tcf-3 mRNAs does not require ERs during their effects by E2 in the mouse uterus (Fig. 1, B and C). The mechanism of ER-independent control of early nonclassical uterine genes is currently unknown. However, it should be noted that one of the early canonical Wnt-signaling genes, such as SFRP-2, has been shown to be regulated by E2 at the level of mRNA without involving any intermediary protein synthesis (9), suggesting posttranslational mechanism, viz. phosphorylation and/or dephosphorylation of unknown proteins could be involved. In this regard, a recently characterized estrogen-binding membrane receptor GPR30 is indeed interesting. Our recent unpublished work indicate that GPR30 is regulated by E2 in the uteri of both wild-type and ERα−/− mice (Ray, S., and S. K. Das, unpublished observations). We are currently exploring a possibility for the role of GPR30 role in estrogenic regulation of ER-independent genes in the mouse uterus.
In our studies, we used ERα−/− mice that were generated by Lubahn et al. (25). In general, the uteri of these null mice showed 5–10% E2 binding as compared with wild type, but failed to exhibit late physiological responses to E2. Although these null mice showed some leakiness in respect to the generation of spliced ER transcripts and proteins (45, 46), their significance in uterine biology is not known. Two smaller transcripts resulted from the disrupted NEO sequence. One of the variants was generated by a frame shift mutation with early termination by a stop codon, whereas the other maintained ER reading frame and encodes a smaller mutant ER (46). In vitro expression studies of the latter form resulted in a smaller protein and detected transactivation function by E2; however, it was completely abolished by ICI 182,780. It should be noted here that gene responses, as we observed by E2 in the presence of ICI 182,780 in uteri of wild-type and ERα−/− mice, suggest that estrogenic responses are indeed ER independent but ligand dependent.
It has been well documented that activated Wnt/β-catenin signaling leads to cross talk with numerous transcription factors (39). Because our previous studies implicated a probable convergence between ERα and Wnt/β-catenin signaling in estrogen-regulated uterine biology, we examined protein-protein interaction between the effectors Tcf/Lef and ERα under the direction of estrogen in the mouse uterus. Our results clearly revealed that the estrogen-dependent activated β-catenin indeed interacted not only with Lef-1/Tcf-3, but also with ERα in a multimeric complex (Fig. 3, A and B). Furthermore, the time-specific interaction of Lef-1 and Tcf-3 toward ERα closely resembles their uterine accumulation under the direction of estrogen (Fig. 2vs. Fig. 3, A and B). In conjunction with this, our dual immunofluorescence studies strongly suggested that estrogen-directed interactions between each of these factors could be achieved primarily in uterine epithelial cells, because activated β-catenin has been shown to be exclusively localized in this cell layer by E2 (10). Overall, these results strongly support a contention that epithelial cell layer is the primary target site for estrogen-directed regulation by these two different classes of transcription factors.
For our analyses, consistent with the above speculation, we selected several estrogen-responsive genes (viz. c-Myc, Cdkn1a, Ltf, Ccnd1, and Mmp7) that are known to be regulated exclusively in uterine epithelial cells, with an exception for Pgr, which showed expression both in epithelial and subluminal stromal cells (8, 26, 27, 29, 30). Indeed, our ChIP analyses clearly revealed that estrogen directs occupancy of both ERα and Lef-1/Tcf-3 in a concomitant fashion at distinct regions of the promoters for c-Myc, Cdkn1a, Ltf, Ccnd1, and Mmp7 (Fig. 5). In this respect, we noted certain regions of the promoters that contain only a single consensus site, but recruited both ERα and Lef-1/Tcf-3, suggesting these two proteins may be occupied at that site in an associated manner (see Fig. 5, at position C for c-Myc, at position A for Cdkn1a, and at position C for Ltf). This speculation is consistent with our observation of protein-protein interaction. However, Pgr appeared to be regulated only by ERα (Fig. 5), suggesting that this gene is probably not a target for Wnt/β-catenin signaling under the direction of E2. Furthermore, it is interesting to note that our analyses revealed that DNA recruitments of Lef-1/Tcf-3 and ERα by E2 detected at 2 h for early genes were absent during the late time point at 24 h (data not shown). Similarly, the recruitments detected at 24 h for late genes were also undetected during the early time point at 2 h (data not shown). These results further emphasize that identified recruitments sites on gene promoters are indeed specific with time for gene regulation of expression by estrogen.
Previous studies have established that both Wnt/β-catenin and ERα signaling events are crucial for estrogen-regulated uterine responses in mice (10, 25). However, the relationship between these two signaling pathways has never been explored in estrogen-regulated uterine biology. Here, we have undertaken for the first time an approach to dissect out the roles of an estrogen-induced individual signaling pathway that is mediated by either ERα or Wnt/β-catenin/Lef-1/Tcf-3 for gene regulation. In this regard, our ChIP analysis, in conjunction with expression studies, after perturbation of individual signaling via adenovirus approaches, revealed that both signaling events cooperate for the regulation of expression via interaction at the level of certain regions of endogenous gene promoters for c-Myc, Cdkn1a, Ltf, and Mmp7 genes (Figs. 6 and 7). It should be noted here that cyclin D1 did not show any cooperative interaction via these transcription factors at the promoter level, although individual signaling appeared to be essential for its expression (Fig. 7, A–C). Overall, these results are consistent with the known regulation via ERα for c-Myc, Cdkn1a, Ltf, Pgr, and Ccnd1 genes (8, 11, 28, 31–33), as well as via the Wnt/β-catenin pathway for Myc, Ccnd1, Cdkn1a, and Mmp7 genes (34–37) in various systems.
In our studies, we have noted an interesting feature, i.e. that both Lef-1 and Tcf-3 are similarly recruited to the late responsive gene promoters Ltf, Ccnd1, and Mmp7 by E2. However, in this regard it should be noted that Lef-1 and Tcf-3 have been shown to possess differential gene regulation acting as activator and repressor, respectively (47). Thus, it is possible that these two factors may potentially balance the regulation of gene transcriptional status.
Our studies clearly revealed for the first time that Ltf gene is indeed regulated by Wnt/β-catenin pathway. In this regard, Lef-1/Tcf-3 was specifically recruited at two distinct regions (at positions B and C). Interestingly, these sites essentially recruited ERα in cooperation with Lef-1/Tcf-3, because perturbations of individual signaling affect the other recruitment in conjunction with the suppression of expression. In addition, the recruitment of ERα by itself in the proximal region (at position A) appeared to be also essential for ERα-dependent gene regulation. This proximal interaction for ERα has previously been reported for gene regulation (11, 32), although the control of additional upstream regulatory regions in gene expression has been previously postulated (32). To a similar notion, the regulation of Mmp7 by ERα is also clearly defined for the first time in this report.
Previous studies have already established that cyclin D1/D2 crucially controls estrogen-dependent uterine growth response (29, 48). Our studies demonstrating the regulation of cyclin D1 gene by each of ERα or Lef-1/Tcf-3 signaling systems, without involving any cooperation at the level of promoter, is consistent with previous observations that either of these pathways directly impacts estrogen-regulated growth response (10, 25).
Our studies strongly suggest that Lef-1/Tcf-3 should impact estrogen-regulated uterine biology. In this regard, homozygous null mice either for Tcf-3 or Lef-1 die during the embryonic or neonatal stages, respectively (49, 50). Thus, uterus-specific conditional knock-out studies will be needed in the future to confirm their specific roles in uterine estrogen-signaling.
Here we attempted to demonstrate the comprehensive dynamic nature of sequential events resulting in transcriptional activation of selective estrogen-regulated uterine genes. Currently, hormone-mediated activation of Wnt/β-catenin and its cooperation with different nuclear transcription factors is considered to be a major area of research for therapeutic manipulation of various endocrine pathologies (39). In this regard, our studies demonstrating estrogen-mediated interactions between ERα and Wnt/β-catenin/Tcf/Lef signaling pathways are likely to contribute to the development of different therapeutic strategies.
MATERIALS AND METHODS
Animals and Injections
Adult CD-1 (Charles River Laboratories, Wilmington, MA) females were housed in our institutional animal care facility according to National Institutes of Health (NIH) and institutional guidelines for laboratory animals. In some studies, wild-type and ER α(−/−) littermate females (C57BL/6J/129/J) were also analyzed in parallel. Both mice were produced by crossing of heterozygous females and males in our animal facility (25). In general, 8- to 10-wk-old adult females were ovariectomized and rested for 10 d before they received any injections. Mice were injected with sesame seed oil (0.1 ml/mouse), 17β-estradiol (E2) (100 ng/mouse), ICI 182,780 (ICI, 500 μg/mouse) or the same dose of ICI 30 min before E2 injection. They were killed at indicated times after the last injection. All of the test agents were dissolved in sesame oil and injected sc (0.1 ml/mouse).
RT-PCR
Procedures for the reverse transcription and comparative PCR followed previously described methods (51, 52) with some modifications. In brief, total RNA was extracted using Trizol according to the manufacturer’s instruction. Reverse transcription with oligo(dT) priming was performed to generate cDNAs from 4 μg total RNA using Superscript II following the instruction provided by the manufacturer. DNA amplification was carried out with Taq DNA polymerase (Invitrogen, San Diego, CA) using the following primers: Tcf-1 (Tcf-7) (233 bp), 5′-GCCAGAAGCAAGGAGTTCAC-3′ and 5′-TACACCAGATCCCAGCATCA-3′; Lef-1 (386 bp), 5′-CTCATCACCTACAGCGACGA-3′ and 5′-TGAGGCTTCACGTGCATTAG-3′; Tcf-3 (Tcf-7l1) (384 bp), 5′-GAGTGCGAAATCCCCAGTTA-3′ and 5′-ATGCATGGCTTCTTGCTCTT-3′; Tcf-4 (Tcf-7l2) (475 bp), 5′-CTCACGCCTCTCATCACGTA-3′ and 5′-TGAATGCATTAAGGGGCTTC-3′, Mmp-7 (222 bp), 5′-CCCGGTACTGTGATGTACCC-3′ and 5′-GAAGAGGGAGACAGGTGCAG-3′; Cdkn1a (268 bp), 5′-ACAACATTCTCCTCGGGATG-3′ and 5′-GGGGTCCCATTAGTGGCTAT-3; Pgr (425 bp), 5′-GGTGGAGGTCGTACAAGCAT-3′ and 5′-AAATTCCACAGCCAGTGTCC-3′; Ltf (306 bp), 5′-CGAAGCACGCACGAATGACAAAGA-3′ and 5′-ATCACACTTGCGCTTCTCCT-3′; Ccnd1 (329 bp), 5′-GCGTACCCTGACACCAATCT-3′ and 5′-CACAACTTCTCGGCAGTCAA-3′; c-Myc (426 bp), 5′-AGAGCTCCTCGAGCTGTTTG-3′ and 5′-ATCGCAGATGAAGCTCTGG- A-3′ rpL7 (246 bp), 5′-TCAATGGAGTAAGCCCAAAG-3′ and 5′CAAGAGACCGAGCAATCAAG-3′. PCR conditions were 94 C for 4 min and then the appropriate number of cycles (as indicated in figures for gene of interests) for linear amplification using 94 C for 30 sec, 55 C for 30 sec, and 72 C for 45 sec, followed by incubation at 72 C for 10 min. Amplified fragments were separated by electrophoresis on 2% agarose gels and visualized by ethidium bromide staining. The intensity of each band was measured by Scion Image (Scion Corp., Frederick, MD), and the abundance of mRNAs for each gene expression was corrected against rPL7.
Antibodies
The affinity-purified polyclonal antibodies for Bip (catalog no. sc-1050), ERα (catalog no. sc-542), actin (catalog no.sc-1615), PR (catalog no. sc-539), Lef-1 (catalog no. sc-8592), Tcf-3 (catalog no. sc-13026) and Wnt-4 (catalog no. sc-5215) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). GFP antibody (A-11122) was purchased from Molecular Probes, Inc. (Eugene, OR). Antibodies for β-catenin (catalog no. 06-734), active β-catenin (catalog no. 05-665) were purchased from Upstate Biotechnology (Temecula, CA). Secondary antibodies for fluorescein isothiocyanate (FITC)-conjugated donkey antigoat (catalog no. 705-095-147), Texas red-conjugated donkey antirabbit (catalog no. 711-075-152), and peroxidase-conjugated donkey antimouse (catalog no. 715-035-150), donkey antirabbit (catalog no. 711-035-152), or donkey antigoat (catalog no. 705-035-147) were purchased from the Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA).
Immunoprecipitation and Western Blotting
These procedures followed the protocol as previously described (53, 54) with some modifications. In brief, whole uterine tissue proteins were extracted by homogenization with Polytron homogenizer in Tissue-PE lysis buffer (catalog no. 786-181, Geno Technology Inc., St. Louis, MO) containing 1× protease arrest (catalog no. 786-108; Genentech). For immunoprecipitation studies, protein extracts (500 μg) were incubated in a buffer [0.1% Triton X-100, 20 mm HEPES (pH 7.5), 150 mm NaCl, 10% glycerol] containing 1 μg primary antibody conjugated with protein A sepharose beads (catalog no. 17-0780-01, Pharmacia, Uppsala, Sweden) for overnight at 4 C with a gentle shaking. The beads were washed three times with the same buffer, and the bound proteins were eluted by boiling the beads in 1× sodium dodecyl sulfate sample buffer for 5 min. After centrifugation at 10,000 × g for 5 min, supernatants were separated by 10% SDS-PAGE, transferred onto immunoblot polyvinylidene difluoride membrane (catalog no. 162-0177, Bio-Rad Laboratories, Inc.), and Western blotted as previously described (53).
Dual Immunofluorescence
This was followed the protocol as previously described by us (51, 53). In brief, formalin-fixed paraffin-embedded sections (6 μm) were deparaffinized, hydrated, and autoclaved for the antigen retrieval. Nonspecific reaction was blocked by incubation in 5% BSA for 30 min at 37 C. Sections were incubated with primary antibodies at 4 C for overnight with anti-ERα antibody (rabbit IgG, 1:200) and anti-Tcf-3 antibody (goat IgG, 1:100) or anti-Lef-1 antibody (goat IgG, 1:100). The sections were washed for 10 min in PBS and then followed incubation with secondary antibodies using Texas red-conjugated antirabbit (1:100) and FITC-conjugated antigoat (1:100) for 1 h at room temperature. The signals were visualized by fluorescence microscopy using a NIKON TS100 inverted microscope (Nikon, Melville, NY) with a Q Imaging digital camera.
Chromatin Immunoprecipitation and PCR Analysis
This procedure was followed as previously described by us (55). In brief, uterine tissues in small pieces (∼0.3 cm) were cross-linked with 1% formaldehyde at room temperature for 15 min and then terminated by addition of 0.1% glycine. Postfixed tissues were resuspended in Tissue-PE Luria Bertani lysis buffer containing 1× protease arrest and subjected to cell lysis by continuous vortex at high speed for 40 min in the presence of acid-washed glass beads on ice. The broken cell suspension was then collected into a new tube and subjected to sonication to derive smaller chromatin fragments of approximately 500 bp. The final lysate was subjected to immunoprecipitation using primary antibodies for ERα, Tcf-3, Lef-1, or normal serum IgG (as control). The bound protein was eluted with immunoprecipitation elution buffer (1% sodium dodecyl sulfate, 0.1 m NaHCO3) and heated at 50 C for 5 h to reverse the cross-linking between DNA and protein. Both DNA and protein were precipitated with 100% ethanol, and the pellet was dissolved in Tris-EDTA buffer in the presence of proteinase K by incubation at 50 C for 30 min. DNA was then extracted once with phenol-chloroform-isoamyl alcohol (24:24:1, vol/vol) and once with chloroform and precipitated with ethanol. Finally, the DNA pellet was dissolved in Tris-EDTA buffer and analyzed by PCR.
In our analysis, we selected six genes c-Myc, Ccnd1, Pgr, Ltf, Cdkn1a, and Mmp7 that are known to be regulated by estrogen in the uterus (8, 26–30). It is well established that two classes of transcription factors ERα or Tcf/Lef primarily regulate gene transcriptions via their respective interactions through defined consensus sites for ERE (estrogen-responsive element)/AP1/SP1 (56–58) or WRE (19–21). However, endogenous regulation of the above mentioned genes in the mouse uterus by estrogen has been poorly understood. Therefore, a computer-based analysis of 10-kb upstream promoter regions for our gene of interests, available at Ensembl mouse site (http://www.ensembl.org/Mus_musculus/index.html) were carried out to identify the predicted responsive regions for ERα and Tcf/Lef proteins using the programs on the web at: http://www.gene-regulation.com/cgi-bin/pub/programs/patch/bin/patch.cgi and http://sdmc.lit.org.sg/ERE-V2/index. Based on above the analysis, several distinct regions (A, B, or C), containing ERα- or Tcf/Lef-responsive sequences on gene promoters have been located. A pair of primers encompassing each distinct region were designed, whereas an arbitrary region located further upstream of gene promoters with no recognizable binding sequences for the transcription factors was chosen for designing the distal primers. Analysis of an estrogen-insensitive gene Actb was used in parallel (as a control) to compare status of ERα or Lef-1/Tcf-3 recruitments in the promoter.
The primers used for PCR were as follows: Ltf (A), 5′-T C T A G G C T G A C T C C G C T C T C-3′ and 5′-T A G A G G T G G G A C A T G G G G T A-3′; Ltf (B), 5′-T C A A G T G G T C A A A C C A A C C A-3′ and 5′-A A C T C T C G G G T G C A A T G A A G-3′; Ltf (C), 5′-TTTGCATGTGTAGTGTGAG GTG-3′ and 5′-C A C A T G C T T A G A G A G A G A C A C A C A-3′; Ltf (distal), 5′-C A T G T G C A T G T A T G T G A G A T G A A-3′ and 5′-A T C C C C T G T C A G T C A G T G C C T T C-3′; Pgr (A), 5′-C C A G C T T G C T C C A G C T A C T T-3′ and 5′-A T A T A G G G G C A G A G G G A G G A-3′; Pgr (B), 5′-G A A T T C C A A C G C C A G A G A T T-3′ and 5′-T C C T C G C A C C C G T A A A T A C T-3′ Pgr (distal), 5′-A C T G T C C A G A A T G C C T C C A C-3′ and 5′-A T C A C C A G G G A G G T G C T A C A-3′; Ccnd1 (A), 5′-A G G T G G A G A A A C A C C A C C A C-3′ and 5′-C G G T T T G C C C A A G A A A A A T A-3′; Ccnd1 (B), 5′-T G A A A T C C G C T C A G G G T A A C-3′ and 5′-G G A C T T G G C T G T T T C T G C T C-3′; Ccnd1 (distal), 5′-A A A T C T C C G C T C T T T G G A-3′ and 5′-A A A T C T C G T G G C A G G A A C T G-3′; Cdkn1a (A), 5′-C A C A G T T G G T C A G G G A C A G A-3′ and 5′-C C T C C C C T C T G G G A A T C T A A-3′; Cdkn1a (B), 5′-C C T A G A A A G C A A GC C T G T G G-3′ and 5′-C G A A G G A A A C A A T G G A T G T G-3′; Cdkn1a (distal), 5′-C T T C A C T A C C C A C C C T G C A T-3′ and 5′-C T T C T C C A T G G C C T T G T C T G-3′; Mmp7 (A), 5′-A A A T G C C A T G T G T T C C T C C T-3′ and 5′-T C A A C A G G C G A T T G T T C T C A-3′; Mmp7 (B), 5′-C A G G T C C T C C T C C T T C C T T C-3′ and 5′-G A A T T C A G G A G G C T G T G G A G-3′; Mmp7 (distal), 5′-C C C A G C A G A C A A A A G A G A G G-3′ and 5′-G G G T C T C T C A C C A A A C C T G A-3′; C-Myc (A), 5′-T G A G C A C A C C T T C C T C T C C T-3′ and 5′-C C C G A T G G C A G A G A G T A C A T-3′; C-Myc (B), 5′-A A G C A T C T T C C C A G A A C C T G-3′ and 5′-T C C C T A G T C T G C G T T T T G C T-3′; c-Myc (C), 5′-T T C C A C C A T C T T C C A A A A G G-3′ and 5′- G C C T C A C T A T G C A G C C A G T T-3′; C-Myc (distal), 5′-T C T C T G T G G T G G C A T A G C T G-3′ and G G G A A G T G A C G A G A G C A A A G-3′; Actb, 5′-T T G A A T G T C C C C A G G A G A A G-3′ and 5′-C C A G A G A A C T T T G C C C T C A C-3′. The anticipated product sizes (bp) were for Ltf: 199 (A), 151 (B), 150 (C), and 249 (distal); Pgr: 191 (A), 151(B) and 222 (distal); Ccnd1: 179 (A), 199 (B) and 300 (distal); Cdkn1a: 192 (A), 186 (B), and 180 (distal); Mmp7: 185 (A), 167 (B), and 167 (distal); C-Myc: 170 (A), 199 (B), 162 (C), and 159 (distal); Actb: 234. PCR conditions were 94 C for 4 min and then 25–30 cycles using 94 C for 30 sec, 55 C for 30 sec, and 72 C for 45 sec, followed by incubation at 72 C for 10 min. PCR products were resolved in 2% agarose gels followed by ethidium bromide staining.
Recombinant Adenoviral Plasmids
The replication-defective adenoviral vectors for Bip-antisense (rAdBip-As), SFRP-2-sense [rAdSFRP-2(S)] and GFP (rAdGFP) were previously described by us (10, 11). The viral packaging of these plasmids was carried out by transfection into 293 cells as described (59). Viral particles were purified through CsCl density gradient centrifugation and stored at −70 C.
In Vivo Delivery of Adenovirus Particles in Mice
This was essentially same as previously described (11). In brief, adenoviral particles were first inoculated directly into uterine lumen of both horns [20 μl solutions in saline containing 5 × 109 plaque-forming units per horn] from the oviduct end just before ovariectomy. They were given rest for 7 d before they received the second inoculum (∼100 μl solution in saline containing 5 × 109 plaque-forming units) through tail vein. They were again rested for an additional 2 d before receiving injections of estradiol-17β (E2 100 ng/mouse) or oil (0.1 ml/mouse) for the indicated times. Uterine tissues were collected for subsequent analysis.
Acknowledgments
We thank Ping Li for her input during the course of the experiments.
This work was supported by National Institutes of Health Grants R01 ES07814 and R01 HD37830.
Disclosure Statement: The authors have nothing to disclose.
Abbreviations
- AP1,
Activator protein 1;
- ChIP,
chromatin immunoprecipitation;
- E2,
17β-estradiol;
- ER,
estrogen receptor;
- ERE,
estrogen responsive element;
- FITC,
fluorescein isothiocyanate;
- GFP,
green fluorescent protein;
- ICI,
ICI 182,780;
- Lef-1,
lymphoid enhancer factor 1;
- SFRP-2,
secreted frizzled related protein-2;
- Tcf-3,
T cell factor 3;
- WRE,
Wnt-responsive element.
Couse JF, Korach KS
Stampfer MJ, Willett WC, Colditz GA, Rosner B, Speizer FE, Hennekens CH
McDonnell DP, Norris JD
Dey SK, Lim H, Das SK, Reese J, Paria BC, Daikoku T, Wang H
Tsai MJ, O’Malley BW
Beato M, Herrlich P, Schütz G
Das SK, Taylor JA, Korach KS, Paria BC, Dey SK, Lubahn DB
Das SK, Tan J, Johnson DC, Dey SK
Das SK, Tan J, Raja S, Halder J, Paria BC, Dey SK
Hou X, Tan Y, Li M, Dey SK, Das SK
Ray S, Hou X, Zhou HE, Wang H, Das SK
Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, Korach KS
Watanabe H, Suzuki A, Kobayashi M, Takahashi E, Itamoto M, Lubahn DB, Handa H, Iguchi T
van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H
Staal FJ, Noort MM, Strous GJ, Clevers HC
Bienz M, Clevers H
van Noort M, Clevers H
van de Wetering M, Oosterwegel M, Dooijes D, Clevers H
Travis A, Amsterdam A, Belanger C, Grosschedl R
Waterman ML, Fischer WH, Jones KA
Oosterwegel M, van de Wetering M, Timmerman J, Kruisbeek A, Destree O, Meijlink F, Clevers H
Korinek V, Barker N, Willert K, Molenaar M, Roose J, Wagenaar G, Markman M, Lamers W, Destree O, Clevers H
Hayashi K, Burghardt RC, Bazer FW, Spencer TE
Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O
Huet-Hudson YM, Andrews GK, Dey SK
Lai MD, Jiang MJ, Wing LY
Yoshida A, Newbold RR, Dixon D
Tong W, Pollard JW
Rudolph-Owen LA, Hulboy DL, Wilson CL, Mudgett J, Matrisian LM
Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M
Liu YH, Teng CT
Watanabe H, Suzuki A, Mizutani T, Khono S, Lubahn S, Handa H, Iguchi T
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler, KW
Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben-Ze’ev A
Kamei J, Toyofuku T, Hori M
Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJ, Rubinfeld B, Polakis P, Matrisian LM
Gustavson MD, Crawford HC, Fingleton B, Matrisian LM
Mulholland DJ, Dedhar S, Coetzee GA, Nelson C C
El-Tanani M, Fernig DG, Barraclough R, Green C, Rudland P
Kouzmenko AP, Takeyama K, Ito S, Furutani T, Sawatsubashi S, Maki A, Suzuki E, Kawasaki Y, Akiyama T, Tabata T, Kato S
Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, Lanyon LE
Chandar N, Saluja R, Lamar PC, Kolman K, Prozialeck WC
Yang F, Li X, Sharma M, Sasaki CY, Longo DL, Lim B, Sun Z
Couse JF, Curtis SW, Washburn TF, Lindzey J, Golding TS, Lubahn DB, Smithies O, Korach KS
Kos M, Denger S, Reid G, Korach KS, Gannon F
Merrill BJ, Gat U, DasGupta R, Fuchs E
Chen B, Pollard JW
van Genderen C, Okamura RM, Farinas I, Quo RG, Parslow TG, Bruhn L, Grosschedl R
Merrill BJ, Pasolli HA, Polak L, Rendl M, García-García MJ, Anderson KV, Fuchs E
Rahman MA, Li M, Li P, Wang H, Dey SK, Das SK
Daikoku T, Tranguch S, Friedman DB, Das SK, Smith DF, Dey SK
Tan J, Raja S, Davis MK, Tawfik O, Dey SK, Das SK
Tan J, Paria BC, Dey SK, Das SK
Ray S, Das SK
Klein-Hitpass L, Ryffel GU, Heitlinger E, Cato AC
Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P
Porter W, Saville B, Hoivik D, Safe S

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}