





Abstract
Somatic synonymous mutations are one of the most frequent genetic variants occurring in the coding region of cancer genomes, while their contributions to cancer development remain largely unknown. To assess whether synonymous mutations involved in post-transcriptional regulation contribute to the genetic etiology of cancers, we collected whole exome data from 8,320 patients across 22 cancer types. By employing our developed algorithm, PIVar, we identified a total of 22,948 posttranscriptionally impaired synonymous SNVs (pisSNVs) spanning 2,042 genes. In addition, 35 RNA binding proteins impacted by these identified pisSNVs were significantly enriched. Remarkably, we discovered markedly elevated ratio of somatic pisSNVs across all 22 cancer types, and a high pisSNV ratio was associated with worse patient survival in five cancer types. Intriguing, several well-established cancer genes, including PTEN, RB1 and PIK3CA, appeared to contribute to tumorigenesis at both protein function and posttranscriptional regulation levels, whereas some pisSNV-hosted genes, including UBR4, EP400 and INTS1, exerted their function during carcinogenesis mainly via posttranscriptional mechanisms. Moreover, we predicted three drugs associated with two pisSNVs, and numerous compounds associated with expression signature of pisSNV-hosted genes. Our study reveals the prevalence and clinical relevance of pisSNVs in cancers, and emphasizes the importance of considering posttranscriptional impaired synonymous mutations in cancer biology.
INTRODUCTION
Somatic synonymous mutations, which do not alter the protein sequences of their host genes (1), are one of the most frequent but rarely investigated genetic changes that occur in the coding regions of human cancer genomes (2). Recent studies have shown that they can act as drivers of cancers by altering RNA splicing, RNA stability and protein translation (3,4), which suggests the existence of uncovered regulatory effects of these ‘silent’ mutations and highlights the significance to adjust our focus beyond the damaging protein-coding mutations (5). Thus, to provide a comprehensive landscape of cancer genome alterations, it is imperative but still challenging to decipher the role of these ‘silent’ mutations genome-wide in pathogenesis of cancers.
RNA-binding proteins (RBPs) play versatile roles in posttranscriptional RNA regulation, including splicing, polyadenylation, mRNA stabilization, RNA structure, subcellular localization and transcription (6–9), and their aberrant expression may lead to chaos within the whole regulation network (10). In several cancer types, the abnormal expression of RBPs was associated with patient prognosis (11,12), and numerous mutations occurred in coding regions of RBPs had been implicated in tumorigenesis and progression, such as KHDRBS1 (13) and ELAVL1 (also known as HuR) (14). Interaction between known cancer driver genes and dysregulation of RBPs were also demonstrated. For example, the oncogenic transcription factor c-Myc upregulated the transcription of three heterogeneous nuclear ribonucleoprotein (hnRNP) proteins to control pyruvate kinase mRNA splicing in cancer cells (15). Additionally, in cancer cells, p53 tumor suppressor protein induced the expression of RNA-binding motif 38 (RBM38), which regulated the stability of targeted mRNAs to promote cell cycle arrest (16). These studies have unveiled the indispensable function of RBPs in carcinogenesis and progression of cancers (17).
It has been demonstrated that RBPs can recognize their RNA substrate via sequence-specific binding motifs (18); therefore, genetic mutations occurred in the binding motifs may disrupt the recognition between RBPs and RNA substrates, resulting in various human diseases (19). For example, a mutation in the 3′ untranslated region of FMR1 decreased neuronal activity-dependent translation of FMRP by disrupting the binding of HuR, leading to developmental delay in patients (20). However, the effect of synonymous single nucleotide variants (SNVs) on RBP-mediated posttranscriptional regulation in human cancers remains unclear.
To test whether synonymous mutations involved in posttranscriptional regulation contribute to the genetic etiology of cancers, we collected whole exome data from 8,320 patients across 22 cancer types. By employing our developed algorithm, PIVar, we identified a substantial number of posttranscriptionally impaired synonymous SNVs (pisSNVs) and observed the clinical relevance of the somatic pisSNV ratio in 8,320 patients across 22 cancer types. The functional effect of these pisSNVs and their host genes, as well as significantly altered subnetworks containing pisSNV-hosted genes, were further analyzed for their co-occurrence and relative contribution to the etiology of cancers.
MATERIALS AND METHODS
Pipeline for detecting posttranscriptionally impaired SNVs (piSNVs)
To evaluate the impact of mutations on posttranscriptional regulation, we developed a heuristic scoring system, PIVar (https://github.com/WeiWenqing/PIVar), which is inspired by RegulomeDB (21) and centered on the disruption of a protein-RNA interaction via alteration of RNA secondary structure and regulation of gene expression, to identify piSNVs. Firstly, we identified the putative regulatory SNV set as those situated in RBP-binding sites detected by CLIP-seq (crosslinking immunoprecipitation sequencing). Then, functional confidence of specific regulatory SNV was categorized based on their impact on RNA expression, RBP binding, alterations of RNA secondary structure (namely riboSNitch) and miRNA binding (Figure 1A, Supplementary Table S1).
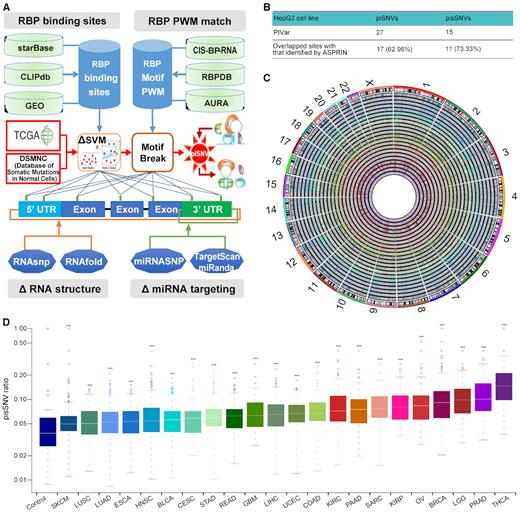
Posttranscriptional impaired synonymous SNVs (pisSNVs) identified in TCGA pan-cancer. (A) Workflow for identifying posttranscriptionally impaired SNVs. (B) Evaluation of the impact of piSNVs identified by PIVar on posttranscriptional regulation through allele-specific binding activity (inferred by ASPRIN (35)) of 103 RBPs based on the CLIP-seq and RNA-seq data of the HepG2 cell line. (C) Genome-wide distribution of pisSNVs identified in 22 TCGA cancer types. The circle adjacent to the karyotypes and the innermost circle show lines representing the distribution of pisSNVs identified in SKCM and THCA, respectively. Other circles from outermost to innermost are arranged according to the order of cancer types listed in (D) (from left to right). (D) Elevated ratio of somatic pisSNVs in TCGA pan-cancer compared with that of control from the DSMNC database (*** P<0.001). For a given sample, the ratio of pisSNV to synonymous SNV was calculated, and then the distribution of pisSNV ratio within each cancer type was compared with that in control samples using Wilcoxon rank-sum test.
In details, expression quantitative trait loci (eQTLs) and RBP binding sites of 112 CLIP-seq datasets derived from 26 human cell lines or tissues were collected from starBase (22), CLIPdb (23) and extracted sequencing data from Gene Expression Omnibus (GEO) (24). As the RNA-binding domains initially determined the binding specificity and preferences of RBPs on RNA substrate via specific sequence motifs (25), all position weight matrices (PWMs) from the catalog of inferred sequence binding preferences of RBPs (25,26) deposited in the AURA database (27) were used to call motif (match score > 0 and P-values < 0.0001) across human transcriptome. For single-base mutations, we employed the RNAsnp (28) with default parameters to estimate the mutation effects on local RNA secondary structure. For insertions and deletions, we evaluated their effects on RNA secondary structure using the minimal free energy generated by RNAfold (29). In addition, we employed LS-GKM (30) and deltaSVM (31) to predict the impact of SNVs on the binding of specific RBPs by calculating the delta SVM scores. Only the SNVs that produced a change of >5 in the gkm-SVM scores, P-values < 0.05 and free energy changes >1 were regarded as SNVs who are likely to affect the binding of RBP. Moreover, we collected response elements for all human microRNAs curated in miRanda (32) and TargetScan v7.0 (33). The mutation effects of specific SNV on microRNA binding were predicted using miRNASNP v2 (34). Finally, SNVs with functional categories 1–2 listed in Supplementary Table S1 were determined to be piSNVs.
To evaluate the efficiency of our workflow, based on the downloaded RNA-seq and CLIP-seq (103 RBPs) data of HepG2 cell line from the ENCODE (Encyclopedia of DNA Elements) database, we compared the piSNV and pisSNVs we identified with allele-specific RBP-RNA interaction sites predicted by ASPRIN (35), which inferred the interactions by observing the allelic preference of RBPs from CLIP-seq as well as RNA-seq experimental data.
Identification of pisSNVs from somatic mutation data of 22 cancer types and human normal cells
We downloaded the somatic mutation data of 8,320 patients across 22 cancer types, including skin cutaneous melanoma (SKCM), lung squamous cell carcinoma (LUSC), lung adenocarcinoma (LUAD), esophageal carcinoma (ESCA), head and neck squamous cell carcinoma (HNSC), bladder urothelial carcinoma (BLCA), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), stomach adenocarcinoma (STAD), rectum adenocarcinoma (READ), glioblastoma multiforme (GBM), liver hepatocellular carcinoma (LIHC), uterine corpus endometrioid carcinoma (UCEC), colon adenocarcinoma (COAD), kidney renal clear cell carcinoma (KIRC), pancreatic adenocarcinoma (PAAD), sarcoma (SARC), kidney renal papillary cell carcinoma (KIRP), ovarian serous cystadenocarcinoma (OV), breast invasive carcinoma (BRCA), brain lower grade glioma (LGG), prostate adenocarcinoma (PRAD), thyroid carcinoma (THCA), from the Broad Institute/The Cancer Genome Atlas (TCGA) data portal (http://gdac.broadinstitute.org/). Taking samples from the DSMNC database (Database of Somatic Mutations in Normal Cells) (36) as a control, we got 0.77 million somatic SNVs occurring in over 579 human normal cells from the DSMNC database. The piSNVs and pisSNVs that could destroy the binding between RNA and RBPs were identified from all these somatic mutation data using PIVar, and the distribution of pisSNV ratio (ratio of pisSNV to synonymous SNV) between control and each cancer type was compared using Wilcoxon rank-sum test. For a given sample, the ratio of posttranscriptionally impaired non-synonymous SNV (pinsSNV) to non-synonymous SNV was also calculated, and then the distribution of pinsSNV ratio within each cancer type was compared with that in pisSNV ratio using Wilcoxon rank-sum test.
Clinical survival analysis
We downloaded corresponding clinical data of each cancer type from cBioPortal (http://www.cbioportal.org/). Then, the function ‘surv_cutpoint’ from R package ‘survminer’ was applied to determine the optimal pisSNV ratio based on disease-free survival (DFS) information of the patients. DFS in patients with high or low pisSNV ratio was compared using the Kaplan–Meier method and the log-rank test. Multivariate Cox proportional regression analysis accounting for age, gender, TNM stage and race of patient was also performed to analyze the association between pisSNV ratio and clinical outcome.
RBP motif enrichment of pisSNV loci and their impact on RBP binding
Many RBPs interact with mRNAs via a limited set of modular RNA-binding domains (37), which initially determine the specificity and preferences of RNA binding with specific sequence motifs (25). Therefore, 247 PWMs from the catalog of the inferred RNA binding motif deposited in the AURA database (27) were used to call motif matches in the transcribed regions of the genome. After the integrative analysis of the RBP motif and CLIP-seq-derived RBP binding for all identified pisSNVs, Fisher exact tests were used to explore the RBPs impacted by pisSNVs (FDR < 0.05 and OR > 1). To further evaluate the impact of pisSNVs on RBP binding, the crystal structures of the RBP-RNA complex (PDB ID: 5EN1 and 2LEC) were downloaded from the Protein Data Bank (https://www.rcsb.org/) and visualized by PyMOL (https://pymol.org).
Chemiluminescent electrophoresis mobility shift assays
To assess the effect of RNA mutation on binding of RBP, electrophoresis mobility shift assays were performed using LightShift Chemiluminescent RNA EMSA kit (Catalog # 20158; Thermo Scientific, Rockford, USA). Two purified RBPs were used in the assay. Of which, PCBP3 was purchased from OriGene Technologies (Catalog # TP329176; Rockville, USA), and PTBP1 was kindly provided by Dr Yuanchao Xue (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China). Then, 200 ng RBP was pre-incubated with 100 μg ml−1 tRNA in 1× RNA EMSA binding buffer for 10 minutes at room temperature. After that, 80 fmol synthesized 3′-biotin-labeled wild-type or point-mutated RNA oligos (Supplementary Table S2) were respectively added to the mixture (15 μl final volume) and incubated for 20 minutes at room temperature. Then, 3.75 μl 5× loading buffer was added to the 15 μl RBP-RNA mixture and immediately loaded into the pre-run TBE polyacrylamide gel, and ran at 100 V for 45–60 min in cooled 0.5× TBE buffer. Samples were then transferred to positively charged nylon membrane (Thermo Scientific, Rockford, USA), and crosslinked with UV-light crosslinking instrument equipped with 254 nm bulbs. The subsequent blocking, washing and detection were performed according to the manufacturer's instructions.
Differential expression analysis
We downloaded the RNA-seq read count data of 22 cancer types and corresponding normal tissues from the Broad Institute. The R package ‘DESeq2’ was used to assay the expression of 2,042 pisSNV-hosted genes and 35 enriched RBPs identified in the previous step, and only genes with fold-change >2 and FDR <0.05 were considered to be differentially expressed genes.
Identification of co-existed protein damaged mutations in pisSNV-hosted genes
All genetic mutations occurred in 2,042 pisSNV-hosted genes across 22 cancer types were curated, and their functional consequences on protein were predicted using Varcards (38). Protein damage mutations were then defined by loss-of-function mutations or missense mutations with a damaging score ≥0.8 predicted using ReVe (39). After that, gene-length corrected occurrence frequency of these genes with protein damaging mutations and posttranscriptional impaired synonymous mutations in all 8,320 cancer patients were analyzed.
Network analysis
We used the HotNet2 workflow (40) to mine significantly mutated subnetworks according to the literature-based Human Protein Reference Database (HPRD) network (http://www.hprd.org/), and the sample frequency of each pisSNV-hosted gene was taken as the network heat score to identify significant subnetworks with default parameters.
KEGG enrichment analysis
To explore the function of pisSNV-hosted gene within each subnetworks, we analyzed KEGG enrichment using the R package ‘clusterProfiler’ (41), with a Bonferroni correction test, and identified significant pathways with FDR values <0.05.
Potential clinical drug analysis
We downloaded somatic mutation data for 1,001 cancer cell lines and natural log IC50 of the dose–response curve for all screened cell line/drug combinations from the Genomics of Drug Sensitivity in Cancer (http://www.cancerrxgene.org/). Next, we used analysis of variance (ANOVA) to identify the associations between posttranscriptionally impaired synonymous mutations and drug responses according to previous publication (42). Cohen's d was used to quantify the effect size, and the resulting P values were corrected by FDR. To further explore the therapeutic effects of pisSNV-hosted genes, the gene expression profiles of each identified pisSNV-hosted gene in each cancer type were compared with drug response signatures listed in the Connectivity Map (CMAP) build 02 (Broad Institute) (43).
RESULTS
Pipeline for detecting posttranscriptionally impaired SNVs (piSNVs)
To investigate the potential impact of genomic mutations on posttranscriptional regulation, we developed PIVar according to the functional confidence of variants based on multi-omic experimental data (Figure 1A). As a pilot study, we first analyzed the mutation data of HepG2 cell line from the ENCODE database using PIVar, and identified 27 piSNVs and 15 pisSNVs in the cell line. A recently developed computational method, ASPRIN (35), could infer RBP-RNA interactions by observing the allelic preference of RBPs from CLIP-seq as well as RNA-seq experimental data, which provided us a method to evaluate the efficiency of our workflow. We used it to analyze allele-specific binding of 103 RBPs based on the CLIP-seq and RNA-seq data from the same cell line, and identified 987 allele-specific RBP–RNA interaction sites in the exon regions. Seventeen (62.96%) piSNVs and 11 (73.33%) pisSNVs obtained through our pipeline were overlapped with the allele-specific RBP–RNA interaction sites identified by ASPRIN (Figure 1B; Supplementary Table S3), which suggests that PIVar was more stringent for identifying the impact of genetic mutations on posttranscriptional regulation network.
Elevated ratio of somatic pisSNVs across cancer types
Inspired by previous studies in which genetic mutations can disrupt the RBP recognition of RNA substrates (20,44) and many RBPs play important roles in tumorigenesis (11–16,35), we then employed PIVar (Figure 1A) to analyze the somatic mutation spectrum of 22 cancer types to explore the correlation between mutations and binding of RBPs. In total, we identified 98,260 nonredundant piSNVs across 22 cancer types that could destroy the binding between mRNA and the corresponding RBP. Synonymous mutations can function as driver mutations in human cancers by disrupting RNA splicing or RBP binding instead of altering the sequence of encoded proteins directly (4); thus, we focused on the previously neglected ‘silent’ mutations and observed a total of 22,948 synonymous piSNVs (pisSNVs) across 22 cancer types (Figure 1C; Supplementary Table S4). Taking samples from the DSMNC database as a control, we observed a significantly higher ratio of pisSNVs in TCGA pan-cancer compared with that of the control (Figure 1D). Also, we observed synonymous mutations had a higher mutation load on posttranscriptional regulation comparing to non-synonymous mutations in 15 cancer types (Supplementary Figure S1). The significant distinction between cancer and normal control samples unveiled the prevalence of posttranscriptionally impaired synonymous mutations in cancer genomes, implying their contribution to cancer etiology.
Clinical relevance of the pisSNV ratio across cancer types
To further identify the clinical relevance of the elevated pisSNV ratio in cancers, we stratified the patients in each cancer type according to the ratio of pisSNV to synonymous SNVs identified in a given sample. We found that DFS of patients with seven cancer types, namely, BLCA, CESE, LUAD, OV, SKCM, STAD and THCA, had a significant correlation with the pisSNV ratio (Figure 2; Supplementary Figure S2), and the patients with a high pisSNV ratio in each cancer type had a worse survival situation than those with a low pisSNV ratio. Additionally, the association between the increased ratio of pisSNV and the DFS of cancer patients remained statistically significant in five cancer types, namely, BLCA, LUAD, OV, SKCM and STAD, even after adjusting for age, gender and TNM stage of patient in the multivariate Cox regression analysis. These results further revealed the contribution of pisSNVs to survival of cancer patients and provided clinical evidence that the pisSNV ratio could serve as a potential prognostic biomarker for several types of cancers.
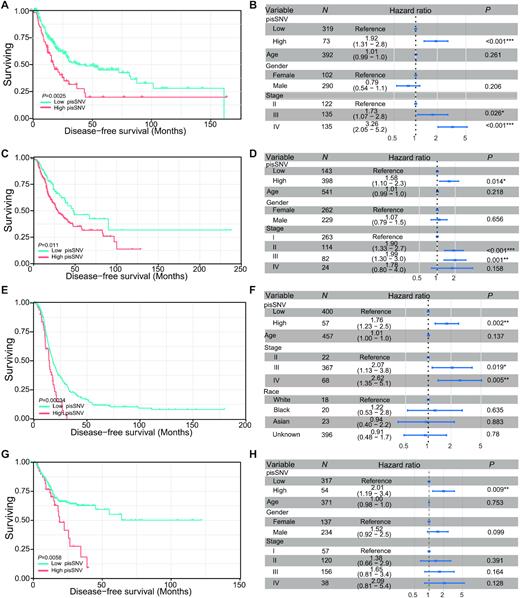
Disease-free survival related to the pisSNV ratio across cancer types. Subjects from the BLCA (A, B), LUAD (C, D), OV (E, F) and STAD (G, H) cohort were stratified according to the pisSNV ratio. Multivariate Cox regression analysis of the pisSNV ratio after taking into account age, gender and TNM stage of patients.
Functional effect of pisSNVs on RBP binding
After obtaining the association between clinical outcome and pisSNV ratio, we wanted to determine the functional effect of these pisSNVs on posttranscriptional regulation, especially on RBP binding. It has been demonstrated that genetic mutations on RNA substrates can disrupt the RBP recognition toward them (20). To provide experimental evidence that pisSNVs disrupt the binding of RBPs, we performed EMSA on three randomly selected pisSNVs who predicted to alter the binding of PCBP3 or PTBP1 (Figure 3A, B; Supplementary Figure S3). As shown in Figure 3A, the binding of PCBP3 to unmutated RNA probe of DAB2, a tumor suppressor by dictating the TGF-β responses of tumor cells (45), was stronger, while the probe with the pisSNV occurring in DAB2 showed visible differences in their binding to PCBP3. Similar results were also found in pisSNV of ZFHX3 or USP9X on binding of PTBP1 (Figure 3B; Supplementary Figure S3).
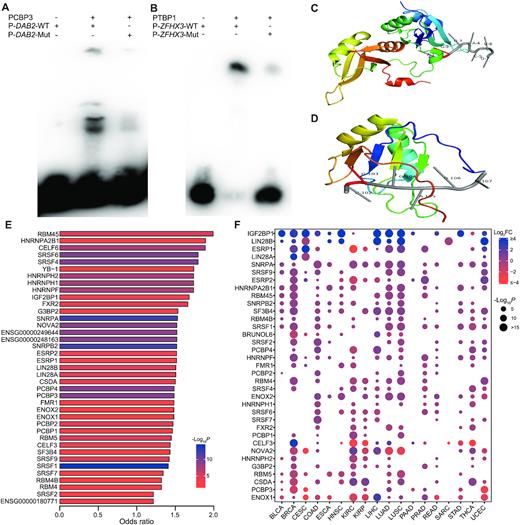
Functional effects of pisSNVs on RBP binding. Electrophoretic mobility shift assays (EMSA) results show the impact of pisSNVs on the recognition of PCBP3 (A) and PTBP1 (B) to their RNA targets. The co-crystal structure of the hnRNPA2B1 (C; PDB ID: 5EN1) or SRSF2 (D; PDB ID: 2LEC)-RNA complex displayed the binding locations of certain RBPs on specific RNA motifs. (E) RBP motif enrichment and CLIP-seq-derived RBP binding site analysis of pisSNVs identified 35 significantly enriched RBPs. (F) Expression of RBPs in different cancer types.
In addition, our analyses of the crystal structure of the RBP-RNA complex supported the effect of some identified pisSNVs on RBP bindings. For example, a pisSNV that occurs in the gene ADCY7 might influence the binding with hnRNP A2/B1, a key player in the posttranscriptional regulation of the maturation, transport, and metabolism of long noncoding RNAs/mRNA (46) and an essential regulator in the development and progression of breast cancer (47). Specifically, the co-crystal structure of the hnRNPA2B1-RNA complex (PDB ID: 5EN1) revealed that hnRNPA2B1 could bind to 5′-AGGACUG-3′ RNA oligonucleotide (46) (Figure 3C). By screening the identified pisSNVs, we found that ADCY7, whose expression significantly correlated with the overall survival of acute myeloid leukemia patients (48), had a mutation from G to A at the second position of the above motif. Another RBP affected by a pisSNV is SRSF2, the RBP that plays an important role in the regulation of alternative splicing events (49), and it was demonstrated that SRSF2 could bind to the 5′-UGGAGU-3′ RNA oligonucleotide (PDB ID: 2LEC) (50) (Figure 3D). We inferred that two pisSNVs in UGGAGU sequence motif residing in DAB2 and ZFHX3 could destroy the binding between SRSF2 and their corresponding RNAs.
To gain an overall insight into the impact of these pisSNVs on RBP binding, we performed motif and binding site enrichment analysis. The integrative analysis of the RBP motif and CLIP-seq-derived RBP binding for all identified pisSNVs revealed 35 significantly enriched RBPs (Figure 3E; Supplementary Table S5). As previous studies demonstrated that abnormal expression of RBPs contributed to cancer development (11,12), we screened the differential expression values of these RBPs between tumor and corresponding normal tissues in different cancer types (Figure 3F). The expression data suggest that dysregulation of these RBPs occurs ubiquitously in cancers. For example, IGF2BP1, a posttranscriptional regulator required for tumor cell proliferation, invasion, and chemoresistance (51), was differentially upregulated in 14 cancer types. ENOX1, a critical mediator of endothelial cell radiosensitization and a potential cancer therapy target (52), was differentially downregulated in 10 cancer types. Collectively, these data provide evidence to support the hypothesis that the identified pisSNVs contribute to cancer pathogenesis by affecting the binding of some cancer-associated RBPs.
The co-occurrence of pisSNV-hosted genes in TCGA pan-cancer
To explore the influence of pisSNVs at the gene level, we summarized the somatic pisSNVs obtained in each cancer type and obtained 2,042 nonredundant pisSNV-hosted genes. A large proportion of pisSNV-hosted genes occurred in a cancer-specific manner, and the top three gene-length corrected frequently occurred pisSNV-hosted genes were LRP1B, LAMA1 and HERC2, which occurred in at least 17 cancer types (Figure 4A). In total of 8,320 patients, the percentage of patients with pisSNVs of LRP1B and LAMA1 was 2.16 and 0.81, respectively. LRP1B was thought to function as a tumor suppressor (53), and microRNA-mediated inactivation of LRP1B could increase the growth and invasive capacity of cancer cells (54). Altered protein expression of LAMA1, posttranscriptionally regulated by miR-202, contributed to cell proliferation and migration of ESCC (55). Our data provide evidence that LRP1B and LAMA1 may contribute to tumorigenesis through posttranscriptional regulation at another level. We also downloaded RNA sequencing data from TCGA to evaluate the differential expression values of these pisSNV-hosted genes between tumor and corresponding normal tissues in different cancer types. From the bubble chart of the top 5% frequently occurred pisSNV-hosted genes (Figure 4B), we found that the majority of them were differentially expressed in each cancer type. For example, IQGAP3 and CENPE were upregulated in 15 cancer types, while ITGA8 and MYH11 was downregulated in 16 and 15 cancer types, respectively, which, from the perspective of expression, reveals the functional relevance of these pisSNV-hosted genes in cancer tumorigenesis. When we summarized the gene-length corrected frequency of occurrence of 2,042 genes based on the status of posttranscriptional impact and/or function damage (Figure 4C), we found that 470 of 8,320 cancer patients carried protein-damaging mutations in LRP1B, the highest frequently occurred pisSNV-hosted gene. Thus, we inferred that LRP1B contributed to tumorigenesis at both the protein function and posttranscriptional regulation levels. This was supported by another evidence that some identified pisSNV-hosted genes are well-established cancer genes with a high deleterious mutation rate, such as PTEN, RB1, COL11A1, PIK3CA, CSMD3 and DNAH5. Interestingly, we found that some pisSNV-hosted gene, such as UBR4, EP400 and INTS1 (Supplementary Table S6), function during carcinogenesis mainly via posttranscriptional manner. The identification of these pisSNV-hosted genes highly expands our understanding of cancer biology.
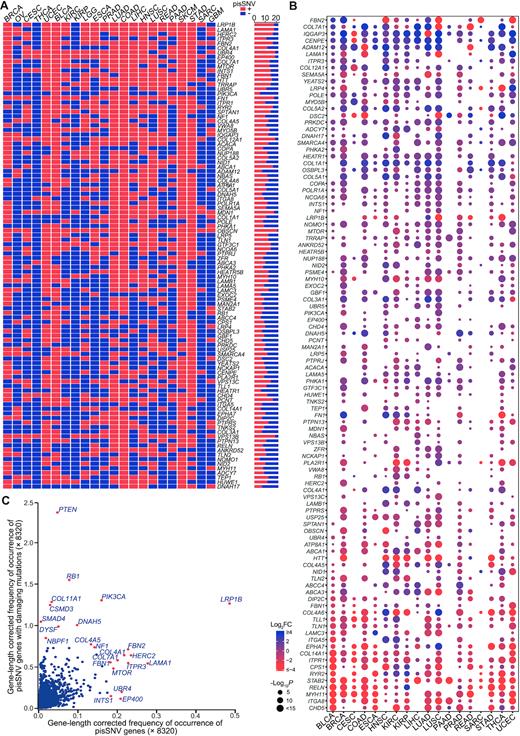
The co-occurrence and expression of pisSNV-hosted genes in TCGA pan-cancer. The co-occurrence (A) and expression (B) of the top 5% (102) gene-length corrected frequently occurred pisSNV-hosted genes in different cancer types. (C) The scatter plot shows the gene-length corrected occurrence frequency of genes with posttranscriptional impaired synonymous mutations and protein damaging mutations.
Significantly altered subnetworks containing pisSNV-hosted genes
To further assess the interactive relationship among the 2,042 pisSNV-hosted genes, we employed HotNet2 (40) to mine the subnetwork in the HPRD network according to the occurrence frequency of each gene, and identified 4 significantly altered subnetworks (Figure 5). Overall, the altered subnetworks included several well-known cancer pathways, such as the phosphoinositide 3-kinase (PI3K)-AKT, mTOR and NOTCH signaling pathways. Subnetwork 1 consisted of ITPR1, ITPR3, RYR2 (2.25% of 8,320 samples), RYR3 and TRPM2, which are involved in thyroid hormone synthesis, pancreatic secretion, and apelin signaling. This subnetwork also contained OBSCN (2.21% of all samples), which co-occurred as pisSNV-hosted genes in >20 cancer types. Subnetwork 2 was related to five cancer types, UCEC, LUAD, COAD, SKCM and STAD, and the genes included were COL1A1, COL4A1, COL4A4, FN1, LAMA5, LAMC1 and LAMA1, which are enriched in PI3K-Akt signaling pathway, suggesting the shared etiology of these cancer types at post-transcriptional level. Subnetwork 3 contained multiple members of the NOTCH signaling pathway in four cancer types, BRCA, UCEC, COAD and SKCM, including NOTCH1, NOTCH2, MAML2, JAG1, JAG2 and MAML3. The last most posttranscriptionally impaired pan-cancer subnetwork consisted of MTOR, RPTOR, CLASP2, CLIP1, RICTOR and CLIP2, which are enriched in the mTOR signaling pathway, AMPK signaling pathway, insulin signaling pathway and PI3K-Akt signaling pathway. Posttranscriptionally impaired synonymous mutations of MTOR, a reported oncogene in multiple cancers (56), co-occurred in 14 cancer types. Interestingly, we also identified some newly cancer-relevant genes within these subnetworks. Our findings indicated that these pisSNV-hosted genes group together with well-established cancer genes, contributing to the carcinogenesis of cancers at the posttranscriptional level.
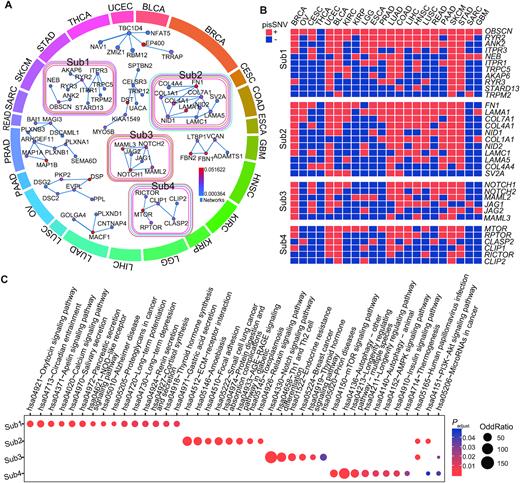
Significantly altered subnetworks and pathways containing pisSNV-hosted genes. (A) HotNet2 consensus subnetworks are arranged according to the enriched cancer types. Colored outlines surrounding each network indicate the cancer types that are enriched, with the color corresponding to the cancer types of the outmost circle. Protein interactions between a subnetwork are derived from the HPRD networks. The co-occurrence (B) and enriched pathways (C) of the pisSNV-hosted genes from different subnetworks.
Therapeutic implications of pisSNVs and pisSNV-hosted genes
To investigate the therapeutic implications of these identified pisSNVs, we analyzed pairwise interactions between mutation data of pisSNVs and dose–response curve from the 1,001 cancer cell lines, and identified two drug-response associated pisSNVs (Figure 6A–C). As shown in Figure 6A, high sensitivity (low IC50 value) for CAY10566, a steroyl-CoA desaturase 1 (SCD1) inhibitor which inhibits the conversion from saturated to monounsaturated, long-chain fatty acyl-CoAs (57), was significantly associated with IGSF3 c.891 G>A mutation. In addition, Ara-G and SGC0946 had higher response to TTN c.22323 C>T mutated cell lines in comparison with wild-type cell lines (Figure 6B). To further explore the potential therapeutic effects of the identified cancer-associated pisSNV-hosted genes, we employed the CMAP workflow (43) to identify clinical drugs based on the expression signature of these pisSNV-hosted genes. In total, 60 compounds were identified, and the majority of them were identified to have an effect on a specific type of cancer (Figure 6D). Two inhibitors of histone deacetylase, namely, MS-275 and vorinostat, were identified as highly ranked compounds with antagonistic effects on gene expression signatures associated with 9 cancer types (Figure 6D). In addition, compound 5182598 and carteolol were found to be able to induce the biological state encoded in the signature associated with five and four cancer types, respectively (Figure 6D). Collectively, the identification of above drugs and compounds might be potentially beneficial for treating patients with specific cancer types.
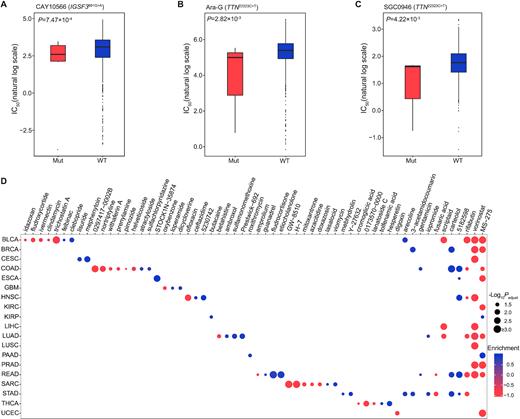
Drugs identified based on pisSNV and expression of pisSNV-hosted genes in different cancer types (A–C) Significant pharmacogenomic interactions for mutant (Mut) versus wild-type (WT) cell lines identified by ANOVA. (D) Drugs identified using CMAP in different cancer types.
DISCUSSION
Recent studies have shown that synonymous mutations could function as cancer drivers by altering RNA splicing, RNA stability and protein translation (3,4,58–60). However, it is still challenging to decipher the role of these mutations genome-wide in the carcinogenesis of cancers. Numerous studies have found that RBPs play an important role in posttranscriptional regulation (6–8,35), and in the cancer genome, abnormal expression of RBPs has a significant effect on cancer phenotype (10–12). Therefore, we speculated that the synonymous mutations present in the cancer genome may disrupt the binding of RBPs by altering the secondary structure of RNA, thereby affecting the translation, transportation or degradation of the RNA and ultimately resulting in the carcinogenesis and progression of cancer. To verify this, we presented a new approach, PIVar, according to the functional confidence of variants based on multi-omic experimental data, for identifying pisSNVs from pan-cancer genome data. When we used the allele-specific binding preference of 103 RBPs derived from the CLIP-seq and RNA-seq data of the HepG2 cell line, we observed that 73.33% of the pisSNVs identified in the cell line could be validated. Also, our EMSA results on three randomly selected pisSNVs provide experimental evidence that the identified pisSNVs could disrupt the binding of their corresponding RBPs. Thus, we provided an efficient and reliable tool for genome-wide deciphering of the role of these synonymous mutations in the development of cancers at the posttranscriptional regulation level.
By employing PIVar, we identified approximately twenty-three thousands nonredundant pisSNVs spanning 2,042 genes across 22 cancer types. Compared with that of control samples from the DSMNC database, an elevated ratio of somatic pisSNVs across cancer types was observed, and a high ratio of pisSNVs could worsen the patients’ survival of five cancer types, including BLCA, LUAD, OV, SKCM and STAD, even when adjusted by multivariate Cox regression. The results provided clinical evidence of the ability of the pisSNV ratio to serve as a potential prognostic biomarker for these five kinds of cancers. It should be noted that some identified pisSNV-hosted genes were well-established cancer-relevant genes with a high deleterious mutation rate, including PTEN, RB1, COL11A1, PIK3CA, CSMD3, DNAH5 and LRP1B. For example, LRP1B, a member of low density lipoprotein receptor-related protein family, had the highest gene-length corrected frequency of occurrence as a pisSNV-hosted gene, while 470 of 8,320 cancer patients was also identified to carry protein damaging mutations of LRP1B. Down-regulation of LRP1B was reported to promote the growth and migration of colon cancer cells (61), and its deletion was associated with acquired chemotherapy resistance to liposomal doxorubicin in high-grade serous ovarian cancers (62). Thus, the pisSNV-hosted genes tend to contribute to tumorigenesis at both the protein functional and posttranscriptional regulation levels. Interestingly, we found that some pisSNV-hosted gene, such as UBR4, EP400 and INTS1 (Supplementary Table S6), might exert their function during carcinogenesis mainly via posttranscriptional mechanisms. The identification of these pisSNV-hosted genes that were not previously well characterized based on missense mutation or expression, highly expands our understanding of cancer biology. It should be mentioned that only impaired structure and posttranscriptional regulation of pisSNV-hosted genes were considered into the current discussion, while some genes may work as transcription factors or epigenetic regulators and exert their function at transcriptional level, thus comprehensive functions of these identified pisSNV-hosted gene and their functional validation is still needed in the future study.
Clinical implication analysis unveiled two drug-response associated pisSNVs and three pisSNV-associated compounds including CAY10566, Ara-G and SGC0946. Of the compounds responding to TTN c.22323 C>T synonymous mutation, Ara-G is a metabolite of nelarabine, which could be used for chemotherapy in T-cell acute lymphoblastic leukemia (63). In addition, SGC0946 can work as an inhibitor of histone lysine methyltransferase for H3K79 and selectively kill mixed lineage leukemia cells (64). Drug susceptibility predictions based on the expression signature of identified pisSNV-hosted genes revealed two highly ranked compounds, that is, MS-275 and vorinostat, with effects on 9 cancer types. MS-275, also known as entinostat and SNDX-275, is a benzamide histone deacetylase inhibitor and was undergoing clinical trials for the treatment of various cancers (65). A recent study suggested that vorinostat (also known as suberanilohydroxamic acid, SAHA) possesses some activity against recurrent glioblastoma multiforme, resulting in a median overall survival of 5.7 months (66). Moreover, combination of vorinostat and carboplatin as well as paclitaxel in the treatment of advanced non-small-cell lung carcinoma (NSCLC) showed improved response rates and increased median progression-free survival and overall survival (67). The identification of these compounds might be beneficial for patients with specific cancer types.
In summary, for the first time, our study reveals the prevalence and clinical relevance of pisSNVs in cancers, and provides valuable resource for future post-transcriptional regulation researches, which may facilitate prognosis of specific cancer types and development of new therapeutic strategies for cancers.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31872237, 31911530148]; National Key Research and Development Program of China [2016YFC0900400]. Funding for open access charge: National Key Research and Development Program of China [2016YFC0900400].
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments