





Abstract
PRMT1, an arginine methyltransferase, plays an important role in numerous cellular processes. In this study, we demonstrate a feedback regulatory loop between PRMT1 and the orphan receptor TR3. Unlike another orphan receptor HNF4, TR3 is not methylated by PRMT1 although they physically interact with each other. By delaying the TR3 protein degradation, PRMT1 binding leads to the elevation of TR3 cellular protein level, thereby enhances the DNA binding and transactivation activity of TR3 in a non-methyltransferase manner. Another coactivator SRC-2 acts synergistically with PRMT1 to regulate TR3 functions. In turn, TR3 binding to the catalytic domain of PRMT1 causes an inhibition of the PRMT1 methyltransferase activity. This repression results in the functional changes in some of PRMT1 substrates, including STAT3 and Sam68. The negative regulation of PRMT1 by TR3 was further confirmed in both TR3-knockdown cells and TR3-knockout mice with the use of an agonist for TR3. Taken together, our study not only identifies a regulatory role of PRMT1, independent on methyltransferase activity, in TR3 transactivation, but also characterizes a novel function of TR3 in the repression of PRMT1 methyltransferase activity.
INTRODUCTION
Arginine methylation, a common post-translational modification mainly occurring in nuclear proteins of eukaryotic cells, is catalyzed by a family of enzymes named protein arginine methyltransfeases (PRMTs) (1). In humans, PRMTs represent a family of 11 known enzymes that utilize S-adenosyl methionine (AdoMet) as a methyl donor (2). The substrates of these PRMTs include glycine arginine-rich (GAR) nucleic acid-binding proteins (3). PRMT1, the first identified arginine methyltransferase, is known to distribute throughout the cell and preferentially binds to GAR motifs within its substrates (4,5). PRMT1 shows a wide spectrum of substrates and is responsible for most of the arginine methylation reaction. RNA-binding proteins, histones and the DNA damage response proteins are known to be PRMT1 substrates (6). PRMT1 methylation has been shown to regulate a variety of cellular processes including transcription, RNA metabolism, translocation, signaling transduction, DNA repair and apoptosis.
Although mice with PRMT1–/– knocked out die shortly after implantation, embryonic stem cells from such embryos are viable under cell culture conditions (7), indicating that PRMT1 is an essential enzyme but dispensable for basic cellular reaction such as gene expression and DNA replication. The major targets of PRMT1 are nuclear proteins such as the orphan receptor HNF4 (8–10). PRMT1 modulates HNF4 transcriptional activity in two ways. On the one hand, it regulates the DNA-binding activity of HNF4; on the other hand, it methylates histones and acts synergistically with other coactivators at the promoters of HNF4 targets (11). PRMT1 methylation on nucleosomal histones also plays an important and dynamic role in nuclear receptor-mediated transcriptional regulation, chromation remodeling and other aspects of gene expression (12,13).
The enzymatic activity of PRMTs can be modulated by multiple manners, such as regulation of their expression, formation of macromolecules, changing accessibility of the enzyme to the substrate, other post-translational modifications and protein–protein interactions. For example, BTG1 and TIS21/BTG2 bind PRMT1 and stimulate its activity toward selected substrates (4). Binding of the tumor suppressor DAL-1 to PRMT3 acts as an inhibitor of its enzymatic activity, both in in vivo reaction and inside the cells (14,15). Protein phosphatase 2A (PP2A) inhibits PRMT1 activity and therefore not only increases the helicase activity of NS3 (a hepatitis C virus protein, which is inhibited by methylation) but also represses interferon-induced signaling through STAT1 to strengthen the cellular defense against viruses (16). It is proposed that some additional yet-to-be-identified factor(s) may multimerize PRMTs in vivo to regulate their activities (17).
Nuclear orphan receptor TR3 (also termed as ‘Nur77’ and ‘NGFI-B’) is a transcription factor that plays key roles in cell proliferation and apoptosis at both transcriptional and post-transcriptional levels (18). More recently, we have found that p300-induced p53 acetylation can be dramatically suppressed by TR3 at the transcriptional level (19). By blocking the acetylation, TR3 downregulates p53 transcriptional activity and leads to a decrease in the transcription level of MDM2 (19). Similarly, TR3 also significantly inhibits the p300-induced RXRα acetylation, resulting in the disassociation of RXRα from DNA, and the translocation of TR3/RXRα from the nucleus to mitochondria to induce apoptosis upon 9-cis retinoic acid stimulation (20). Further analysis revealed that TR3 negatively regulates the acetylation and transcriptional activity of many p300-regulated transcription factors through physical interaction (21).
In this study we investigated whether TR3 is subjected to PRMT1 methylation just like the other orphan receptor HNF4. Our results indicated that TR3 is not a substrate of PRMT1. However, PRMT1 could interact with TR3 and resulted in elevation of its cellular protein level, thus promoted the DNA binding and transactivation activity of TR3. On the other hand, we unexpectedly found that accumulated TR3 in turn inhibited PRMT1 methyltransferase activity by masking its catalytic domain. We further confirmed the inhibitory effect of TR3 on PRMT1 methyltransferase activity in both TR3-knockdown cells and TR3-knockout (KO) mice with the use of an agonist for TR3. Together, our study uncovers a novel molecular mechanism by which PRMT1 and TR3 form a feedback regulatory loop to modulate their functions mutually.
MATERIALS AND METHODS
Cell culture and transfection
Human embryonic kidney cells HEK293T (obtained from the ATCC, USA) and human hepatocellular liver carcinoma cells HepG2 (purchased from Cell Biology Institute, Shang-Hai, China) were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere of 5% CO2 at 37°C. Transfection was performed using calcium phosphate precipitation method as described previously (19).
Preparation of cytosolic and nuclear fractions
Harvested cells were suspended in 100 μl Buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.15% NP-40) containing 1% protease inhibitor, followed by a 15-min incubation. Homogenates were centrifuged at 12 000 g for 1 min at 4°C, and the supernatant (cytosolic fraction) was stored at –80°C. The pellet was washed with 1 ml Buffer A, then resuspended in 150 μl Buffer B (20 mM HEPES, pH 7.9, 0.4 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% NP-40) containing 1% protease inhibitor and sonicated at 4°C. Cellular debris was removed by centrifugation at 12 000 g for 30 min at 4°C and the supernatant (nuclear fraction) was stored at –80°C.
Immunoprecipitation and western blotting (Co-IP assay)
Cells were transfected with various plasmids as required and harvested at 36 h post-transfection. Cell lysate preparation, immunoprecipitation and western blotting were performed as described previously (21). Briefly, cell lysates were incubated with the appropriate antibody for 2 h, and subsequently incubated with protein A-Sepharose beads for 1 h. The protein–antibody complexes that were recovered on beads were subjected to western blot analysis using different antibodies as required after separation by SDS-PAGE. The immunoreactive products were then detected by using Enhanced chemiluminescence kit (Amersham).
Eletrophoretic mobility shift assay (EMSA)
For the in vitro EMSA, GST fusion proteins of PRMT1, TR3 and its deletion mutants as indicated were expressed in BL21 cells, and EMSA was performed as described previously (20,22) with biotin-labeled NurRE oligonucleotide (5′-GATCCGTGACCTTTATTCTCAAAGGTCA-3′; Invitrogen, Shanghai, China) (23).
For the in vivo EMSA, cells were transfected with different expression vectors as required, and nuclear extracts were prepared as described previously (24). EMSA was performed as described above.
Chromatin immunoprecipitation (ChIP) assay
Cells were cross-linked by adding formaldehyde directly to culture medium to a final concentration of 1% and incubated for 10 min at 37°C. Pellets were suspended in 200 μl of SDS lysis buffer (1% SDS, 10 mM EDTA and 50 mM Tris, pH 8.1), then sonicated. After centrifugation, 10% of the total supernatant was saved as total input control. The remaining supernatant was diluted by 10-fold in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7mM Tris–HCl, pH 8.1, 167 mM NaCl), and then divided into two parts, incubating with or without 2 μg of anti-TR3 antibody (Santa Cruz) overnight at 4°C. After immunoprecipitation, 60 μl of salmon sperm DNA/Protein-A agarose was added, and incubated for another 1 h at 4°C. Sepharose beads were washed sequentially in low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 150 mM NaCl) once, high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 500 mM NaCl) once, LiCl immune complex wash buffer (0.25 M LiCl, 1% IGEPAL CA630, 1% deoxycholic acid, 1 mM EDTA, 10 mM Tris, pH 8.1) once, and 1×TE for two times. Beads were then extracted with 500-μl elution buffer (1% SDS, 0.1 M NaHCO3). 20 μl 5 M NaCl and 2 μl of RNase A (0.5 mg/ml) were added and heated at 65°C for 5 h to reverse the formaldehyde cross-linking.
For quantitative PCR (qPCR), the primer sequences were showed as below:
Cyclin D2 promoter: Sense: 5′ CCTCCCTCTTCTTCCTCTGC 3′; antisense: 5′ TCCTAATCCTCCTGCCCTTG 3′
GAPDH promoter: Sense: 5′ GAGATGCCAGGAGCCAGGAGATG 3′; antisense: 5' GAAACAGGAGGACTTTGGGAACG 3′
In vitro methylation assay
PRMT1, GAR, TR3 and its deletions were prepared as recombinant glutathione S-transferase (GST) fusion protein and eluted from glutathione-agarose beads with 20 mM glutathione. Myc-PRMT1, Myc-Sam68, and Flag-STAT3 were expressed in 293T cells and immunoprecipitated with anti-Myc (Roche) and anti-Flag (Sigma) antibodies. Ten micrograms of substrate proteins were respectively incubated with 1 μg of GST-PRMT1 or immunoprecipitated Myc-PRMT1 and 6 μl of S-adenosyl-l-[methyl-3H]methionine (Amersham) or 600 μM noradioactive S-adenosylmethionine (NEB) in 30 μl of methyltransferase reaction buffer [20 mM Tris–HCl (pH 8.0), 200 mM NaCl, 0.4 mM EDTA] at 30°C for 2 h. Reactions were stopped by adding 30 μl of 2×SDS-PAGE sample buffer, followed by heating at 100°C for 10 min. Samples were loaded on SDS-polyacrylamide gels and analyzed by Coomassie blue staining and western blotting. For fluorography, gels were treated with EN3HANCE (Perkin-Elmer Life Sciences) as per the manufacturer's instructions, dried and exposed at –80°C for 2 weeks.
Luciferase assay
Cells were transfected with pGL3-NurRE, pGL3-promoter vector or APRE luciferase reporter genes, β-galactosidase (β-gal) and other different expression vectors as required. After transfection, luciferase activity was normalized for transfection efficiency using corresponding β-gal activity. The ratios of luciferase/β-gal activity were used as indicators for transcriptional activity of different promoters.
GST pull-down assay
GST pull-down assay was performed as described previously (19). Approximately 1 μg of GST or GST-TR3 immobilized to 10 μl of glutathione-Sepharose 4B was incubated with 0.5–1.0 μg of His-PRMT1 in 500 μl of modified GBT buffer (10% glycerol, 50 mM HEPES-NaOH (pH 8.0), 170 mM KCl, 7.5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 1% Triton X-100) containing 1% bovine serum albumin. After extensively washing with GBT buffer, the bound proteins were fractionated by 10% SDS-PAGE and subjected to Coomassie brilliant blue staining and western blot.
In vivo ubiquitination assay
Cells were transfected with expression vectors as required. After transfection, 20% of cells were used for western blot analysis; the other cells were lysed in Buffer A (6 M guanidine HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris/HCl, pH 8.0, 10 mM β-mercaptoethanol, 5 mM imidazole, 0.2% Triton X-100 and fresh protease inhibitors). Lysates were mildly sonicated to reduce viscosity, and ubiquitinated proteins were bound for 4 h at room temperature in 50 µl of nickel-nitrilotriacetic acid-agarose (Ni–NTA-agarose; QIAGEN, Valencia, CA). The beads were successively washed once for 5 min at room temperature with 1 ml of each of Buffer A, Buffer B (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris/HCl, pH 8.0, 10 mM β-mercaptoethanol, 5 mM imidazole), Buffer C (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris/HCl, pH 6.3, 10 mM β-mercaptoethanol, 5 mM imidazole). The bound proteins were fractionated by 10% SDS-PAGE and subjected to western blotting.
5-Bromo-2-deoxyuridine (BrdU) incorporation assay
Cells were transfected with expression vectors as required and incubated with BrdU (20 μM; Sigma) for 2 h. After washing with PBS, harvested cells were fixed with 4% paraformaldehyde for 30 min at room temperature and then incubated with PBS containing 0.1% saponin (Sigma) for another 10 min. The cells were resuspended in the buffer (0.1 M NaAc, pH=5.2, 5 M MgSO4) containing 30 μg DNase I and incubated for 30 min. After blocking for 30 min at 37°C, the cells were incubated with anti-BrdU antibody and then PE-linked anti-mouse antibody. Finally, cells were analyzed by flow cytometer (Beckman Coulter).
Immunofluorescent staining and microscopic observation
Cells that seeded on cover glass overnight were fixed in 4% paraformaldehyde. To stain exogenous sam68, cells were incubated with anti-Myc antibody, followed by Texas red-conjugated secondary antibody. Nucleus was stained by DAPI (Roche). Stained cells were visualized with confocal microscope (Leica TCS SP2 SE).
RESULTS
PRMT1 binds to TR3 and elevates its cellular protein level
Although the arginine methyltransferase PRMT1 displays a broad spectrum of substrate specificity (6), most of its substrates contain glycine- and arginine-rich (GAR) sequences including multiple arginines in RGG or RXR contexts (3,25,26). Similar to 53BP1 and hnRNP K, two known methylation substrates of PRMT1, TR3 also contains a ‘GRRGR’ motif within its DNA-binding domain (Figure 1A), suggesting that it might be a potential target of PRMT1. To test this possibility, we constructed several GST-fusion proteins, including full-length TR3, TR3(DBD) (DBD, DNA-binding domain), TR3(TAD + DBD) (TAD, transactivation domain) and TR3(LBD) (LBD, ligand-binding domain) (Figure 1A). When these proteins were incubated with PRMT1 that was immunoprecipitated from transfected cells, we could not detect any methylation signal (Figure 1B). On the other hand, the methylation of GST-GAR (GST fusion protein containing the glycine- and arginine-rich N-terminal region of fibrillarin) by PRMT1 was clearly observed in the control experiment (Figure 1B). Together, these results suggest that TR3 is not a methylation substrate of PRMT1 as we previously thought.
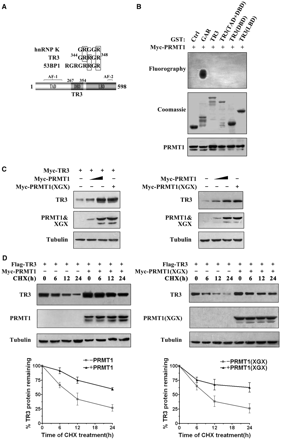
Upregulation of TR3 expression by PRMT1 is irrelevant to PRMT1 methylation activity. (A) Schematic presentation of the functional domains of TR3 (Bottom) with the comparison of its potential methylation site with those in hnRNP K and 53BP1 (Top). (B) In vitro PRMT1 methylation assay. GST-TR3 and its truncation mutants were separately incubated with PRMT1 protein immunoprecipitated from 293T cells. GAR was used as a positive control. (C) Effect of PRMT1 and its point mutant PRMT1 (XGX) on TR3 expression. Increased Myc-PRMT1 or Myc-PRMT1 (XGX) was transfected into 293T cells with or without Myc-TR3. The expression level of TR3 was assayed by western blotting with anti-myc or -TR3 antibody. Tubulin was used as the loading control. (D) Effect of PRMT1 and its point mutant on TR3 stability. Flag-TR3 with or without PRMT1/PRMT1(XGX) was transfected into 293T cells. The transfected cells were then treated with CHX (100 μg/ml) for the indicated times. TR3 expression levels were showed by western blotting using anti-Flag antibody (upper panel), and quantified by densitometry (down panel). Error bars indicate standard deviations (SD) of three independent experiments. (E) PRMT1 attenuated the ubiquitination of TR3. 293T cells were transiently transfected with Flag-TR3, His-ubiquitin or Myc-PRMT1 and subjected to the in vivo ubiquitination assay. (F) PRMT1 interacts with TR3. Flag-TR3, with or without Myc-PRMT1 or PRMT1(XGX) was transfected into 293T cells. Cell lysates were immunoprecipitated with anti-Myc antibody. The immunoprecipitates were then analyzed by western blotting using anti-Flag antibody for TR3 protein. (G) TR3 binds to PRMT1 in vivo. Cell lysates from 293T cells were immunoprecipitated with anti-PRMT1 antibody. The immunoprecipitates were then analyzed by western blotting using anti-TR3 antibody. (H) GST pull-down assay for determination of PRMT1-TR3 interaction in vitro. GST-TR3 was incubated with His-PRMT1 protein, and then subjected to Coomassie brilliant blue staining and western blotting with anti-His antibody to show TR3 and PRMT1 protein, respectively.
Unexpectedly, we found that PRMT1 could dramatically increase the cellular level of exogenous TR3 protein in cotransfected 293T cells (Figure 1C, left panel). Such effect seemed not dependent on the TR3 expression vector, as PRMT1 also elevated the endogenous level of TR3 protein in a dose-dependent manner (Figure 1C, right panel). In addition, we noted that the ability of PRMT1 to upregulate TR3 protein level was not correlated with its methyltransferase activity. When PRMT1(XGX), a point mutant deficient in methyltransferase activity (27), was cotransfected into cells with TR3, it could still enhance the cellular level of TR3 as effectively as the wild-type PRMT1 (Figure 1C). Therefore, the presence of PRMT1 protein, regardless of its methyltransferase activity, imposes a positive regulation on the cellular level of TR3 protein. In contrast, another arginine methyltransferase CARM1 had no effect on the protein level of TR3 (Supplementary Figure A), indicating a specific role of PRMT1 on regulation of TR3 level.
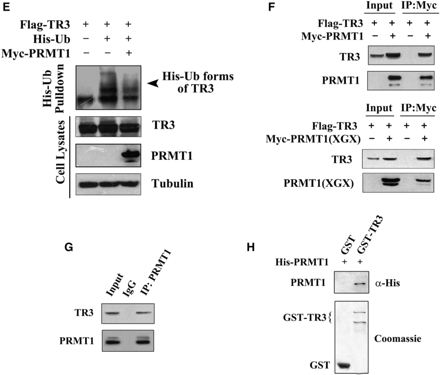
The increased TR3 level seen in PRMT1 transfected cells could be a result of delayed protein degradation. To verify this, we treated 293T cells with cycloheximide (CHX), a protein synthesis inhibitor, to block the de novo TR3 synthesis and monitored its lifetime at different time points by western blotting. As shown in Figure 1D, the half-life time (the time required for a protein to be degraded to 50%) of TR3 was slightly <12 h in 293T cells that transfected with TR3. With the coexpression of PRMT1 or PRMT1(XGX), the half-life time of TR3 protein was significantly prolonged to be >24 h (Figure 1D). It has been known that ubiquitin can target TR3 and induces its degradation (22). However, addition of PRMT1 abolished ubiquitin-induced TR3 degradation (Figure 1E). Therefore, the presence of PRMT1 could delay the degradation process of TR3 protein through ubiquitination pathway. As a result, the total cellular level of TR3 was elevated.
Based on the above results, we speculated that PRMT1 may physically interact with TR3. Therefore, 293T cells were cotransfected with Flag-TR3 and Myc-PRMT1, and subjected to Co-immunoprecipitation (Co-IP) assay. As we expected, Flag-TR3 could readily be detected by anti-Flag antibody once Myc-tagged PRMT1 or PRMT1(XGX) was immunoprecipitated by the anti-Myc antibody (Figure 1F). The physical interaction between PRMT1 and TR3 was further confirmed in vivo by Co-IP assay, where TR3 was detected in PRMT1-precipitated lysates (Figure 1G), and also in vitro by GST pull-down assay, in which a strong and specific binding of His-PRMT1 to GST-TR3 was detected (Figure 1H). Clearly, PRMT1 can directly interact with TR3 in vivo and in vitro.
PRMT1 enhances the DNA-binding ability of TR3
We went on to dissect the domain of TR3 that interacts with PRMT1 with the use of various TR3 truncation mutants (Figure 2A, top). Co-IP assays revealed that deletion of the C-terminus from TR3 (TR3/D2) abolished its binding to PRMT1 (Figure 2A, bottom), indicating that the C-terminus is a crucial region for TR3-PRMT1 interaction. Further mapping analysis demonstrated that LBD (aa 440–598) is the predominant PRMT1-binding region, although the DBD also showed a very weak interaction with PRMT1 (Figure 2A, bottom).
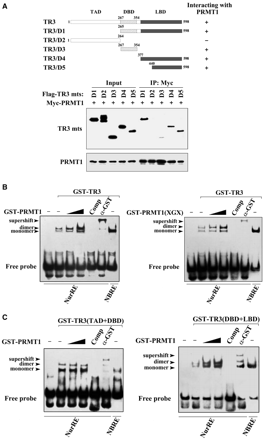
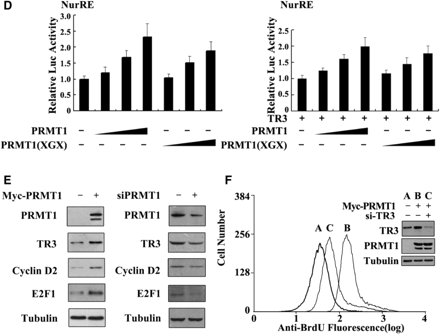
PRMT1 promotes the DNA binding and transactivation of TR3. (A) Determination of PRMT1-binding sites in TR3. Schematic diagrams depict different TR3 deletion mutants used in the domain mapping experiments (Top). 293T cells were transfected with Myc-PRMT1 and different Flag-TR3 deletion constructs as indicated. Cell lysates were immunoprecipitated with anti-Myc antibody. The immunoprecipitates were analyzed by western blotting using anti-Flag antibody for TR3 proteins. (B, C) Effect of PRMT1 on TR3 DNA binding. GST-TR3 and its deletion mutants, including TAD + DBD and DBD + LBD, were incubated with GST-PRMT1 or its point mutant PRMT1(XGX), respectively, and then subjected to the EMSA assay with the NurRE (for TR3 dimer binding) or NBRE (for TR3 monomer binding) as a probe. Specificity of the complexes was assessed by competition with 200-fold excess of homologous unlabeled oligonucleotides or with anti-GST antibody to show the supershifted band. (D) Effect of PRMT1 on the transactivation of TR3. Increasing amount of PRMT1 and its mutant PRMT1(XGX), together with or without TR3 were transfected into 293T cells. NurRE-luciferase reporter gene and β-gal gene expression vector were transfected simultaneously. Reporter gene activity was determined and normalized in relation to the cotransfected β-gal activity. Error bars indicate standard deviations (SD) of three independent experiments. (E) Effect of PRMT1 on the expression levels of E2F1 and Cyclin D2. Myc-PRMT1 or si-PRMT1 was transfected into 293T cells. The expression levels of PRMT1, TR3 and TR3-dependent downstream factors Cyclin D2 and E2F1 were assayed by western blotting with anti-PRMT1, -TR3, -Cyclin D2 or –E2F1 antibodies. (F) PRMT1 regulates cell proliferation through TR3. Different expression plasmids, including Myc-PRMT1 and si-TR3, were transfected into 293T cells as indicated. The cells were then subjected to FACS as described in ‘Materials and methods’ Section. TR3 and PRMT1 expression levels were showed by western blotting using anti-TR3 and anti-Myc antibodies (inset).
Next, we carried out EMSA assay to assess whether PRMT1 enhances TR3 binding to its cognate element by using the TR3 response element (NurRE) as a probe (22). As shown in Figure 2B, GST-TR3 could bind to the NurRE, forming a dimeric complex, and this complex was completely dissociated by the addition of a competitor or showed a supershift band by incubation with GST antibody. When GST-TR3 was preincubated with GST-PRMT1, its binding to DNA was enhanced and became stronger when the amount of PRMT1 was increased (Figure 2B, left). Similar results were observed when PRMT1(XGX) mutant protein was used (Figure 2B, right), indicating that PRMT1 methyltransferase activity is not involved in the regulation of TR3 DNA binding. As a negative control, the complex of TR3 was not influenced by incubation with GST (Supplementary Figure B). We further used TR3 truncation mutants to perform the parallel experiments. Due to the existence of the DBD, both TR3(TAD + DBD) and TR3(DBD + LBD) all bound to the NurRE, respectively (Figure 2C). However, only the NurRE-binding activity of TR3(DBD + LBD), but not TR3(TAD + DBD), could be enhanced upon incubation with PRMT1 (Figure 2C). Since PRMT1 mainly interacts with LBD but not TAD or DBD (Figure 2A), this result strongly suggests that the interaction of PRMT1 with TR3 plays a positive regulatory role in TR3 DNA-binding activity.
It is expected that the enhanced DNA-binding activity of TR3 by PRMT1 would consequently affect the transactivation of TR3, we therefore measured TR3 transactivation in transient transfection cells by luciferase assay, in which a construct containing two TR3 homodimer-binding sites (NurRE) linked to luciferase reporter gene (pGL3-promoter) (19) was used. As shown in Figure 2D, PRMT1 and PRMT1(XGX) could activate the transcriptional activity of reporter gene in a dose-dependent manner, in the presence of endogenous or exogenous TR3, which showed a good consistency with the effect of PRMT1 on TR3 DNA binding. As a negative control, PRMT1 and PRMT1(XGX) did not showed any effect on the activation of pGL3-promoter vector, a nonrelated reporter gene (Supplementary Figure C), suggesting a selective activation of PRMT1 for TR3 transactivation. Since PRMT1 enhanced the transactivation of TR3, it is of interest to determine whether PRMT1 influences the expression levels of TR3 downstream factors, such as E2F1 and Cyclin D2 (28,29). Consistent with our speculation, the expression levels of both E2F1 and Cyclin D2 were upregulated by overexpression of PRMT1, but downregulated by knockdown of endogenous PRMT1 (Figure 2E). Since both E2F1 and Cyclin D2 play important role in cell proliferation, we finally evaluate the biological relevant of the upregulation of TR3 transactivation by PRMT1 with the method of BrdU incorporation assay. As shown in Figure 2F, PRMT1 transfected 293T cells exhibited much stronger BrdU incorporation compared with the control, and the effect of PRMT1 was impaired by the transfection of si-TR3. This result suggests that PRMT1 stimulats the proliferation of 293T cells through enhancing the activity of TR3.
SRC-2 acts synergistically with PRMT1 to regulate TR3
PRMT1 generally functions together with its coactivator to achieve synergistic enhancement of nuclear receptor function. Since steroid receptor coactivators (SRCs) have been shown to be involved in TR3 transactivation (42), we tested whether SRCs and PRMT1 could act synergistically to regulate TR3. As shown in Figure 3A, all three SRC members (SRC-1, -2 and -3) had very little effect (gray bars) on TR3-activated reporter gene activity in the absence of PRMT1. However, with the co-presence of PRMT1 and SRC-2 but not SRC-1 or SRC-3 displayed a strong synergistic effect to stimulate TR3 transactivation (black bars). As a negative control, SRC-2 did not influence the transactivation of pGL3-promoter vector, no matter whether PRMT1 was present or not (Supplementary Figure D).
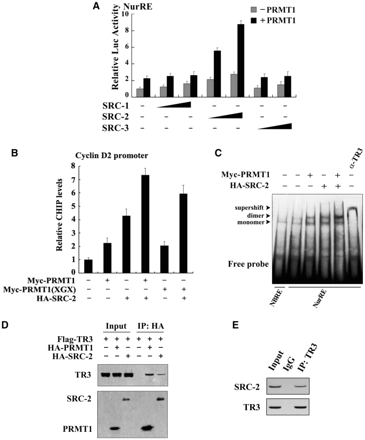
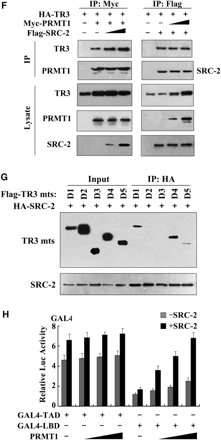
SRC-2 exerts a synergistic effect with PRMT1 on TR3 transactivation. (A) Effect of SRCs on TR3 transactivation. Increasing amount of SRCs, including SRC-1, SRC-2 and SRC-3, with or without PRMT1 were transfected into 293T cells. The reporter gene activity of NurRE was determined. (B) PRMT1 and SRC-2 enhanced the binding of TR3 to Cyclin D2 promoter. Myc-PRMT1, Myc-PRMT1 (XGX) and HA-SRC-2 were transfected into 293T cells as indicated. The binding of TR3 to Cyclin D2 promoter was determined by ChIP assay using anti-TR3 antibodies, and then quantified by qPCR. (C) Effect of PRMT1/SRC-2 on endogenous TR3 binding to the NurRE. Myc-PRMT1 and HA-SRC-2 were transfected into 293T cells as indicated. Nuclear extracts were prepared, with or without incubated with anti-TR3 antibody, and then subjected to the EMSA assay with the NurRE or NBRE as a probe. (D) Both PRMT1 and SRC-2 bound to TR3. 293T cells were transfected with Flag-TR3, HA-PRMT1 and HA-SRC-2. Cell lysates were immunoprecipitated with anti-HA antibody. The immunoprecipitates were then analyzed by western blotting using anti-Flag antibody to show the interaction of TR3 with PRMT1 and SRC-2, respectively. (E) SRC-2 interacted with TR3 in vivo. 293T cells were lysated and immunoprecipitated with anti-TR3 antibodies. The pellets were subjected to western blotting using anti-SRC-2 antibody. IgG was set as a negative control. (F) SRC-2 but not PRMT1 enhanced TR3-PRMT1 interaction. 293T cells were transfected with different plasmids as indicated. After transfection, cells lysates were immuoprecipitated with anti-Myc or anti-Flag antibodies, and then the immunoprecipitates were subjected to western blotting. (G) Determination of SRC-2-binding sites in TR3. 293T cells were transfected with HA-SRC-2 and different Flag-TR3 deletion constructs as indicated in Figure 2A. Cell lysates were immunoprecipitated with anti-HA antibody. The immunoprecipitates were analyzed by western blotting using anti-Flag antibody for TR3 proteins. (H) Synergistic effect of SRC-2 with PRMT1 on TR3 transactivation. 293T cells were transfected with increasing amount of PRMT1, together with GAL4 reporter gene and GAL4-TAD or GAL4-LBD in the absence or presence of SRC-2 as indicated. The luciferase activity was then determined.
To further determine the synergistic role of SRC-2, we carried out qChIP assay (quantitative chromatin immunoprecipitation) with the use of cyclin D2 promoter that contains a TR3 response element (29). Although either SRC-2 or PRMT1 could help TR3 targeting to the cyclin D2 promoter, this binding was enhanced when both coactivators were used together (Figure 3B, lane 4 versus lane 2 or lane 3). Similar result was also observed when SRC-2 and PRMT1(XGX) were used (lane 6 versus lane 5). However, as a negative control, PRMT1 and SRC-2 showed no effect on nonrelated gene GAPDH (Supplementary Figure E). Moreover, in the in vivo EMSA assay, the complex formed in nuclear extracts was TR3 protein, as this complex was supershifted when nuclear extracts were incubated with anti-TR3 antibody (Figure 3C). Similarly, the amount of TR3-bound NurRE increased following the presence of PRMT1 or SRC-2, and reached a maximum when both were presented. This result further confirmed that SRC-2 exerts a synergistic effect with PRMT1 to activate TR3 transactivation through promoting TR3 DNA binding.
The above results suggest that SRC-2 may also bind to TR3 in a similar way as PRMT1. We thus conducted a Co-IP assay to test this possibility. Indeed, when HA-tagged SRC-2 or PRMT1 was immunoprecipetated, the Flag-TR3 could be detected (Figure 3D). As reported by other group that TR3 and SRC-2 bound to each other in vitro (30), we also confirmed the interaction between TR3 and SRC-2 in vivo (Figure 3E). To further evaluate the mutual influence on the binding with TR3, different amount of SRC-2 or PRMT1 were transfected into 293T cells. As shown in Figure 3F, with the increasing expression of SRC-2, the interaction between PRMT1 and TR3 became much stronger. In contrast, PRMT1 could not enhance the interaction between TR3 and SRC-2. Again, this result supports SRC-2 synergistic effect on TR3-PRMT1 interaction. We went on to determine the region of TR3 that interacts with SRC-2 by coexpressing different truncation mutants of TR3 in 293T cells. Co-IP assays showed that TR3/D1 and TR3/D4, but not TR3/D2 and TR3/D3, retained the ability to interact with SRC-2 (Figure 3G). Compared with the result of PRMT1 interaction with TR3, we conclude that PRMT1 and SRC-2 all bind to the LBD of TR3, with PRMT1 mainly targeting to aa 440–598 and SRC-2 mainly targeting to aa 377–440.
The fact that both PRMT1 and SRC-2 bind to the LBD of TR3 suggests that the LBD might function as the major TR3 transactivation domain in response to PRMT1 and SRC-2. To test this possibility, the assay for luciferase activity was performed. 293T cells were transfected with a chimeric GAL4-TR3, GAL4-TR3(TAD) (containing the AF-1 domain) or GAL4-TR3(LBD) (containing the AF-2 domain), together with a construct containing five GAL4 response elements linked to a luciferase reporter gene (pGAL4). Consistent with the previous report that transactivation of AF-1 was more powerful than AF-2 (30), cells transfected with TR3(TAD) showed a higher luciferase activity than those transfected with TR3(LBD) in the absence of PRMT1/SRC-2 (Figure 3H, bar 1 versus bar 9). Cotransfection of PRMT1 slightly increased their transactivation activity (Figure 3H, gray bars). However, cotransfection of both SRC-2 and PRMT1 led to a significant activation on TR3(LBD) but not TR3(TAD) (Figure 3H, black bars). This finding thus confirms a coordinate effect of PRMT1 and SRC-2 on TR3 transactivation, and demonstrates the role of LBD as their major mediator. In addition, we noted that transcriptional activity of GAL4-TAD was still different in the absence or presence of SRC-2, although endogenous PRMT1 was inhibited by its siRNA (Supplementary Figure F). This result suggests that in the absence of SRC-2, other cofactors might be involved in the activation of GAL4-TAD, while in the presence of SRC-2, these cofactors might bridge SRC-2 targeting to the TAD of TR3, so that SRC-2 still activates the GAL4-TAD.
TR3 inhibits PRMT1 methyltransferase activity
PRMT1 is the only known enzyme that mediates asymmetric dimethylation of Arg3 on Histone H4 (H4 R3) (31,32). We did observe the methylation of H4 R3 by PRMT1 in vitro with AdoMet as the methyl donor (Figure 4A). However, when different PRMT1 truncation mutants were assay in vitro, no methylation signals could be detected, even with the PRMT1Δ5 mutant that had intact catalytic domain and dimerization arm (Figure 4A). These results indicate that an intact PRMT1 is critical for its methylation activity.
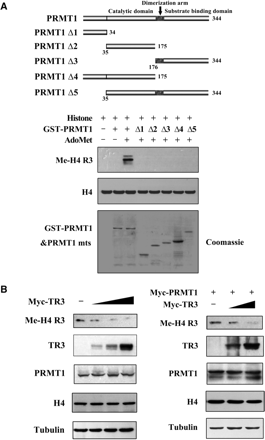
TR3 inhibits PRMT1 methyltransferase activity. (A) Full-length PRMT1 but not its truncation mutants methylate H4 R3 in vitro. GST-PRMT1 and its mutants (Top) were incubated with Histone and AdoMet, respectively. The reaction mixtures were resolved on SDS-PAGE, and visualized by anti-methyl-H4 R3 antibody. (B, C) TR3 exerted an inhibitory effect on the methylation of H4 R3 in vivo. Myc-TR3 (B) or siRNA-TR3 (C) was transfected into 293T cells with or without PRMT1. The in vivo methylation of H4 R3 was examined. (D) Effect of TR3 on the in vitro methylation of GAR by PRMT1. The methylation of GAR in vitro was determined with anti-dimethyl-arginine asymmetric antibody (ASYM24). (E) TR3 interacts with PRMT1 truncation mutants. Different GST-PRMT1 mutants were separately incubated with Flag-TR3 protein immunoprecipitated from 293T cells to detect their interactions by GST pull-down assay. (F) Effect of PRMT1 mutants on TR3 transactivational activity. Different PRMT1 truncation mutants were transfected into 293T cells. The luciferase activity of NurRE reporter gene was determined. (G) Effect of PRMT1 mutants on TR3 expression levels. Different PRMT1 truncation mutants together with Flag-TR3 were transfected into 293T cells. The expression levels of TR3 were determined by western blotting. (H) Effect of TR3 mutants on PRMT1-mediated H4 R3 methylation. Different TR3 mutants as indicated were transfected into 293T cells. The in vivo methylation of H4 R3 was examined.
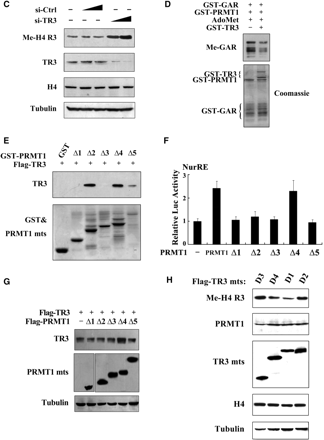
On the other hand, we are thinking why PRMT1 cannot methylate TR3, although TR3 contains a glycine- and arginine-rich motif ‘GRRGR’ in its DBD; and why PRMT1 activates TR3 transactivational activity in a non-methyltransferase fashion? One of possibilities might be that TR3 in turn has an ability to affect PRMT1 methyltransferase activity. To test this, TR3 was introduced into cells and the methylation of H4 R3 was examined. As shown in Figure 4B, although TR3 did not alter the expression level of either endogenous or exogenous PRMT1, it effectively inhibited the methylation level of H4 R3 in a dose-dependent manner. However, overexpression of TR3 could not affect the methylation level of H3 R17 induced by CARM1 specifically (Supplementary Figure G), indicating a specific role of TR3 on PRMT1-induced methylation. Furthermore, when siRNA was introduced into cells to knock down endogenous TR3 levels, the methylation level of H4 R3 was greatly increased (Figure 4C). Similarly, the PRMT1-induced GAR methylation could also be suppressed by TR3 in vitro (Figure 4D). These results firmly demonstrated that TR3 has a unique characteristic to inhibit PRMT1 methyltransferase activity.
PRMT1 contains two important domains, the catalytic domain and the substrate-binding domain (12). Some of PRMT1 substrates, such as Histone H4, STAT3 or Sam68, were found to bind the substrate-binding domain of PRMT1 (Supplementary Figure H–J). However, in the current case, we unexpectedly found that TR3 interacted with all the catalytic domain-containing mutants (PRMT1Δ2, Δ4 and Δ5), but not those catalytic domain-absent mutants (PRMT1Δ1 and Δ3) (Figure 4E), suggesting the importance of catalytic domain in their interaction. We further analyzed the effect of PRMT1 truncation mutants on TR3 transactivation activity and expression level. Although Δ2, Δ4 and Δ5 all interacted with TR3, only Δ4 was found to upregulate both the transactivation and expression level of TR3 (Figure 4F and G), demonstrating a key role for the N-terminus of PRMT1 in the regulation of TR3 functions. Finally, the methylation activity of PRMT1 could only be suppressed by the TR3 deletion mutants (D1 and D4) that possess the ability to bind PRMT1 (Figure 4H). Taken together, our data reveal that TR3 directly targets to the catalytic domain of PRMT1 to inhibit its methyltransferase activity.
TR3 negatively regulates PRMT1-methylated proteins
Next, we investigated whether TR3 could negatively regulate PRMT1-mediated arginine methylation on a broad spectrum of substrates under physiological conditions. We examined the PRMT1-induced asymmetric dimethylation in total cell lysates by using specific anti-dimethyl-arginine asymmetric antibody. We could observe multiple methylated protein bands upon the expression of PRMT1. However, coexpression of TR3 led to a decrease in the methylation level of these proteins in a dose-dependent manner (Figure 5A). Therefore, TR3 indeed inhibits PRMT1-induced methylation of numerous cellular proteins.
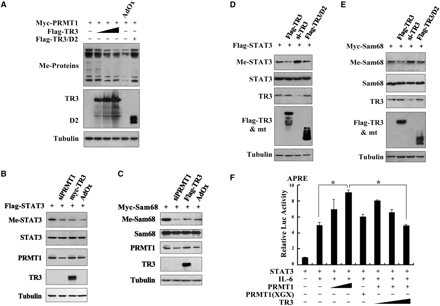
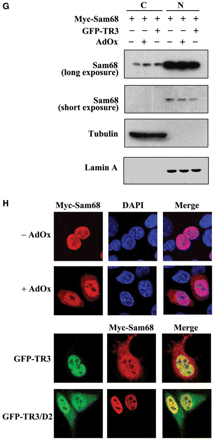
TR3 attenuated PRMT1-induced methylation of STAT3 and Sam68. (A) TR3 inhibited the methylation activity of PRMT1 on a broad spectrum of substrates. Myc-PRMT1, with increasing amount of Flag-TR3 or TR3/D2, was transfected into 293T cells. Cell lysates were prepared and then subjected to western blotting. The methylation of proteins was detected by anti-dimethyl-Arginine asymmetric antibody (ASYM24). AdOx was used as a positive control. (B, D) TR3 attenuated PRMT1-induced methylation of STAT3. Flag-STAT3, siPRMT1, Myc-TR3, TR3/D2 and si-TR3 were transfected into 293T cells as indicated, and then immunoprecipitated with anti-Flag antibody. The methylation of STAT3 was determined with anti-dimethyl-arginine asymmetric antibody (ASYM24). AdOx treatment was set as a control. (C, E) TR3 abolished PRMT1-induced methylation of Sam68. Myc-Sam68, siPRMT1, Flag-TR3, TR3/D2 and si-TR3 were transfected into 293T cells as indicated. The methylation of Sam68 was determined. (F) Effect of TR3 on PRMT1-induced transactivation of STAT3. Increasing amount of PRMT1 or its mutant PRMT1(XGX), together with Flag-STAT3 or Flag-TR3 were transfected into 293T cells. After transfection, cells were treated with IL6 (10 ng/ml) for 30 min. The luciferase activity of APRE reporter gene was determined. *P < 0.05 by two-tailed Student's t-test. (G) Effect of TR3 on localization of Sam68. Myc-Sam68 with or without GFP-TR3 was transfected into 293T cells. Nuclear and cytoplasmic fractions were prepared and subjected to western blotting with anti-Myc antibody. Tubulin and Lamin A were used to quantify the amount of protein loaded in the cytoplasm and nucleus. (H) Confocal analysis of Sam68 subcellular location regulated by TR3 and its deletion mutant. Myc-Sam68, GFP-TR3 and TR3/D2 were transfected into 293T cells at different combinations as indicated. Cells were immunostained by anti-Myc antibody followed by Texas Red-conjugated secondary antibody to detect Sam68. Stained cells were visualized with the confocal microscope. AdOx was used as a positive control.
It is expected that TR3 suppression on PRMT1-dependent methylation would affect the functions of PRMT1 substrates. We analyzed the effect of TR3 on Sam68 and STAT3, two proteins known to be methylated by PRMT1 (33,34). Both STAT3 and Sam68 displayed a PRMT1-dependent methylation, which could be abolished by the transfection of siRNA against PRMT1 in 293T cells (Figure 5B and C). As we expected, TR3 exerted an attenuated effect on methylation of these two proteins as effectively as AdOx, a global methylation inhibitor (Figure 5B and C). Conversely, this methylation was enhanced when si-TR3 was introduced into the same cell line (Figure 5D and E). We also noted that TR3 mutant (TR3/D2) which did not interact with PRMT1 lost its ability to regulate PRMT1-induced methylation of STAT3 and Sam68 (Figure 5D and E).
STAT3 is a transcription factor and its activity toward the reporter gene APRE (35) could be enhanced by PRMT1 instead of PRMT1(XGX) in the presence of IL-6 (Figure 5F). However, such enhancement was significantly repressed by TR3 in a dose-dependent manner (Figure 5F). Sam68 is an RNA-binding protein and methylated Sam68 would be imported into the nucleus to exert its function (34). Similar to AdOx, TR3 could effectively block the nuclear entry of Sam68, resulting in the cytoplasmic accumulation of Sam68, detected by confocal microscopy and western blotting (Figure 5G and H). Taken together, our data demonstrated that TR3, through the inhibition of PRMT1 methylation activity, negatively regulates the functions of PRMT1 targets.
The role of TR3 agonist in PRMT1 methyltransferase activity
Very recently, we screened a bank of natural products from fungi and found an agonist for TR3, called cytosporone B (Csn-B) (36). We expected that Csn-B would have similar inhibitory effect as TR3 on PRMT1 methyltransferase activity. Co-IP assays revealed that the TR3-PRMT1 interaction was enhanced in response to Csn-B (Figure 6A). Although it upregulated the expression level of endogenous TR3 but not that of PRMT1, Csn-B indeed repressed the PRMT1-induced methylation of H4 R3 in a dose-dependent manner (Figure 6B). Such inhibitory effect was abolished when a siRNA against TR3 was introduced into cells to inhibit endogenous TR3 expression (Figure 6C), suggesting that the repression of PRMT1 methyltransferase activity by Csn-B is directly mediated by TR3. In addition, we also compared the occurrence between Csn-B-induced TR3 expression and TR3-inhibited PRMT1 methyltransferase activity at different time-points. The result showed that TR3 expression reached the peak at 4 h after cells were treated with Csn-B, and it was at the same time-point when the maximal inhibition of H4 R3 methylation was achieved (Figure 6D). This result further supports the conclusion that it is the agonist-induced TR3 that contributes to the inhibition of PRMT1 methyltransferase activity.
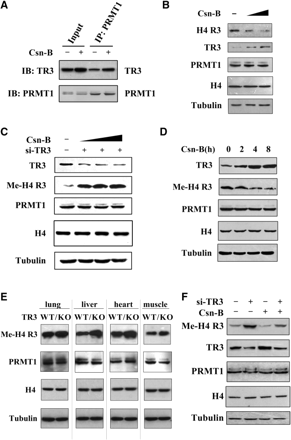
The role of TR3 agonist Csn-B in both cells and mice. (A) Csn-B enhanced TR3 interaction with PRMT1. HepG2 cells were treated with Csn-B (10 μM) for 5 h, and then the cell lysates were prepared. Co-IP assay was performed as described in Figure 1E. (B) Csn-B inhibited PRMT1 methylation on H4 R3 in HepG2 cells. Cells were treated with increased doses of Csn-B, and then the in vivo methylation of H4 R3 was examined with anti-methyl-H4 R3 antibody. The expression levels of PRMT1, TR3 and Histone H4 were determined by western blotting. (C) Inhibition of Csn-B on PRMT1 methylation was abolished by siRNA against TR3. HepG2 cells were transfected with si-TR3 and then treated with increased doses of Csn-B. The in vivo methylation of H4 R3 was examined with anti-methyl-H4 R3 antibody. (D) A correlation between increased TR3 expression level and reduced PRMT1 methylation activity upon Csn-B treatment. HepG2 cells were treated with Csn-B for the indicated times. The in vivo methylation of H4 R3 was examined with anti-methyl-H4 R3 antibody. (E) Comparison of methylation activity of H4 R3 between wild-type C57 mice and TR3–/– C57 mice. Different tissues, including lung, liver, heart and muscle, were taken from the mice. Protein lysates were prepared, separated by SDS-PAGE and probed with anti-methyl-H4 R3, anti-PRMT1, Histone H4 and anti-Tubulin antibodies, respectively. (F) Effect of Csn-B on PRMT1 methyltransferase activity in nude mice. Nude mice were devised into four groups: lane 1, the mice were injected with gastric cancer BGC-823 cells; lane 2, the mice were injected with expressing siRNA-TR3 BGC-823 cells; lane 3, the mice were administrated with the dose of 50 ng Csn-B/g bodyweight intraperitoneally every 3 days for 4 weeks; lane 4, the mice expressing si-TR3 were administrated with Csn-B the same as the lane 3 group. Four weeks later, the mice were executed and the formed tumors were taken. Protein lysates were prepared, separated by SDS-PAGE and probed with anti-methyl-H4 R3, -PRMT1, -TR3, Histone H4 and -Tubulin antibodies, respectively.
Finally, we confirmed the negative regulation of TR3 on PRMT1 methyltransferase activity in different tissues of wildtype (WT) and TR3 KO C57 mice. In WT mice, different methylation levels of H4 R3 were detected in lung, liver, heart and muscle. In TR3 KO mice, the methylation levels of H4 R3 were higher in each tissue as compare to that in WT mice. We further analyzed the effect of TR3 agonist on PRMT1 methyltransferase activity in the xenograft of nude mice. The tumor samples were obtained from our previous experiment (36). Western blot analysis showed that Csn-B increased the TR3 expression level in the xenograft, resulting in the decrease of H4 R3 methylation (Figure 6F). Conversely, introducing siRNA against TR3 reversed the methylation of H4 R3 to even a higher level than that of the control regardless of the presence of Csn-B (Figure 6F). These results demonstrated the negative correlation between TR3 expression and PRMT1 methylation activity in vivo, and revealed the possibility to use TR3 agonist as a candidate inhibitor of PRMT1.
DISCUSSION
Although arginine methylation represents an emerging mechanism to regulate protein functions, how PRMT1 regulates its methylation substrates or how its methyltransferase activity is regulated by other factors are still largely unknown. In the current study, we established a novel feedback regulatory loop between PRMT1 and the orphan receptor TR3 (Figure 7). Although PRMT1 does not methylate TR3, it interacts with TR3 and promotes the DNA binding and transactivation activity of TR3. On the other hand, by binding to the catalytic region of PRMT1, TR3 inhibits the methyltransferase activity of PRMT1.
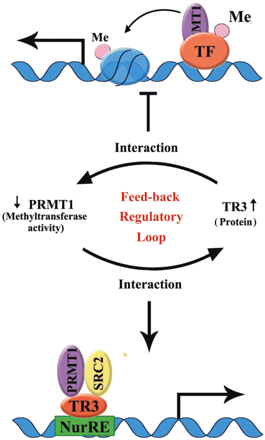
A working model to illustrate the feedback regulatory loop between PRMT1 methyltransferase activity and TR3 protein level.
PRMT1 has been demonstrated as an important arginine-specific histone methyltransferase. It can methylate histone H4 at Arg-3 in vivo and in vitro (37,31,32), thereby enhancing transcriptional activation by nuclear receptors (32,38). PRMT1 is recruited to promoters of specific genes through direct interaction with DNA-binding transcription factors, such as p53 and YY1 (39,40), or by binding to other coactivators, such as the p160 coactivator GRIP1, which associates directly with nuclear receptors and many other types of DNA-binding transcription factors (38). Previous studies suggest that the enzymic activity is required for promoter-associated PRMT1 to stimulate transcriptional activation (31,41). However, our finding that the PRMT1(XGX) mutant deficient of methyltransferase activity could also promote the DNA binding of TR3 and contributed to transcriptional activation suggests that the methyltransferase activity of PRMT1 is not necessary for its coactivator function, at least in the case of TR3. Moreover, we found that PRMT1 truncation mutants that had lost the ability to methylate H4 R3 still retained the ability to enhance TR3 transactivational activity, further suggesting that activation of TR3 transactivation is not associated with PRMT1 methyltransferase activity. Therefore, it is likely that the methyltransferase activity of PRMT1 and its ability to bind nuclear receptors both contribute to its coactivator function.
SRCs have been shown to act as coactivators to facilitate nuclear receptor DNA binding (42). We observed a synergistic effect of SRC-2 and PRMT1 on TR3 transactivation. Both coactivators interacted with the LBD of TR3 and either one could enhance TR3 binding to the cyclin D2 promoter that contains a TR3 response element. Although we cannot decide which coactivator played a main role in TR3 transactivation, according to the fact that SRC-2 could not upregulate TR3 expression levels (data not shown), we propose that PRMT1 may first interact with TR3 to induce its accumulation, thereby increasing its DNA-binding capacity. Then, SRC-2, via an unknown mechanism that may involve PRMT1, bound to TR3 to exert synergistic effect, and finally co-initiate the transcription of TR3 target genes. Therefore, PRMT1 may be a device for specific activation of NurRE-containing promoters.
So far, litter is known about how the methyltransferase activity of PRMT1 is regulated. The members of PRMT family have been found to interact with proteins that are often not methyl-accepting substrates but can modulate the methyltransferase activity of PRMTs through distinct mechanisms. For example, tumor suppressor DAL-1/4 functions as an inhibitor of PRMT3 activity through interaction (14). By contrast, it regulates PRMT5 activity by either repressing or enhancing protein methylation (15). PRMT1 has been identified as an interacting protein of the immediate early gene product BTG1 (B-cell translocation gene 1), which also modulates PRMT1 activity toward selected substrates (4,43). In addition, hCAF1, a partner of BTG1 and BTG2, was also found to regulate PRMT1 activity in a substrate-dependent manner (44). Our current study revealed that, TR3, another immediate early gene product (45), also plays a regulatory function on PRMT1 activity.
The mechanism by which TR3 regulates PRMT1 activity is through protein–protein interaction. The critical step might be the binding of TR3 to the catalytic domain of PRMT1, which causes the loss of its methyltransferase activity. The binding region of TR3 on PRMT1 is different from that of Histone H4, Sam68 and STAT3, the latter bound to the substrate-binding domain of PRMT1. This may explain why PRMT1 cannot methylate TR3, although TR3 contains a glycine- and arginine-rich motif ‘GRRGR’ in its DBD. On the other hand, although only full-length PRMT1 effectively methylates H4 R3, our results indicated that the N-terminus of PRMT1 plays an important role in regulation of TR3 function, including expression levels and transactivation activity of TR3. In fact, N-terminus are the major regions where at least 11 human PRMTs differ from each other (2), and has been reported to be important for recognizing its target substrates (46). It seems that the functions of this N-terminus may depend on the nature of the protein substrate involved.
Repression of PRMT1 activity by its interaction partner would indirectly affect the functions of its substrates. The Sam68 RNA-binding protein is methylated in vivo by PRMT1, and inhibition of Sam68 methylation by deletion of the methylation sites or use of methylation inhibitor causes accumulation of Sam68 in the cytoplasm and prevents Sam68-mediated export of HIV RNAs (34). We also observed reduced Sam68 nuclear localization when TR3 was expressed. Similarly, the transcriptional activity of STAT3 induced by PRMT1 methylation was also inhibited by TR3. These two events could all be attributed to the repression of PRMT1 methylation activity by TR3. Therefore, PRMT1 may provide a platform for TR3 to regulate other protein functions.
One particular interesting finding from our study is that an agonist for TR3 can inhibit methyltransferase activity of PRMT1 through activating TR3. Data from cell cultures and mice both indicated the existence of a negative correlation between TR3 expression level and PRMT1 methyltransferase activity. This finding is significant for the therapeutic application of TR3 agonist, because many diseases have been shown to be correlated with aberrant arginine methylation. For example, asymmetric dimethylarginine (ADMA) is synthesized when arginine residues in proteins are methylated by the action of PRMTs (47,2). ADMA inhibits nitric oxide syntheses (NOS) (48), and therefore has the potential to produce detrimental biological effects, particularly in the cardiovascular system. Recently, several studies have suggested that the plasma concentrations of ADMA provide a marker of risk for endothelial dysfunction and cardiovascular disease (48–51). Therefore, it is of interesting to further investigate the role of TR3 agonist through regulation of methyltransferase activity, which may lead to new therapeutic applications.
FUNDING
The National Natural Science Foundation of China (30425014, 30570936, 30630070); grants from the ‘973’ Project (2007CB914402) and the ‘863’ project (2006AA09Z410) of the Ministry of Science and Technology; grant from the Ministry of Education (706036); and Key project of Science and Technology of Fujian province (2007Y0033). Funding for open access charge: 973 project (2007CB914402) of the Ministry of Science and Technology of China.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Dr Uta-Maria Bauer (Philipps-University of Marburg) for plasmids of Myc-PRMT1 and Myc-PRMT1 (XGX); Dr Jörg Kobarg (Laboratório Nacional de Luz Síncrotron) for plasmids of GST-PRMT1 and its deletions; Dr Michael R. Stallcup (University of Southern California) for plasmid of HA-PRMT1; Dr Mark T. Bedford (The University of Texas M.D. Anderson Cancer Center) for plasmid of GST-GAR; Dr Shih-Ming HUANG (National Defense Medical Center, Taipei) for plasmid of HA-SRC-2; Dr Edward Seto (University of South Florida) for plasmid of siPRMT1; Dr Micheal David (University of California) for plasmid of His-PRMT1; and Dr Stéphane Richard (McGill University) for plasmid of Myc-Sam68.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments