





Abstract
Influenza A virus (IAV) genomes are composed of eight single-stranded RNA segments that are coated by viral nucleoprotein (NP) molecules. Classically, the interaction between NP and viral RNA (vRNA) is depicted as a uniform pattern of ‘beads on a string’. Using high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP), we identified the vRNA binding profiles of NP for two H1N1 IAV strains in virions. Contrary to the prevailing model for vRNA packaging, NP does not bind vRNA uniformly in the A/WSN/1933 and A/California/07/2009 strains, but instead each vRNA segment exhibits a unique binding profile, containing sites that are enriched or poor in NP association. Intriguingly, both H1N1 strains have similar yet distinct NP binding profiles despite extensive sequence conservation. Peaks identified by HITS-CLIP were verified as true NP binding sites based on insensitivity to DNA antisense oligonucleotide-mediated RNase H digestion. Moreover, nucleotide content analysis of NP peaks revealed that these sites are relatively G-rich and U-poor compared to the genome-wide nucleotide content, indicating an as-yet unidentified sequence bias for NP association in vivo. Taken together, our genome-wide study of NP–vRNA interaction has implications for the understanding of influenza vRNA architecture and genome packaging.
INTRODUCTION
Influenza A viruses (IAV) pose a substantial public health burden through frequent epidemics and occasional pandemics in humans. IAV contain eight, negative-sense, single-stranded RNA segments that are named according to the primarily encoded protein: PB2, PB1, PA, HA, NP (nucleoprotein), NA, M and NS (listed from longest to shortest segment). The segments range in size from ∼0.9 to 2.3 kb with a total genome size of approximately 13.5 kb. All viral RNA (vRNA) genome segments contain 13 and 12 conserved nucleotides at the 5′ and 3′ termini, respectively, that show partial complementarity and anneal with each other to form a ‘panhandle’ structure, which associates with the viral RNA-dependent RNA polymerase composed of PB2, PB1 and PA. Mass spectrometry analysis of purified influenza virions indicated that ∼66 NP molecules are present for each packaged polymerase, which suggests that one NP molecule covers ∼26 bases, assuming a total of ∼13.5 kb in the IAV genome (1). A comparable estimate for the NP–vRNA ratio was obtained from in vivo reconstitution assays of viral ribonucleoprotein (vRNP) complexes (2). Cryo-electron microscopy (EM) reconstructions indicated that vRNPs are double-helical hairpin structures with two anti-parallel strands (3,4). However, the resolution of EM, which relies on class averaging, or mass spectrometry is unable to capture the precise structure of vRNA associated to NP. Therefore, the universal depiction of vRNP structures in textbooks is of a uniform random binding of NP over the entire length of each vRNA segment (5–10) (Figure 1A). This ‘beads on a string’ model is supported by in vitro observations indicating that NP binds RNA with no sequence specificity (11). However, this model neglects the possibility that some regions of vRNA segments may be devoid of NP to produce functional secondary structures.
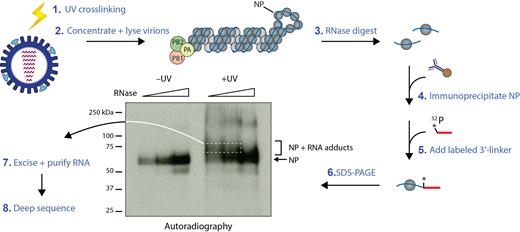
Schematic representation of the HITS-CLIP protocol for influenza virions. Culture supernatant containing intact influenza virus particles were subjected to UV light irradiation, and virions were subsequently harvested by ultracentrifugation on a 30% sucrose cushion to obtain highly pure samples. Virions were then lysed, subjected to a partial RNase A digestion and immunoprecipitation using an anti-NP antibody. Actual autoradiograph of a representative NP HITS-CLIP with IAV strain A/WSN/1933 is shown. Only following UV treatment (+UV), crosslinked NP–RNA adducts (bracket) appear, which migrate slower in the gel than monomeric NP (arrow) and contain viral RNA footprints of NP present in virions. Size selection was performed by excising these slower migrating bands in the +UV sample (white dashed box), followed by isolation of RNA and preparation of deep sequencing libraries. Sequencing was performed on the Illumina NextSeq500 platform, and reads were mapped to influenza reference genomes.
The segmented composition of IAV genomes promotes reassortment, whereby genome segments from multiple strains are exchanged inside a co-infected cell. Reassortment is the process underlying the evolution of new lineages that may give rise to novel influenza pandemics. Notably, not all permutations of segment reassortment occur at equal frequency and certain reassortment events are observed at a higher rate (12). The underlying mechanism of reassortment bias is not completely understood, but it can be assumed that infectious particles with some segment combinations are not formed due to improper vRNP packaging into virions. Incorporation of all eight vRNPs into a mature virion is necessary for the production of fully infectious IAV (13,14), and assembly of vRNPs occurs en route to the plasma membrane (15,16). An expanding body of evidence suggests that the process of vRNP assembly is reliant on RNA-RNA interactions between segments (17–22), as in vitro RNA binding assays indicated that multiple sites within the coding regions of vRNA segments can form RNA-RNA interactions (20,23). However, as these experiments were performed on naked in vitro transcribed RNA in the absence of NP, it is unclear as to whether these regions are indeed free of NP association in vivo and available for RNA–RNA contacts. Thus, a detailed map of NP binding sites on the IAV genome is required to identify areas that are exposed and potentially capable of engaging in RNA–RNA interactions.
In this study, we sought to define the binding profile of NP for each vRNA segment using high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP), which allows the identification of genome/transcriptome-wide RNA-binding sites for a RNA-binding protein of interest (24,25). Using this methodology, we mapped NP association to all eight vRNA segments for two H1N1 IAV strains. Contrary to published vRNP structure schematics, we observed a distinct pattern of NP association to vRNA that is neither random nor uniform. Interestingly, the nucleotide content of NP binding sites on vRNA is relatively guanine-rich and uracil-poor compared to the overall IAV genome, suggesting that this bias in nucleotide composition may be an important determinant of NP binding. These data suggest modifications to the prevailing model of vRNP structure and have substantial implications for our understanding of IAV replication and assembly.
MATERIALS AND METHODS
Cells and viruses
MDCK cells were cultured in Eagle's Minimum Essential Medium (EMEM) supplemented with 10% FBS, l-glutamine and penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. Rescue of recombinant A/WSN/1933 (H1N1) and A/California/07/2009 (H1N1) strains were previously described (15,26).
HITS-CLIP and deep sequencing data analysis
Two confluent T175 flasks of MDCK cells were washed twice with phosphate-buffered saline (PBS) and infected at a dilution of 1:10,000 with the indicated virus in serum-free EMEM containing TCPK-trypsin (Worthington Biochemicals). At 96 h post-infection, 40 ml of the culture medium containing ∼107 infectious virus particle per ml was harvested and cellular debris was pelleted by centrifugation at 2000 × g for 20 min. UV light irradiation at 254 nm (400 mJ/cm2 followed by 200 mJ/cm2) was performed on clarified culture medium. Crosslinked virus supernatant was layered onto a 30% sucrose-NTE (100 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA) cushion and centrifuged at 200 000 × g for 2 h at 4°C. HITS-CLIP was carried out as previously described with minor modifications to adjust the protocol to influenza virions (27). Briefly, virus particles concentrated from 25 ml of culture supernatant were resuspended in 300 μl PXL buffer (1× PBS, 1% NP40, 0.5% deoxycholate, 0.1% SDS), followed by DNase and RNase treatment. For each viral strain sample, partial RNase digestions were carried out for 5 min at 37°C with three aliquots of 100 μl viral lysate and a tenfold dilution series of RNase A (0.25, 0.025 and 0.0025 μg total amount of enzyme, respectively). Following antibodies were used for immunoprecipitating influenza NP variants of different strains: mouse monoclonal HB65 antibody (a kind gift from Dr John Yewell, NIAID NIH) for A/WSN/1933; mouse monoclonal antibody MAB8251 (Millipore) for A/WSN/1933 and H1N1pdm (A/California/07/2009). For each IP reaction, 25 μl of antibody-Dynabeads Protein G complexes were used. Ligation of 5′ and 3′ adapters, RT reaction and first-round PCR amplification step were carried out as described (27). The first-round PCR products were then converted into an Illumina-compatible deep sequencing library using the NEBNext Ultra DNA Library Prep Kit (NEB), and deep sequencing was carried out using Illumina's NextSeq platform. Data analysis was performed as described (27) using the NovoAlign alignment program and mapping the reads to reference genomes available from the NCBI database. CIMS analysis was performed as described (27).
For deep sequencing of total RNA from virions (‘input’ samples), first and second cDNA strands were synthesized using the NEBNext mRNA Library Prep Kit (NEB). Double-stranded cDNAs were then converted into deep sequencing libraries using Illumina's Nextera XT DNA Library Prep Kit. Sequencing results were displayed on the Integrated Genome Viewer (28). Deep sequencing data were deposited in the Sequence Read Archive under accession nos. SRR5647930–SRR5647936. Supplementary Table S3 lists the number of mapped reads and the SRA accession number of each sample.
Pearson correlation analysis
HITS-CLIP data were normalized to the highest peak of each segment. The normalized read depth at each nucleotide position was compared between the two H1N1 strains using the Prism 6 software (GraphPad). The Pearson correlation coefficients and corresponding p-values determined between IAV strains for each segment are presented in Supplementary Table S1. In general, Pearson coefficients (r) range from 1 to –1, where r ≥ 0.7 signifies a high positive correlation, 0.5 ≤ r < 0.7 a moderate positive correlation, 0.3 ≤ r < 0.5 a low positive correlation, and r < 0.3 is a negligible correlation (29).
Peak calling using MACS
The peak-finding algorithm MACS was used to predict NP binding sites (30). For each HITS-CLIP experiment, a P-score was chosen that exhibited the best performance for calling NP peaks. Non-peaks were defined as regions not called as peaks by MACS and having a HITS-CLIP sequencing coverage of <5% of the maximum peak height of each experiment (also see Supplementary Table S2). Mean peak widths were calculated using the coordinates obtained from MACS analysis, omitting apparent double peaks that were called as a single peak by the algorithm.
To analyze the nucleotide compositions of peaks and non-peaks, the sequences for peaks called by MACS were retrieved and the percentages of A, U, G and C were calculated based on the lengths of the peaks. The same calculations were performed for non-peak regions.
vRNA accessibility assay using DNA ASO-mediated RNase H digestion
Virions from 25 ml culture supernatant were harvested by ultracentrifugation (see above) and resuspended in 100 μl Digestion buffer (50 mM HEPES pH 7.5, 75 mM NaCl, 3 mM MgCl2, 10 mM DTT, 1% NP-40 and 0.05% Triton X-100). Viral lysate was then diluted five-fold in Digestion buffer containing no Triton X-100. To examine accessibility of vRNA regions, DNA ASOs complementary to segments NP, HA and NS of A/WSN/1933 (position within the segment shown in Figure 3A, Supplementary Figure S3A and B) and RNase H were added to the lysate. For each reaction, 100 μl lysate was incubated with 5 μM ASO (final concentration) and 5 units RNase H for 15 min at 37°C. Following ASOs were used: 5′-ctcatgctctaccgactgagctagccgggc-3′ (negative control); 5′-ccttgaactgagaagcagatact-3′ (ASO #1); 5′-tgtatggatctgccgtagccagtg-3′ (ASO #2); 5′-tttgaatgatgcaacttaccagag-3′ (ASO #3); 5′-aggaataaatatctagaagaacat-3′ (ASO #4); 5′-tcactggtgcttttggtctccctg-3′ (ASO #5); 5′-gacttaaatgtgaagaatctgtac-3′ (ASO #6); 5′-ggattcatacccaaagctgacca-3′ (ASO #7); 5′-tgacacaatactcgagaagaatgt-3′ (ASO #8); 5′-cagactgaagataacagagaatag-3′ (ASO #9); 5′-agagattcgcttggagaa-3′ (ASO #10). Following digestion, RNA was purified using Trizol, resolved on a formaldehyde-containing 1.5% agarose gel, and transferred onto a nylon membrane for Northern blot analysis. For ASO-mediated RNase H digestion using naked RNA, 1 μg of purified viral RNA was incubated for 3 min at 95°C in the presence of 5 μM ASO and then chilled on ice prior to adding 5 units RNase H for 15 min at 37°C. The same buffer conditions as for vRNP substrates were used. Probes for NP, HA, NS and PB2 vRNAs were generated by random priming of linearized pHW2000-NP, pHW2000-HA, pHW2000-NS and pHW2000-PB2 plasmids, respectively.
Nucleotide variability analysis
The variability of HA and NA was determined from 385 and 353 H1N1 sequences, respectively, spanning the years 1918–2016. To prevent bias from only recent strains, an attempt was made to analyze sequences spanning the last 90 years with similar distribution from older strains. The precise breakdown was 193 and 149 strains from 1918–1994, 61 and 79 strains from 1995–2005, and 131 and 125 strains from 2006–2016 for HA and NA, respectively. Sequences were obtained from NCBI Influenza Virus Resource (https://www.ncbi.nlm.nih.gov/genome/viruses/variation/flu), GISAID (http://platform.gisaid.org), and FluDB (http://www.fludb.org). We employed an overlapping 50-nucleotide sliding window analysis of nucleotide variability as described (31,32).
RESULTS
Identifying NP–vRNA association in virions by HITS-CLIP
To map regions of vRNA that are occupied by NP, we carried out HITS-CLIP with isolated influenza virions (Figure 1). After UV irradiation of clarified culture medium containing virions, we isolated the virions by ultracentrifugation over a 30% sucrose cushion. This purification approach has previously been used for obtaining pure virion preparations for EM studies (22,33). Notably, UV irradiation of intact virions generates covalent crosslinks between NP and directly bound vRNA regions that occur in virions and thus preserves only bona fide interactions. Following limited RNase A digestion of viral lysate to produce vRNA footprints of NP, immunoprecipitation of NP was carried out and a radioactively labeled 3′ linker was added to the NP–vRNA adducts. We resolved these crosslinked products by SDS-gel electrophoresis, which was then visualized by autoradiography (Figure 1). As UV crosslinking was performed prior to viral lysate preparation, the NP–vRNA adducts represent the RNA fragments bound to NP in intact virions and were not formed non-specifically post lysis as a result of RNA reassociation. Importantly, these adducts were only present in the UV-treated sample (bracketed region of autoradiograph in Figure 1) and absent in the –UV sample, further underlining the fact that the RNA-NP adducts indeed represent bona fide RNA-NP interactions. The crosslinked products migrate as a smear above the NP monomers due to the variable lengths of associated vRNA fragments resulting from partial RNase digestion; these were excised and subjected to high-throughput sequencing to identify in vivo NP binding sites in virions. In the non-crosslinked sample, we observed only short labeled RNA fragments that did not retard NP migration and remained bound to NP despite the denaturing conditions of SDS-PAGE. PCR amplification of these short fragments for generating a deep sequencing library resulted in primer dimer formation only, suggesting that these fragments primarily consist of the radiolabeled adapter. Taken together, the specific immunoprecipitation of NP coupled to size selection of NP-bound RNAs exclude the contamination by RNAs associated with polymerase components or other host RNA-binding proteins.
We performed HITS-CLIP on a lab-adapted H1N1 strain, A/WSN/1933 (WSN), as well as on the 2009 H1N1 pandemic (H1N1pdm) strain (A/California/07/2009) (Figure 2). Surprisingly, the NP binding profiles of both H1N1 strains were not consistent with the current model for vRNA–NP packaging in that NP association with vRNA was (i) not regularly spaced and (ii) absent in extended regions of several segments, such as segments PA and HA (Figure 2C and D). Moreover, peak heights across the segments differed markedly, suggesting that NP binding is more abundant in certain regions than in others. For example, NP association within the HA segment is more pronounced towards the 5′ and 3′ termini of the segment in both strains (Figure 2D).
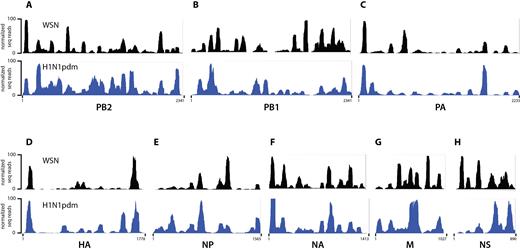
NP binding profile determined by HITS-CLIP for A/WSN/1933 (WSN, black) and A/California/07/2009 (H1N1pdm, blue) strains. (A–H) IGV tracks of all eight IAV segments are shown; their names and nucleotide lengths are indicated at the bottom of each track. Abundance of HITS-CLIP reads (y-axis) were normalized against the highest peak in each individual vRNA segment and arbitrarily set to 100. Profiles of both H1N1 viruses do not adhere to the classical model of uniform and random association of NP with vRNA, but exhibit specific NP peaks as well as regions not enriched for NP. Both H1N1 strains have a similar yet distinct NP binding profile, reflected by a Pearson correlation coefficient of 0.411, indicative of a moderate positive correlation.
Even though both IAV strains are of the H1N1 subtype, their NP binding profiles displayed similarities, but also clear differences in NP association with vRNA. For example, the 5′ region of the NS segment in WSN contains two prominent peaks that are absent in the H1N1pdm strain (Figure 2H). Conversely, some peaks are found in H1N1pdm that are absent in WSN, such as in the center of the PB1 segment or in the 3′ region of the HA segment (Figure 2B and D). The Pearson correlation coefficient for both HITS-CLIP data across all segments was 0.411, indicative of a moderate positive correlation. The strength of the inter-strain correlation varied among genome segments (Supplementary Table S1). For example, NP binding profiles for the PA and HA segments were moderately correlated (r = 0.663 and 0.685, respectively), whereas those for the PB1 and NS segments showed poor correlations (r = 0.074 and 0.097, respectively). Correlation between NP–vRNA binding profiles was not based on host origin of H1N1pdm segments, since the PB1 segment is derived from a human H3N2 strain, while the HA, NP, and NS segments originated from the classical swine lineage (34). Furthermore, the thirteen positively charged amino acids that make up the NP RNA-binding groove (Y148, R150, R152, R156, R174, R175, K184, R195, R199, R213, R214, R221 and R236) were identical between H1N1pdm and WSN strains (35–38), indicating that the RNA-binding domains of the two NP variants do not account for the difference in NP–vRNA binding profiles observed between these strains. Taken together, our data suggest that the NP–vRNA binding profiles of H1N1 viruses are similar, but not identical.
Validation of NP binding sites identified by HITS-CLIP
To ascertain that the NP binding profiles inferred from this method were specific and not random, we performed a second independent biological replicate for the WSN strain. We observed a nearly identical NP–vRNA binding profile between experiments (Pearson correlation coefficient r = 0.798) (Figure 3A, Supplementary Figure S1, Supplementary Table S1). Moreover, as we used two different anti-NP specific antibodies in our HITS-CLIP assays for the WSN and H1N1pdm strains, we verified that the differences in NP binding profiles are not due to some epitopes on NP being masked in one strain and not the other. For this, we repeated HITS-CLIP for the WSN strain using the same antibody that was used for the H1N1pdm strain (see Materials and Methods for details). Both antibodies generated highly overlapping NP binding profiles with a Pearson correlation coefficient of 0.9291 for the WSN strain and a lower correlation of 0.3080 between WSN and H1N1pdm strains (Supplementary Figure S1, Table S1), demonstrating that the differences in the NP binding profiles between the two strains are not due to epitope exclusion. Furthermore, RNA-seq of total RNA from purified virions (‘RNA input’) revealed complete sequencing coverage of the vRNA segments and did not show any sequencing bias for particular vRNA regions (Figure 3A bottom panel, Supplementary Figure S2). As the peaks observed in the HITS-CLIP experiments are not evident in the input sample, this observation supports the notion that the HITS-CLIP peaks are not caused by sequencing bias. The reproducible HITS-CLIP profile between replicates strongly suggests that NP binding to vRNA is neither uniform nor random, but rather specific to certain regions of all segments.
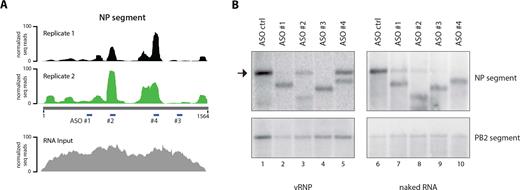
Validation of NP binding sites identified by HITS-CLIP. (A) The NP HITS-CLIP results from two independent biological replicates are shown for the NP segment as a representative. The NP binding profiles of the replicates are highly reproducible, evidenced by a Pearson correlation coefficient of 0.798 (please also see Supplementary Table S1 for details). The RNA-seq track of total RNA input is shown in the bottom panel, which does not display the same peaks observed in the HITS-CLIP experiments. Note that the 5′ and 3′ ends of the segments are underrepresented due to the fact that tagmentation was used for library preparation. Sites targeted by DNA ASO #1–4 used in RNase H digestion assays are indicated. (B) DNA ASO-mediated RNase H digestion assay. vRNA accessibility assay was performed on WSN viral lysate containing vRNA segments complexed with NP. DNA ASO #1–4 were used to target regions of the NP segment depicted in A. Northern blot analysis for NP segment was carried out to examine accessibility as determined by RNase H digestion. NP peaks identified by HITS-CLIP are greatly protected from degradation, demonstrating NP binding at regions targeted by ASO #2 and 4, while predicted NP-depleted regions targeted by ASO #1 and 3 are not. The same assay was performed with purified naked viral RNA (lanes 6–10), resulting in robust RNase H-mediated degradation by all ASOs. Unlike viral lysate, addition of ASO #1 to naked RNA results in partial segment degradation (compare lanes 2 and 6), probably due to formation of secondary structures of naked RNA, which interferes with the accessibility of ASOs. Arrow indicates full-length/non-degraded NP segment. Northern blot against the PB2 segment is shown as a negative control.
The exact RNA-protein crosslink sites can be mapped at nucleotide resolution by computationally searching the deep sequence reads for so-called crosslink-induced mutation sites (CIMS), which are deletions and point mutations in cDNAs caused by reverse transcriptase errors at the exact nucleotide where amino acids crosslinked to RNA. We examined the distribution of all CIMS across all NP peaks in our HITS-CLIP data set for the WSN strain and observed that the majority of CIMS are located between nucleotides -35 and +6 from the peak center (Supplementary Figure S3C). The presence of CIMS over NP peaks supports the notion that our HITS-CLIP data indeed uncovered NP binding sites.
To verify that the NP-enriched vRNA regions identified by HITS-CLIP are indeed NP binding sites in virions, we examined whether the peak sequences are tightly associated with NP and thus not accessible for hybridization. For this, we used DNA antisense oligonucleotides (ASOs) complementary to vRNA regions that either exhibit strong NP association (peaks) or are relatively depleted of NP. Accessible regions are able to base pair with complementary ASOs to form DNA–RNA hybrids and are subsequently induced for cleavage in the presence of RNase H. In contrast, inaccessible regions due to strong NP association would not result in RNase H-mediated degradation, as they are protected by NP. ASO #1 and #3 target non-peak regions, whereas ASO #2 and #4 were complementary to peaks of the NP segment (position within segment indicated in Figure 3A). Consistent with our HITS-CLIP data, addition of ASOs #2 and #4 inefficiently induced cleavage of the NP segment when added to non-crosslinked viral lysate containing vRNA complexed with NP (Figure 3B). RNase H digestion was not completely inhibited because the harsh conditions of virion lysis may have partially disrupted the NP–vRNA interactions and allowed a proportion of NP-bound regions to dissociate. On the other hand, ASOs #1 and #3 readily formed DNA–RNA hybrids and induced complete cleavage of the NP segment, as shown by Northern blot analysis (Figure 3B, left panel). To rule out the possibility that the ASOs targeting NP binding sites are ineffectual for RNase H-mediated digestion, we performed this assay also with purified naked viral RNA. As shown in Figure 3B (right panel), addition of ASOs #2 and #4 induced RNase H-mediated cleavage of naked RNA (compare lanes 3 to 8 and 5 to 10). We also tested vRNA accessibility for segments HA and NS (Supplementary Figure S3A and B), and observed that the accessibility of these segments for hybridization is also reflected in the genome-wide NP binding profile. Taken together, these results verify that the peaks identified by HITS-CLIP represent areas of vRNA that are less accessible compared to non-peak regions and likely NP binding sites in virions.
NP binding sites are relatively G-rich and U-depleted
We next searched for the determinant of NP binding to vRNA and first probed for a conserved sequence motif in NP peaks. The peak-finding algorithm MACS (30) was used to call NP binding sites in our HITS-CLIP data and to obtain nucleotide coordinates of each called peak. Non-peaks were defined as regions of low NP coverage with less than 5% of the maximum peak height of each experiment (see Materials and Methods for additional details). Figure 4A shows a representative BED file for called peaks and non-peaks of the PB2 segment of WSN. Interestingly, we observed that all peaks for both IAV strains have a similar width (average of 62.5 nts and 72.7 nts for WSN and H1N1pdm, respectively; Supplementary Table S2). We analyzed the peak vRNA sequences for nucleotide motif enrichment using the MEME suite (39,40), but found no obvious motif, confirming previous observations that NP binds vRNA without sequence specificity (11).
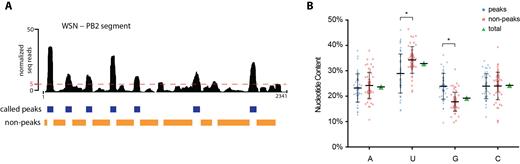
Analysis of nucleotide contents of NP peaks and non-peaks. (A) The peak-finding algorithm MACS was used to predict NP binding sites. The WSN PB2 segment is shown as a representative. The BED track (blue) underneath the HITS-CLIP profile indicates the called peaks. Non-peaks were defined as regions not called as peaks in MACS and those containing less than 5% read coverage of the maximum peak height for each HITS-CLIP experiment (orange BED track). The dashed red line indicates the 5%-threshold. For mean peak widths, see Supplementary Table S2. (B) Nucleotide compositions of each WSN peak defined by MACS (blue) and non-peak (orange) are graphed as a scatter plot as a percentage of the corresponding sequence. Mean and standard deviations are shown. The overall genome-wide nucleotide composition of WSN vRNA segments is displayed in green. A statistically significant difference, denoted by an asterisk, in the G and U content between peaks and non-peaks was observed (two-way ANOVA analysis, P-value < 0.0001). No statistically significant differences were observed for the percentages of A and C bases. All peak and non-peak regions from WSN segments are plotted as a representative; H1N1pdm peaks show the same trend in that peaks are G-rich and U-poor compared to the overall genome-wide nucleotide content (Supplementary Table S2).
We next considered whether evolutionary conservation rates could underlie NP binding specificity. We examined whether conserved (present in both strains) and non-conserved (present only in one strain) NP peaks differ in sequence conservation, rationalizing that NP binding sites might be subject to different forces of directional or diversifying selection. WSN and H1N1pdm are H1N1 viruses separated by over 70 years, and the error-prone viral polymerase is known to generate an average of ∼10−5 substitutions per nucleotide site and infectious cycle (41–45). This sequence evolution of the HA and NA segments over seasonal epidemics is known as antigenic drift and is carefully documented for vaccine production. We compared the nucleotide diversity of these two segments from hundreds of deposited IAV genomes using an overlapping sliding window algorithm (31,32) for H1N1 viruses spanning the past 70 years (see Materials and Methods for details). The sequences of most conserved peaks in the HA segment were relatively evolutionarily conserved (∼80–90% nucleotide identity), while non-conserved peaks involved regions of both low and high sequence variability (Supplementary Figure S4A). In contrast, the nucleotide variability pattern of conserved and non-conserved peaks for the NA segment was less consistent, showing both low and high nucleotide variability (Supplementary Figure S4B). Thus, the absence of a clear sequence motif and pattern of selection at the nucleotide level suggests that any evolutionary forces governing the relationship between vRNA and NP binding are acting on features beyond the primary nucleotide sequences.
We next analyzed the nucleotide contents of peak and non-peak regions in both IAV strains and compared them to each other as well as to the overall genome-wide content (Figure 4B, Supplementary Table S2). Interestingly, peaks of the WSN strain were significantly enriched in guanine (G) bases compared to non-peaks (23.9% compared to 17.5%), while uracil (U) bases were relatively depleted (28.9% compared to 34.2%). The average genome-wide percentages of these bases lay between these values (G: 19.2%; U: 32.9%). These differences were statistically significant based on a two-way ANOVA analysis (P-value < 0.0001). The content of adenine (A) and cytosine (C) bases did not vary between peaks and non-peaks, and were maintained at approximately 23% in both populations. This significant trend of G-enrichment and U-depletion for NP binding sites was also observed in the H1N1pdm strain (Supplementary Table S2). Importantly, UV crosslinking, as performed in CLIP experiments, does not induce a nucleotide bias that would explain these observations (46). Taken together, these data suggest that NP molecules preferably associate with relatively G-rich and U-poor regions of vRNA in vivo. This is the first known sequence bias to be described for NP association to vRNA.
DISCUSSION
We employed HITS-CLIP methodology to delineate the first genome-wide maps of NP-association with vRNA for two H1N1 strains of IAV (Figure 1). Notably, we present evidence that the ‘beads on a string’ model for vRNA packaging requires revision, as NP decorates the vRNA segments non-uniformly (Figures 2 and 3). Furthermore, we uncovered a preference in nucleotide content for NP binding in vivo, as binding sites are relatively enriched in guanines and depleted for uracils compared to the genome-wide percentages of these bases (Figure 4B), suggesting that vRNA nucleotide content may be a contributing factor for the degree of NP association. These parameters for nucleotide content may help establish a predictive algorithm for defining NP–vRNA association for a given segment solely based on sequence.
Based on our results, we propose a revised model for vRNA architecture, where certain regions of vRNA are tightly associated with NP (‘peaks’) while other regions may dynamically associate and disassociate from NP, producing regions free of NP (Figure 5B). The presence of NP-free regions observed in HITS-CLIP is consistent with a previous cryo-EM study that suggested that some regions of vRNA are exposed due to spacing of the RNA-binding domains of NP within the helical reconstruction of vRNPs (4). Interestingly, even closely related H1N1 IAV strains appear to have distinct non-peak regions, while their overall NP binding profile is moderately similar. This observation raises the question of how different the HITS-CLIP profiles might be for evolutionarily distant strains, such as those of another subtype. As UV-crosslinking of RNA to protein is dependent on the right geometry of their interaction, we cannot rule out the possibility that some of these apparently non-peak regions are genuine but undetected NP binding sites. We also considered the possibility that some non-peak regions may be occupied by host RNA-binding proteins incorporated into virions (1), and therefore are not enriched during our procedure. However, given that results from our vRNA accessibility assays with non-crosslinked viral lysates agree with our HITS-CLIP profiles and show that the tested NP-free regions are indeed accessible (Figure 3B, Supplementary Figure S3A and SB), we do not favor these alternative possibilities. A recent publication identified the host RNA-binding protein DAI (DNA-dependent activator of IFN-regulatory factors) associated with vRNA inside infected cells (47); no apparent overlap between DAI-associated regions and NP binding sites was observed. Further experiments examining the association of host RNA-binding proteins to vRNA will elucidate their role during influenza virus replication and complement our results on the accessibility of non-peak regions.
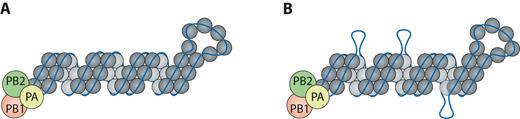
Proposed revised model for NP–vRNA packaging. Based on our data, not all regions of the influenza genome are coated uniformly by NP as previously suggested (A). (B) Instead, some regions may be tightly associated with NP (peaks), while other regions (non-peaks) may provide a more dynamic association with NP and reveal regions free of NP (shown here as loops).
The segmented nature of the IAV genome provides challenges for selective assembly as well as an evolutionary advantage to the virus by facilitating genetic reassortment. It also contributes to the emergence of novel pandemic strains, as evidenced by co-segregating vRNA segments (48–50). Understanding reassortment between IAV is critical for predicting emerging influenza pandemics. Since a genetic reassortment bias has been described in the literature, our HITS-CLIP data may provide some insight into this phenomenon. Previously, in vitro binding assays have identified specific areas of vRNA segments that can interact with other segments (17–21). However, these studies were performed on naked RNA in the absence of NP. Based on the distinct association of NP to vRNA segments, the addition of NP might alter the availability of segment regions capable of base pairing. It is tempting to speculate that some of the identified non-peak regions may participate in RNA-RNA interactions with other segments, which have been proposed to contribute to selective assembly of all eight vRNA segments into virions. In this vein, the greater uracil abundance in relatively unbound regions and their propensity to form wobble base pairs (51) may help mediate vRNA–vRNA interactions between regions of imperfect Watson–Crick complementarity. Therefore, conserved non-peak regions may not only provide insight into the vRNA regions mediating direct RNA-RNA interactions, but also help predict the reassortment potential of certain vRNA segments.
Previous UV-crosslinking experiments indicated that the polymerase complex interacts with the 5′ and 3′ ends of vRNA segments (52,53). Recent crystal structures of the polymerase complex with short RNAs demonstrated that nucleotides 1–10 of the 5′ end form a compact stem-loop within a deep pocket at the interface of PB1 and PA, while nucleotides 1–9 of the 3′ end bind to the surface of the polymerase (54–56), suggesting that the vRNA termini should be devoid of NP, as they are occupied by the polymerase complex. Unexpectedly, we found that not all 5′ and 3′ ends lack NP association. For example, the 3′ end of the NP segment of the H1N1pdm strain harbors a strong NP peak (Figure 2E). Moreover, based on previous studies demonstrating that the 5′ and 3′ non-coding sequences are important for vRNP packaging, we expected the vRNA ends to be free of NP as well to promote intersegmental interactions required for virion assembly and association with the viral polymerase complex (5,12,57). Unexpectedly, the packaging and bundling sequences at the 5′ and 3′ ends of all vRNA segments were not consistently devoid of NP binding. A possible explanation for this observation may be that the ends of each vRNA segment have a unique function during influenza virus assembly, and thus not all segments require free ends. Additional studies are needed to resolve these discrepancies and determine the importance of the variability observed in NP binding at the vRNA ends.
In summary, our data suggest a novel structure of influenza vRNPs, necessitating the current model of IAV genome assembly and packaging to be revisited. Our revised model has significant implications for identifying the biophysical constraints and potential for IAV genome reassortment. Furthermore, our study describes a novel application for HITS-CLIP methodology that could be applied to examine replication and assembly of all negative-sense RNA viruses.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank members of the Lakdawala laboratory for critical discussion.
FUNDING
National Institute of Health [U01AI124302 to D.J.S. and V.S.C.].
Conflict of interest statement. S.S.L. and N.L. are named inventors on a pending patent application describing the use of antisense oligonucleotides against specific NP binding sites as therapeutics.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments