





Abstract
We report a new web server, aLeaves (http://aleaves.cdb.riken.jp/), for homologue collection from diverse animal genomes. In molecular comparative studies involving multiple species, orthology identification is the basis on which most subsequent biological analyses rely. It can be achieved most accurately by explicit phylogenetic inference. More and more species are subjected to large-scale sequencing, but the resultant resources are scattered in independent project-based, and multi-species, but separate, web sites. This complicates data access and is becoming a serious barrier to the comprehensiveness of molecular phylogenetic analysis. aLeaves, launched to overcome this difficulty, collects sequences similar to an input query sequence from various data sources. The collected sequences can be passed on to the MAFFT sequence alignment server (http://mafft.cbrc.jp/alignment/server/), which has been significantly improved in interactivity. This update enables to switch between (i) sequence selection using the Archaeopteryx tree viewer, (ii) multiple sequence alignment and (iii) tree inference. This can be performed as a loop until one reaches a sensible data set, which minimizes redundancy for better visibility and handling in phylogenetic inference while covering relevant taxa. The work flow achieved by the seamless link between aLeaves and MAFFT provides a convenient online platform to address various questions in zoology and evolutionary biology.
INTRODUCTION
In any cross-species comparison at the molecular level, identification of orthology and paralogy is the basis on which most subsequent analyses rely (1). The most reliable approach for distinguishing orthologues from paralogues is by explicit phylogenetic inference. Some databases host genome-wide sets of molecular phylogenetic trees for individual gene families (‘phylomes’) (2–4). However, those existing databases provide only phylomes for a limited number of species with genome-wide sequence resources, and cannot fully accommodate biologists’ daily demands for custom data sets, sometimes including organisms without genome-wide information or sequences identified on their own. To achieve smooth custom analyses from sequence collection to tree inference, several tools have been developed (5–8). However, they require program installation on local systems or/and elaborate database maintenance and allow no handy interface for refining data sets.
Moreover, a large amount of molecular sequence data has been produced, thanks to recent developments in sequencing technologies. In fact, the resultant resources for protein-coding gene sets are not integrated into a single archive, such as GenBank (9) managed by NCBI (10), but are scattered in separate databases including independent project-based web sites. For example, GenBank does not host genome-wide data of many species with sequenced genomes including Xenopus tropicalis, Danio rerio and Ciona intestinalis, all of which are available at Ensembl (http://www.ensembl.org/) (2). Conversely, Ensembl does not host many invertebrate species with sequenced genomes that are hosted at EnsemblGenomes (http://www.ensemblgenomes.org/), such as those of many insects including Apis melifera (honey bee). Resources for other species that are not in either NCBI or Ensembl are also deposited in project-based individual web sites, such as the site of Joint Genome Institute (http://www.jgi.doe.gov/; e.g. for Capitella teleta). The heavily scattered data deposition complicates data access and is becoming a serious barrier to large-scale sequence comparison and molecular phylogenetic analyses.
To accommodate the demand for building high-coverage phylogenetic trees by a wide range of biologists including laboratory workers with specific interest in particular molecules, we have established a new web server, aLeaves (èılí:vz; http://aleaves.cdb.riken.jp/), which provides a simple and biologist-oriented interface and easy-to-interpret output. Sequences collected by aLeaves can readily be passed on to multiple sequence alignment (MSA) and phylogenetic tree inference on the MAFFT server upgraded to enhance its function through the coordination with aLeaves. Here we introduce the seamless work flow achieved by these two web servers.
COLLECTING SEQUENCES ON THE ALEAVES SERVER
The aLeaves server accepts a protein sequence query to run a BLASTP search based on NCBI BLAST (11) over pre-compiled databases covering multiple species (Figures 1 and 2). Users can set (or unset) low complexity filtering, the threshold for search hits and the number of sequences to retrieve (Figure 1). The databases include genome project-based datasets covering ∼100 metazoan species (Table 1), as well as sequences from the widely used public database GenBank. The latter category contains biologically annotated sequences submitted by individual researchers and serves as an indispensable information source in analyses focusing on particular targets. As explained above, the taxonomic coverage of aLeaves is wider than that of Ensembl or NCBI Genome, and none of other currently available tools can explore such a comprehensive list of diverse metazoan species’ genomes online in a single search. We do not perform any gene prediction on genome assemblies, but instead adopt data sets that already exist as protein sequences. The list of databases available at aLeaves will be enriched by incorporating emerging information from more species with frequent updates. Updates of the aLeaves server will be announced in the external blog site linked from the ‘History’ page. The ‘Help’ page of aLeaves serves step-by-step tutorial of its function as well as frequently asked questions with answers for them.
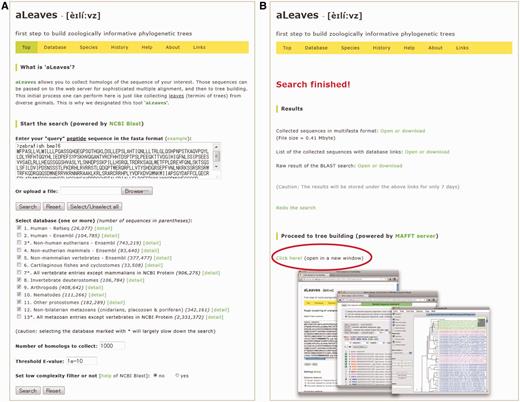
Overview of the interface of the aLeaves web server. (A) The ‘Top’ page of the aLeaves server, containing the search interface. (B) The results page shown when a search and sequence collection is completed. The ‘Proceed to tree building’ section (red oval) provides a gateway to the MAFFT server for the rest of the process from data set refinement to molecular phylogenetic tree inference.
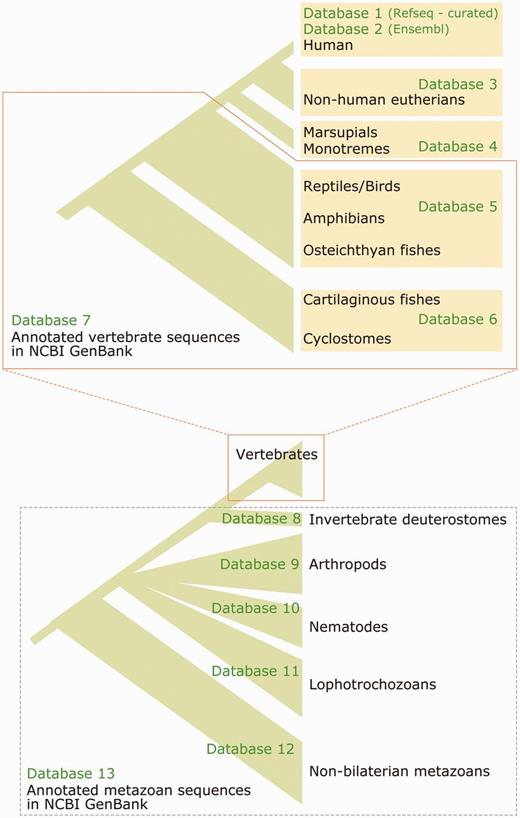
Phylogenetic coverage of the compiled databases available at the aLeaves web server. Numbering of the databases (Database 1–13) corresponds to that in the aLeaves server (http://aleaves.cdb.riken.jp/aleaves/database.html).
Sources of genome-wide protein data sets available at aLeaves but not available at Ensemble-based or NCBI-based sites
| Species name | English common name | Phylum | Database ID at aLeavesa | URL |
|---|---|---|---|---|
| Callorhinchus milii | Elephant shark (or ghost shark) | Chordata | 6 | http://people.inf.ethz.ch/cdessimo/Cmilii/ |
| Oikopleura dioica | Chordata | 8 | http://www.genoscope.cns.fr/externe/ GenomeBrowser/Oikopleura/ | |
| Branchiostoma floridae | Amphioxus | Chordata | 8 | http://genome.jgi-psf.org/Brafl1/ |
| Meloidogyne incognita | Phytoparasitic root-knot nematode | Nematoda | 10 | http://www.inra.fr/meloidogyne_incognita/ |
| Schistosoma japonicum | Parasitic flatworm | Platyhelminthes | 11 | http://lifecenter.sgst.cn/schistosoma/cn/ schistosomaCnIndexPage.do |
| Capitella teleta | Polychaete worm | Annelida | 11 | http://genome.jgi-psf.org/Capca1/ |
| Helobdella robusta | Leech | Annelida | 11 | http://genome.jgi-psf.org/Helro1/ |
| Pinctada fucata | Pearl oyster | Mollusca | 11 | http://marinegenomics.oist.jp/ |
| Crassostrea gigas | Pacific oyster | Mollusca | 11 | http://gigadb.org/pacific_oyster/ |
| Acropora digitifera | Okinawan staghorn coral | Cnidaria | 12 | http://marinegenomics.oist.jp/ |
| Species name | English common name | Phylum | Database ID at aLeavesa | URL |
|---|---|---|---|---|
| Callorhinchus milii | Elephant shark (or ghost shark) | Chordata | 6 | http://people.inf.ethz.ch/cdessimo/Cmilii/ |
| Oikopleura dioica | Chordata | 8 | http://www.genoscope.cns.fr/externe/ GenomeBrowser/Oikopleura/ | |
| Branchiostoma floridae | Amphioxus | Chordata | 8 | http://genome.jgi-psf.org/Brafl1/ |
| Meloidogyne incognita | Phytoparasitic root-knot nematode | Nematoda | 10 | http://www.inra.fr/meloidogyne_incognita/ |
| Schistosoma japonicum | Parasitic flatworm | Platyhelminthes | 11 | http://lifecenter.sgst.cn/schistosoma/cn/ schistosomaCnIndexPage.do |
| Capitella teleta | Polychaete worm | Annelida | 11 | http://genome.jgi-psf.org/Capca1/ |
| Helobdella robusta | Leech | Annelida | 11 | http://genome.jgi-psf.org/Helro1/ |
| Pinctada fucata | Pearl oyster | Mollusca | 11 | http://marinegenomics.oist.jp/ |
| Crassostrea gigas | Pacific oyster | Mollusca | 11 | http://gigadb.org/pacific_oyster/ |
| Acropora digitifera | Okinawan staghorn coral | Cnidaria | 12 | http://marinegenomics.oist.jp/ |
These species are available at aLeaves but not available at ‘Ensembl', ‘EnsemblGenomes Metazoa' or NCBI Genome (as of April 8, 2013). The complete list of species available at aLeaves is found at http://aleaves.cdb.riken.jp/aleaves/species.html.
aThe detail of the aLeaves databases is found in Figure 2 (also see http://aleaves.cdb.riken.jp/aleaves/database.html).
Sources of genome-wide protein data sets available at aLeaves but not available at Ensemble-based or NCBI-based sites
| Species name | English common name | Phylum | Database ID at aLeavesa | URL |
|---|---|---|---|---|
| Callorhinchus milii | Elephant shark (or ghost shark) | Chordata | 6 | http://people.inf.ethz.ch/cdessimo/Cmilii/ |
| Oikopleura dioica | Chordata | 8 | http://www.genoscope.cns.fr/externe/ GenomeBrowser/Oikopleura/ | |
| Branchiostoma floridae | Amphioxus | Chordata | 8 | http://genome.jgi-psf.org/Brafl1/ |
| Meloidogyne incognita | Phytoparasitic root-knot nematode | Nematoda | 10 | http://www.inra.fr/meloidogyne_incognita/ |
| Schistosoma japonicum | Parasitic flatworm | Platyhelminthes | 11 | http://lifecenter.sgst.cn/schistosoma/cn/ schistosomaCnIndexPage.do |
| Capitella teleta | Polychaete worm | Annelida | 11 | http://genome.jgi-psf.org/Capca1/ |
| Helobdella robusta | Leech | Annelida | 11 | http://genome.jgi-psf.org/Helro1/ |
| Pinctada fucata | Pearl oyster | Mollusca | 11 | http://marinegenomics.oist.jp/ |
| Crassostrea gigas | Pacific oyster | Mollusca | 11 | http://gigadb.org/pacific_oyster/ |
| Acropora digitifera | Okinawan staghorn coral | Cnidaria | 12 | http://marinegenomics.oist.jp/ |
| Species name | English common name | Phylum | Database ID at aLeavesa | URL |
|---|---|---|---|---|
| Callorhinchus milii | Elephant shark (or ghost shark) | Chordata | 6 | http://people.inf.ethz.ch/cdessimo/Cmilii/ |
| Oikopleura dioica | Chordata | 8 | http://www.genoscope.cns.fr/externe/ GenomeBrowser/Oikopleura/ | |
| Branchiostoma floridae | Amphioxus | Chordata | 8 | http://genome.jgi-psf.org/Brafl1/ |
| Meloidogyne incognita | Phytoparasitic root-knot nematode | Nematoda | 10 | http://www.inra.fr/meloidogyne_incognita/ |
| Schistosoma japonicum | Parasitic flatworm | Platyhelminthes | 11 | http://lifecenter.sgst.cn/schistosoma/cn/ schistosomaCnIndexPage.do |
| Capitella teleta | Polychaete worm | Annelida | 11 | http://genome.jgi-psf.org/Capca1/ |
| Helobdella robusta | Leech | Annelida | 11 | http://genome.jgi-psf.org/Helro1/ |
| Pinctada fucata | Pearl oyster | Mollusca | 11 | http://marinegenomics.oist.jp/ |
| Crassostrea gigas | Pacific oyster | Mollusca | 11 | http://gigadb.org/pacific_oyster/ |
| Acropora digitifera | Okinawan staghorn coral | Cnidaria | 12 | http://marinegenomics.oist.jp/ |
These species are available at aLeaves but not available at ‘Ensembl', ‘EnsemblGenomes Metazoa' or NCBI Genome (as of April 8, 2013). The complete list of species available at aLeaves is found at http://aleaves.cdb.riken.jp/aleaves/species.html.
aThe detail of the aLeaves databases is found in Figure 2 (also see http://aleaves.cdb.riken.jp/aleaves/database.html).
After the BLASTP search, the specified numbers of sequences are retrieved in form of a multi-fasta file, sorted by E-value, and downloadable from the results page. The output of collected sequences has species identifiers (‘Species ID’; for example, ‘HOMSA’ for Homo sapiens) for sequences derived from genome-wide databases and category identifiers (‘Category ID’; for example, ‘ART’ for arthropods) for sequences derived from the GenBank database (the lists of the identifiers are available at http://aleaves.cdb.riken.jp/aleaves/species.html). The results page also provides a link to the sequence clustering functions on the MAFFT server (12) where subsequent analyses are performed.
ALIGNMENT AND TREE INFERENCE ON THE MAFFT SERVER
The sequences transferred from the aLeaves server are first subjected to all-to-all pairwise comparison implemented in the MAFFT program (13,14). Several different methods, a rapid one based on the number of shared k-mers (15) and more rigorous ones based on local or global pairwise alignment scores (16–18), can be selected in this step, according to the data size. The pairwise distances are used for initial clustering by UPGMA or minimal linkage. The resulting tree is intended to roughly visualize the tree-like relationship for even large numbers of sequences and to provide visual guidance for the next steps, and is displayed with a Java applet version of the Archaeopteryx phylogenetic tree visualization and analysis tool (19,20). By using the new features described below, users can select any subset of the collected sequences. Then, an MSA of the selected sequences is calculated by MAFFT and subjected to phylogenetic tree inference with the neighbour-joining (NJ) method (21).
One highlight of the new features of the MAFFT server is the communication between Archaeopteryx and the sequence data set refinement process in the web browser for user-friendly sequence selection (Figure 3). A useful practice in evolutionary analysis is the detailed (re)analyses of the phylogenetic relationships of the sequences making up one or more subtree of a larger, previously inferred but relatively inaccurate, tree. One advantage of this approach is that such an analysis is usually more accurate because moderately divergent sequences can be aligned more precisely than larger sets of too divergent sequences—inaccurate MSAs are one of the main sources of errors in phylogenetic analysis. Furthermore, the removal of redundant uninteresting sequences allows for analysis with more accurate but more time-consuming approaches, and simplifies visual analysis. While manual selection of sequences and realignment is possible, it is labour intensive and error prone. Therefore, we modified Archaeopteryx in such a way that one can select or unselect, via its graphical user interface, individual sequences as well as complete sets of sequences included in particular subtrees (by clicking on their common root node) for reanalysis. This improvement should dramatically ease the manual process to select or unselect sequences from a preliminary data set often containing truncated, redundant or phylogenetically too distant sequences. To further facilitate visual tree analysis, species identifiers and category identifiers given by aLeaves are automatically recognized and used for species-specific colouring of sequence labels in trees visualized in Archaeopteryx (Figure 3). This function allows users to get a rough overview of the molecular phylogeny and in this initial step, delete distantly related sequences that can complicate and slow down the subsequent steps.
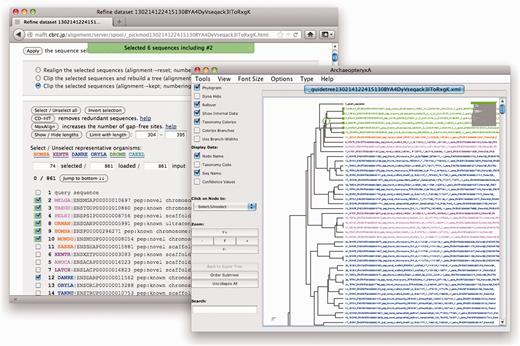
Sequence data set refinement at the MAFFT web server through Archaeopteryx. Shown as inset is a view of the Archaeopteryx applet, in which a single node containing six sequences is selected (highlighted in bright green with parent node marked by a circle). The parental web browser window shows an HTML page with a list of sequences in the present data set, in which the six sequences selected in Archaeopteryx are newly selected with ticks on the left. The colouring of the different sequences indicates their taxonomic categorization (detailed in the ‘Species’ page of the aLeaves server).
Two additional tools, CD-HIT (22) and MaxAlign (23), have been adopted in the MAFFT service to help the data refinement process. CD-HIT is used to exclude redundant sequences. MaxAlign is used to exclude short amino acid sequences that are inconvenient for phylogenetic analyses. Even genome-based databases have such sequences mainly because of incomplete sequencing or gene misidentification. Users can check the result of these automatic processes and decide whether they accept the result in the next cycle. Whenever preferable, users can also secondarily add sequences of their choice to the data set using the ‘New sequences’ input form in the MAFFT web page. Refinement of sequence data set is a seemingly simple but virtually tedious process in molecular phylogenetic analyses. Retrieval of similar sequences from various databases usually results in a bulky data set, which can obscure the target of the analysis. In contrast, if one reduces too many sequences, the resultant data set may miss a crucial subset of molecular phylogeny, causing artefacts, such as ‘hidden paralogy’ (24). The MAFFT server with the enhancement of user-controlled interactive function for sequence selection provides a solution to minimize the risk of handling data sets that tend to be unnecessarily huge initially.
The refined sequence set can then be aligned, and the aligned sequences can be subjected to molecular phylogenetic tree inference with the NJ method (21). For distance calculation in this step, several measures can be selected: the Poisson correction (25), maximum-likelihood (ML) estimation with the JTT model (26) and ML estimation with the WAG model (27). We modified the MOLPHY package (28) to consider rate heterogeneity across sites with the discrete Γ model (29) and use it in the ML distance estimation. Bootstrap analysis (30,31) is also available. To show a relationship among the selected sequences, at present, this service supports the NJ method, but does not support the ML method because of too much load on the server. After checking the NJ tree, users can download the MSA in the multi-fasta format and use it for tree inference with the ML or other methods outside the server. The sequences are also downloadable to apply different MSA methods other than MAFFT.
At any step in the process, one can go back to the data set refinement page with a single click and delete sequences through the Archaeopteryx applet. While a user switches between these different steps, previous versions of the data sets are stored and shown in the ‘History’ section of the HTML page, and a particular version of interest can be easily restored on demand.
EXAMPLES
Insect wingless
Wingless (Wg) is the invertebrate orthologue of vertebrate Wnt1 genes (32,33). Using the amino acid sequence of the Drosophila melanogaster Wg gene (AAF52501 in NCBI Protein), a search was performed at aLeaves with the default parameter settings in all the databases covering invertebrates (Database 8–13), as well as Database 1 and 5 covering vertebrates as key phylogenetic landmarks. The search resulted in a data set with 1000 sequences, which included not only vertebrate Wnt1 and invertebrate Wingless but also phylogenetically distant sequences, such as Wnt4 (http://aleaves.cdb.riken.jp/aleaves/sample/wg/). This data set was then subjected to clustering with 6-mer and the ‘UPGMA’ method, which allowed to visualize a preliminary tree in <15 s with only a few clicks. Many identical Wg sequences are redundantly deposited mainly in GenBank by individual researchers because of long-standing interest in this gene. In this particular situation, the application of CD-HIT effectively deleted more than half of the initial sequences. Further refinement of the data set was demonstrated by identifying a subtree including all arthropod Wg genes. Because some sequences with truncated N- or C-ends limited the number of gap-free sites in the alignment, they were excluded from the data set. Finally, 36 arthropod sequences, including chelicerates (spiders and tick) as outgroup, were retained in the data set harbouring 276 gap-free sites (http://mafft.cbrc.jp/alignment/server/spool/aleaves_example_wingless.html). The resultant tree therein did not indicate any gene duplication inside the arthropod lineage, and its tree topology was largely consistent with arthropod species phylogeny proposed with larger data sets (34).
Vertebrate Hox14
The second example is the molecular phylogeny of vertebrate Hox14 genes (35). Hox genes are an intensively studied group of metazoan regulatory genes (36), and above all, Hox14 marks a unique phylogenetic feature that this group of genes has not been identified in the genomes of any traditional laboratory vertebrate species (37,38). As a query, the amino acid sequence of Neoceratodus forsteri (Australian lungfish) HoxA14 gene (CBY85303 in NCBI Protein) was used, and the search at the aLeaves server was performed in the all databases (Database 1–13) with the default setting. The resultant data set contained previously documented Hox14 members and other Hox genes as well as non-Hox genes, such as Cdx (http://aleaves.cdb.riken.jp/aleaves/sample/hox14/). After a clustering of the sequences in the initial data set, a subtree containing the previously documented Hox14 members was identified in the preliminary tree shown in Archaeopteryx. No sequence other than the previously documented Hox14 members was identified inside this subtree, in the existing protein-coding gene sets available for all the genomes of the species covered by aLeaves except one species, namely coelacanth Latimeria (http://mafft.cbrc.jp/alignment/server/spool/aleaves_example_hox14.html). The tree topology and the non-identification of any Hox14 member in the genomes of most vertebrates were consistent with the results of a previous study (37). The unified platform based on genome resources of dozens of species at aLeaves facilitates the identification of homologues of users’ particular interest.
Vertebrate CTCF
CCCTC-binding factor (CTCF) is a Zinc finger containing transcription factor regulating chromatin compartments by marking particular genomic regions as insulators (39). A search at the aLeaves server was performed using the human CTCF amino acid sequence (AAB07788 in NCBI Protein) with the default parameter setting in the Database 1, 4, 5 and 7 covering major vertebrate lineages and Database 8 containing their potential orthologues of invertebrate deuterostomes as outgroup. The search resulted in 1000 sequences as requested (http://aleaves.cdb.riken.jp/aleaves/sample/ctcf/), but the data set contained many sequences that seemed phylogenetically distant from CTCF.
The entire set of the collected sequences was subjected to clustering as in the above examples. To create a data set focused on vertebrate CTCF genes, we first excluded many mammalian Zinc finger protein (ZNF) sequences apparently distantly related from CTCF, by identifying a subtree consisting almost completely exclusively of abundant mammalian sequences including ZNF484 and ZNF180. The identification of the uninteresting subtree, which was recognized as a paraphyletic group with initial rooting, was facilitated by using the ‘Root/Reroot’ function of Archaeopteryx, and this operation deleted >650 sequences out of the initial 1000. To further reduce the number of sequences in the data set, CD-HIT was applied to retain 285 sequences. Clustering was performed again with ‘accurate (global)’ distance calculation and the ‘minimum linkage’ method to more carefully assess the relationships among the sequences in the data set. The resultant tree in Archaeopteryx allowed the identification of the monophyletic group consisting of vertebrate CTCF genes and their close relative, CTCF-like (or BORIS), rooted by their immediate outgroup (invertebrate deuterostomes). The data set of this subtree containing 58 sequences was subjected to MSA, but the resulting MSA did not contain any gap-free sites. As applying MaxAlign turned out to delete the invertebrate outgroup, which is necessary for rooting, we thus decided not to rely on the automated process but to exclude individual sequences in the data set that are reducing gap-free sites with careful inspection. As demonstrated here, depending on the nature of data sets and aims, users can choose more sensible strategies among diverse implemented functions to refine data sets.
For this example, the 30 sequences remaining after the final careful refinement were aligned again, and using the 405 gap-free sites, an NJ tree was inferred (http://mafft.cbrc.jp/alignment/server/spool/aleaves_example_ctcf.html). The resultant tree topology was largely consistent with those in previous studies (40) and Ensembl Tree view (http://www.ensembl.org/Homo_sapiens/Gene/Compara_Tree?g=ENSG00000102974), and the data set derived from aLeaves covered some additional species. On demand, some of the sequences can further be deleted, and conversely, additional sequences determined by one’s own can easily be integrated into the MSA for more customized tree based on amino acid sites of the user’s choice. The major difference between these results was the phylogenetic timing of the gene duplication between CTCF and CTCF-like (BORIS). This question can be further scrutinized by downloading the data set file and applying other phylogenetic tree inference methods on it.
CONCLUSION
The two web servers, aLeaves and MAFFT, provide a unique online platform to explore metazoan gene family trees on demand. It is expected to handle demands on two extreme ends—automated processing of a large sequence dataset from diverse genomes and highly interactive analysis with manual inspections in molecular phylogenetics—that are impossible to reconcile with other existing tools. The framework of the aLeaves server is planned to expand in the future, which includes addition of more organisms inside as well as outside Metazoa.
FUNDING
Center for Developmental Biology (CDB), RIKEN (to S.K.); Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to K.K.); National Institutes of Health NIMG [R01GM101457 to C.M.Z.]. Funding for open access charge: Center for Developmental Biology, RIKEN, Japan (the first author’s affiliation).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Naoyuki Iwabe, Yuichiro Hara, Nathalie Feiner and Miyuki Noro for testing earlier versions of aLeaves and valuable comments. The authors also thank Hiroshi Suga, Kei-ichi Kuma and Go Sasaki for providing a part of the tree inference programs.

{kind=link}
{kind=link}
{kind=link}
Comments