





Abstract
Human ALT cancers show high mutation rates in ATRX and DAXX. Although it is well known that the absence of ATRX/DAXX disrupts H3.3 deposition at heterochromatin, its impact on H3.3 deposition and post-translational modification in the global genome remains unclear. Here, we explore the dynamics of phosphorylated H3.3 serine 31 (H3.3S31ph) in human ALT cancer cells. While H3.3S31ph is found only at pericentric satellite DNA repeats during mitosis in most somatic human cells, a high level of H3.3S31ph is detected on the entire chromosome in ALT cells, attributable to an elevated CHK1 activity in these cells. Drug inhibition of CHK1 activity during mitosis and expression of mutant H3.3S31A in these ALT cells result in a decrease in H3.3S31ph levels accompanied with increased levels of phosphorylated H2AX serine 139 on chromosome arms and at the telomeres. Furthermore, the inhibition of CHK1 activity in these cells also reduces cell viability. Our findings suggest a novel role of CHK1 as an H3.3S31 kinase, and that CHK1-mediated H3.3S31ph plays an important role in the maintenance of chromatin integrity and cell survival in ALT cancer cells.
INTRODUCTION
Telomeres are specialized DNA structures that protect chromosome ends from degradation and illegitimate recombination (1,2). In human cells, telomeric DNA is shortened with every cell division due to end replication problems, limiting their proliferative potential. For this reason, the long-term proliferation of tumors requires continual maintenance of telomere length. To achieve this, the majority of human cancers re-express the telomerase enzyme. However, a subset of human cancers utilizes a DNA recombination-mediated mechanism known as Alternative Lengthening of Telomeres (ALT) (3–5). Telomerase-null ALT cancer cells generally contain extensive genomic instability, as indicated by severe chromosomal fragmentation, frequent micronucleation, a high basal level of DNA damage foci and elevated DNA damage response (DDR) signaling in the absence of exogenous damage (6,7).
Recently, it has been shown that the Alpha Thalassemia Mental Retardation X-linked (ATRX) gene is inactivated in 90% of in vitro immortalized ALT cell lines (6), while loss of wild-type ATRX expression in somatic cell hybrids correlates with the activation of ALT mechanism (8). Furthermore, mutations in ATRX have been detected in many ALT tumors, including pancreatic neuroendocrine tumors, neuroblastomas and medulloblastomas (9–12), suggesting that ATRX acts as a suppressor of the ALT pathway. ATRX associates with Death-associated protein 6 (DAXX) to function as a histone chaperone complex that deposits histone variant H3.3 in heterochromatin, including telomeres and pericentric satellite DNA repeats (13–20). The binding of ATRX at the pericentric heterochromatin depends on the interaction of the ATRX ADD (ATRX-DNMT3-DNMT3L) domain with the H3 N-terminal tail that is trimethylated on lysine 9 and unmethylated on lysine 4 (21,22). ATRX is required for maintaining transcription repression (17,19). Recent studies also suggest that it is important for the resolution of stalled replication forks and re-chromatinization of repaired DNA (23–28). Consistent with this, ATRX-deficient ALT cells show highly elevated DDR signaling, evidenced by high levels of phosphorylated histone variant H2AX on Ser139 (γH2AX), a DNA damage marker and activation of the DNA damage proteins ATM and CHK2 (6,26,27).
The deposition of histone variants by specific chaperones together with associated histone post-translational modifications (PTMs) can significantly impact chromatin structure and function. Although it is clear that loss of ATRX function results in a failure to deposit H3.3 in heterochromatin (6,8,9,12), whether this leads to further aberrant H3.3 loading and/or PTMs in other genomic regions is unknown. To investigate this, we examined the dynamics of H3.3 Serine 31 phosphorylation (H3.3S31ph) in ATRX-deficient ALT cancer cells. Serine 31 is unique to H3.3 (canonical H3.1 and H3.2 have an alanine in the corresponding position) and is highly conserved in H3.3. In mammalian cells, H3.3S31ph occurs during mitosis and is a chromatin mark associated with heterochromatin (29). In somatic cells, H3.3S31ph is enriched at pericentric satellite DNA repeats of metaphase chromosomes, with no enrichment on chromosome arms (29), while in pluripotent mouse embryonic stem (ES) cells, it localizes at telomeres (14). Unlike the phosphorylation of the two Serine residues 10 and 28 on canonical H3, the protein kinase mediating H3.3S31 phosphorylation has not been identified to date.
In this study, we report an extremely high level and extensive spreading of H3.3S31ph across the entire chromosome during mitosis in the human ALT cancer cell lines—in sharp contrast to the previously reported pericentric and telomeric localization of H3.3S31ph (14,29). This aberrant pattern of H3.3S31ph is driven by a high level of activated CHK1 serine/threonine kinase. As CHK1 is activated by persistent DNA damage and genome instability, our findings link H3.3S31ph to the DDR pathway. In the human ALT cell lines, drug inhibition of CHK1 activity during mitosis and expression of mutant H3.3S31A not only reduces H3.3S31ph level on the chromosomes but also leads to increases in γH2AX levels on the chromosome arms and at the telomeres. The inhibition of CHK1 activity also affects cell viability. Our data suggests a role for CHK1-mediated H3.3S31ph in chromatin maintenance and cell survival in ALT cancer cells.
Although previous studies have identified CHK1 as a histone kinase phosphorylating H3S10 and T11 (30,31), the biological significance of CHK1-associated histone phosphorylation remains largely unknown. Our findings that up-regulated CHK1 activity accounts for the strong H3.3S31 phosphorylation in ALT cancer cells, and that this H3.3S31ph mark is important for the maintenance of chromatin integrity and cell survival of these cells, altogether provide an insight into the regulatory role of CHK1 in chromatin metabolism. We propose that CHK1-mediated H3.3S31ph may serve as a prominent hallmark for ALT cancer cells and that CHK1 signaling, in particular its role in mediating H3.3S31 phosphorylation, represents a promising new therapeutic target for treatment of ALT cancers.
MATERIALS AND METHODS
Cell cultures
Human telomerase-positive cell lines HT1080 (fibrosarcoma), A549 (lung adenocarcinoma), HT29 (colorectal cancer), HeLa (cervical cancer) and ALT-positive cell lines Saos-2 (osteosarcoma), U2OS (osteosarcoma), G292 (osteosarcoma), KMST6 (immortalized fibroblast), SUSM1 (immortalized fibroblast), SKLU1 (lung adenocarcinoma), GM847 (SV40-transformed fibroblasts) and W138-VA13/2RA (SV40-transformed fibroblasts) were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 10% FCS+ 1% Penicillin/Streptomycin. ESmar10 mouse ES cells (32) were cultured in DMEM with 12% heat-inactivated FCS (v/v) and 1000 units/ml leukemic inhibitory factor, 0.1 mM β-mercaptoethanol and 100 μg/ml Zeocin (33–35).
Antibodies and inhibitors
Primary antibodies used were rabbit antibodies against ATRX (sc-15408, Santa Cruz), H3.3 (09–838, Millipore Merck), phosphorylated H3.3S31 (ab2889, Abcam; Active Motif, 39637; H3.3S31ph) (Abcam; Active Motif), CHK1 Ser317 (12302, Cell Signaling Technology), CHK2 Thr68 (abe343, Cell Signaling Technology), CHK1 (NBP1–19210, Novus Biologicals) and CHK2 (05–649, Millipore Merck); mouse monoclonal antibodies against γH2AX Ser139 (05–636, Millipore Merck), and myc tag (05–724, Millipore Merck), tubulin (Roche), and human anti centromere CREST serum (34). CHK1 inhibitor UCN-01 (Millipore Merck) and SB218078 (Millipore Merck) were used at a concentration of 1 μM and 5 μM, respectively (36). CHK2 inhibitor II hydrate (Sigma Aldrich) was used at a final concentration of 250 μM.
Cell-cycle analysis
Cells were arrested with 9 μM of RO-3306 (Sigma-Aldrich) for 17 h prior to release from the G2/M block by washing three times in phosphate buffered saline (PBS). For FACS analysis, cells were harvested and washed in ice-cold PBS and blocked in 1% BSA (w/v) before fixing in absolute ethanol overnight. Cells were then stained with propidium iodide and analyzed on a FACS Calibur Analyzer (BD Biosciences) with ModFit software (Verity).
siRNA depletion of ATRX and CHK1
siRNA oligonucleotides specific against ATRX and CHK1 were transfected using the Lipofectamine 3000 (Invitrogen). As controls, scramble siRNA oligonucleotides (Invitrogen) were included in the experiments.
Chk1 siRNA oligonucleotide
5′ CCCUCAUACAUUGAUAAAUTT 3′ and 5′ AUUUAUCAAUGUAUGAGGGTT 3′
ATRX siRNA oligonucleotide set #1
5′ GGA AGU UCC ACA AGA UAA AGA 3′ and 5′ UUU AUC UUG UGG AAC UUC CUG 3′
ATRX siRNA oligonucleotide set #2
5′ GAC AGA AGA UAA AGA UAA ACC 3′ and 5′ UUU AUC UUU AUC UUC UGU CUU 3′
Real-time PCR analysis
RNA was prepared according to the manufacturer's protocol (Roche) after 48 h of transfection and treated with DNAse using the Promega RQ1 reagent (Promega). cDNA was synthesized using the cDNA Reverse Transcriptase kit (Life Technologies). The level of CHK1 mRNA was quantitated with the FastStart DNA Green Sybr using the LightCycler (Roche). As an internal control, primers specific for Actin was used in real-time PCR analysis. Fold changes were calculated according to the manufacturer's instructions.
The DNA oligonucleotides used in real-time PCR analysis include:
CHK1 set#1: 5′ GCC TGA AAG AGA CTT GTG AGA AGT TGG G 3′ and 5′ TCC ATC ACC CTT AGA AAG CCG GAA 3′
CHK1 set #2: 5′ GTG AGA AGT TGG GCT ATC AAT GG 3′ and 5′ GAA CTC CAA TCC ATC ACC CTT AG 3′
Actin: 5′ GGC ATC CTC ACC CTG AAG TA 3′ and 5′ GGG GTG TTG AAG GTC TCA AA 3′
Over-expression of wild-type mycH3.3 and mutant H3.3S31 DNA constructs
The construction of wild-type mycH3.3 DNA plasmid in pcDNA4/TO/myc HisA DNA vector backbone has previously been described (14). mycH3.3S31A fragment was synthesized from the IDT gBlock Gene Fragments, followed by subsequent cloning into the pcDNA4/TO/myc HisA vector (Life Technologies). Transfection reaction was set up using the Lipofectamine 3000 system (Invitrogen) according to the manufacturer's instruction. Cells were transfected a second time 48 h after the first round of transfection and harvested for analysis 48 h after the second transfection.
Cell extracts and western blot analysis
Cells were lysed in cold RIPA buffer (150 mM NaCl, 50 mM Tris-HCl at pH 7.5, 0.25% sodium deoxycholate, 0.1% NP40, 1 mM NaF, 1 mM sodium orthovanadate and protease inhibitor) with Benzonase (Sigma), followed by 10 s pulse sonication. The lysate was collected after 10 min centrifugation at 12 000 rpm and boiled in sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer prior to SDS-PAGE and western blotting. Intensities of protein bands were quantitated using the Image J Software. Relative protein levels are expressed as a ratio relative to the tubulin loading control.
Immunofluorescence analysis
Cells were treated with Colcemid (Life Technologies) or Nocodazole (Sigma) at a final concentration of 0.1 μg/ml and 40 ng/ml, respectively, for 30 to 45 min before being harvested for immunofluorescence analysis (33–35,37). Cells were subjected to hypotonic treatment in 0.075 M KCl, cytospun on slides and incubated in KCM buffer (120 mM KCl, 20 mM NaCl, 10 mM Tris-HCl pH7.2, 0.5 mM EDTA, 0.1% (v/v) Triton X-100 and protease inhibitor). Slides were blocked in KCM buffer containing 1% BSA (w/v), incubated with relevant primary and secondary antibodies at 37°C for 1 h. After each round of antibody incubation, slides were washed three times in KCM buffer. Slides were then fixed in KCM with 4% (v/v) formaldehyde and mounted with Vectashield (Vector Lab) supplemented with DAPI at 250 ng/ml. Images were collected using a Zeiss Imager M2 fluorescence microscope linked to an AxioCam MRm CCD camera system. Microscopy analyses were processed using the Zen software 2011 (Carl Zeiss Microscopy).
Immunoprecipitation and in vitro kinase assay
For immunoprecipitation, cells were lysed in ice-cold lysis buffer (50 mM HEPES pH8.0, 600 mM KCl, 0.5% NP-40, 0.1 mM DTT, complete protease inhibitor, 25 mM β-glycerophosphate, 50 mM NaF, 1 mM Na2VO4) for 5 min, followed by brief sonication. The lysates were pre-cleared with Protein A Agarose beads prior to incubation with antibodies against CHK1 or CHK1S317ph and Protein A Agarose beads overnight at 4°C with constant agitation. Beads were washed three times in ice-cold lysis buffer and twice in in vitro kinase assay buffer (60 mM HEPES pH7.4, 30 mM MgCl2, 30 mM MnCl2, 12 mM DTT, 5% glycerol, protease inhibitor cocktail, 0.2 mM ATP). The immunoprecipitated CHK1 was then incubated with H3.3 recombinant protein in kinase buffer, with or without the presence of CHK1 inhibitor SB218078 at 37°C for 1 h. The reaction was terminated by the addition of 2X SDS-PAGE sample buffer. The samples were then subjected to SDS-PAGE and western blot analysis. For in vitro kinase reaction of recombinant GST-tagged CHK1 protein (Sapphire Biosciences), GST-CHK1 protein was incubated with recombinant H3.1 or H3.3 protein in the presence of Magnesium/ATP cocktail (Millipore Merck) for 1 h at 37°C. The samples were then subjected to SDS-PAGE and western blot analysis.
RESULTS
ALT cancer cells show intense and aberrant H3.3S31ph staining on chromosome arms at mitosis
Considering the importance of ATRX/DAXX in H3.3 deposition and maintenance of heterochromatin at telomeres, it is not surprising that ALT cancer cells show severe telomere chromatin dysfunction. However, little is known on how ALT pathway activation impacts H3.3 distribution. We hypothesized that the loss of ATRX could impact on H3.3 deposition and PTM profile in ALT cells. To investigate this, we analyzed H3.3S31ph distribution in ALT cancer cell lines, as H3.3S31ph has been shown to serve as a reliable marker for heterochromatin in mammalian cells (14,29). Immunofluorescence analysis was performed using an antibody specific for H3.3S31ph (Figure 1 and Supplementary Figure S1) (14). As expected, telomerase-positive mouse ES cells show H3.3S31ph enrichment at telomeres of metaphase chromosomes (Figure 1A) (13,14), whereas human telomerase-positive A549 (Figure 1B), HT29 cells and HT1080 cells (Supplementary Figure S2; quantitation of H3.3S31ph intensity is shown in Supplementary Figure S3) display H3.3S31ph staining at pericentric satellite DNA repeats. In contrast, ALT cancer cell lines including U2OS, W138-VA13/2RA and GM847 show strong H3.3S31ph staining—not only on pericentric DNA repeats but also along the entire chromosome arms (Figure 1C–F). Co-incubation with an H3.3S31ph blocking peptide removes H3.3S31ph signals on the chromosomes, suggesting that the antibody specifically targets H3.3S31ph (Supplementary Figure S1). Immunofluorescence images of GM847 cells were also taken with a lower exposure time period to show that the level of H3.3S31ph at pericentric DNA repeats are higher than those on chromosome arms (Figure 1F; this differential enrichment in H3.3S31ph staining is detectable by reducing the H3.3S31ph image acquisition time by 4-fold). There is no further enrichment of H3.3S31ph at the telomeres, when compared to the staining intensities on the chromosome arms.
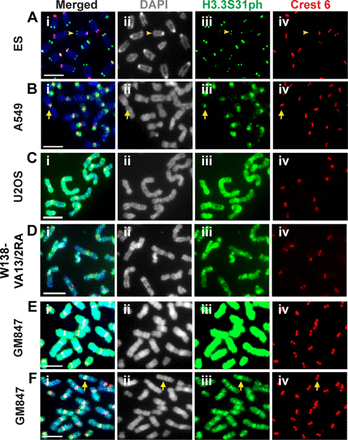
Localization of H3.3S31ph on the chromosome arms in human ALT cancer cell lines. Immunofluorescence analysis was performed using antibodies against H3.3S31ph (green) and centromeres (human Crest antibody; red). (A) H3.3S31ph localizes at the telomeres in mouse ES cells (arrowhead). (B) In telomerase-positive A549 cells, H3.3S31ph is enriched at the pericentric satellite DNA repeats (arrow). No significant enrichment is observed on the chromosome arms. (C–D) In ALT-positive U2OS and W138-VA13/2RA cancer cells, H3.3S31ph is found at extremely high levels at the pericentric DNA repeats and across the chromosome arms. No clear enrichment of H3.3S31ph was found at the telomeres when compared to signal on the chromosome arms. (E–F) In GM847 ALT cancer cells, a high level of H3.3S31ph is also detected on the pericentric DNA repeats and chromosome arms. A lower exposure (F) indicates a slightly higher intensity of H3.3S31ph at the pericentric DNA repeats (arrow). Representative images of 50 chromosome spreads are shown. Scale bar = 5 μm.
To our knowledge, high levels and aberrant distribution of H3.3S31ph on chromosome arms have not been previously described. To explore if it serves as a hallmark of cancer cells that utilize the ALT pathway, we extended our analyses to other ALT cell lines and determined whether aberrant H3.3S31ph staining correlated with loss of ATRX expression (Supplementary Figure S2). Among the ALT cell lines tested, all show high levels and spreading of H3.3S31ph on chromosome arms except for SKLU1 cells. It is interesting that SKLU1 is the only ALT line with a normal expression and cellular distribution of ATRX (6). Although a positive ATRX protein band is also detected in G292 (Supplementary Figure S4A), these cells have been shown in a previous study to have an abnormal immunostaining pattern of ATRX (6). Consistently, our immunofluorescence analysis also shows an abnormal ATRX distribution pattern (data not shown), indicating that the function of ATRX is likely to be defective in these cells.
To test if the aberrant distribution of H3.3S31ph is associated with a loss of ATRX function, siRNA depletion of ATRX was performed in A549 and SKLU1 cells (Supplementary Figure S4B). After 48 h of transfection, we observe an increased spreading of H3.3S31ph on chromosome arms in both cell lines (Supplementary Figure S4C), however, the level of H3.3S31ph staining on the chromosomes is less intense than the staining patterns in other ATRX-null ALT cancer cell lines (Figure 1 and Supplementary Figure S2). Although loss of ATRX function can induce H3.3S31ph, it is unlikely the sole factor that drives the abnormally high H3.3S31ph signals in ALT cells. We have also examined the overall level of H3.3 protein in these ALT cell lines in both asynchronous and mitotic enriched cell population. There is no significant increase in H3.3 protein levels in these ALT cells, when compared to that in A549 cells (Supplementary Figure S5). This indicates that the abundance of S31 phosphorylation on chromosome arms in the ALT cells is unlikely caused by a difference in H3.3 expression levels. Together, our data show that H3.3S31ph is up-regulated and re-distributed in ALT cancer cells. This intense and aberrant distribution pattern of H3.3S31ph is not caused by a change in H3.3 protein level and can in part be induced by a loss of ATRX expression (Supplementary Figure S4).
A delay in mitotic progression contributes to the elevated and extensive H3.3S31ph staining on metaphase chromosome arms in ALT cancer cells
Given that H3.3S31 phosphorylation occurs during mitosis (29), it is possible that the aberrant H3.3S31ph pattern could be a result of prolonged mitotic progression in ALT cancer cells. Indeed, persistent DNA damage and genomic instability in ALT cells (6,7) evidenced by fragmented chromosomes (Supplementary Figure S6) could negatively impact mitotic progression. This would lengthen the time available for a putative H3.3 kinase to promote phosphorylation of H3.3S31. In asynchronous ALT cancer cell populations, we found by FACS analysis the percentage of G2/M cells (mitotic index) in telomerase-positive A549 cells to be 15%, whereas mitotic indices in U2OS and GM847 ALT cells are as high as 24% (Figure 2A). To examine the kinetics of mitotic progression of ALT cells, we blocked A549, U2OS and GM847 cells at G2 by 17 h treatment with RO-3306 (a CDK1 inhibitor widely used to synchronize cells in G2/M phase (38)), followed by a release into mitosis and G1 phase. Upon release from G2/M arrest, the majority of A549 cells progress through mitosis into G1 within 2 to 3 h, whereas U2OS and GM847 only exit mitosis 5 h after release from G2/M (Figure 2B). These data show that ALT cell lines exhibit a marked delay in mitotic progression.
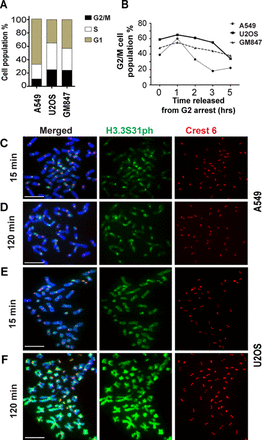
Association of prolonged mitotic progression with the aberrant H3.3S31ph staining pattern in ALT cancer cell lines. (A) FACS analysis was performed to determine the cell-cycle profiles of A549, U2OS and GM847. The ALT cancer cell lines U2OS and GM847 showed higher percentages of G2/M cell population when compared to telomerase-positive A549 cells (24% versus 15%). (B) Human A549, U2OS and GM847 cells were arrested at G2/M boundary by treatment with RO-3306. FACS analysis was performed on these cells following their release into mitosis and G1. In A549, most cells exited mitosis by 3 h post release into mitosis. In contrast, U2OS and GM847 only exited mitosis 5 h post-release; at 3 h post-release, 45–55% of cells still remained at mitosis. Representative graphs of two independent experiments are shown. (C–F) Immunofluorescence analysis was performed on A549 and U2OS cells released from G2 arrest and subjected to subsequent Colcemid treatment (either 15 min or 2 h treatment). In A549 cells (C–D) subjected to Colcemid mitotic arrest, H3.3S31ph was enriched at pericentric DNA repeats, with no staining on chromosome arms. An additional 2 h mitotic arrest did not lead to significant increase in H3.3S31ph staining. In contrast, U2OS (F) ALT cancer cells showed high levels of H3.3S31ph staining at both the pericentric DNA repeats and chromosome arms, even with only 15 min of Colcemid treatment. Representative images of 50 chromosome spreads are shown. Scale bar = 10 μm.
Next, we examined whether this prolonged progression through mitosis contributed to the aberrant H3.3S31ph staining in ALT cells. If so, one would expect a gradual spreading of H3.3S31ph along the chromosome arms in ALT cells. To test this, cells were synchronized at the G2/M boundary with RO-3306, released and blocked at mitosis for different time periods with Colcemid. In A549 cells, the majority of the mitotic chromosome spreads show specific staining of H3.3S31ph at pericentric satellite DNA repeats 15 min post-G2/M release (Figure 2C). After 2 h of Colcemid treatment, no significant increase in H3.3S31ph on pericentric DNA repeat regions or chromosome arms of A549 cells is detected (Figure 2D). In contrast, U2OS ALT cells show intense H3.3S31ph staining on both pericentric DNA repeats and chromosome arms as early as 15 min after being released from G2/M arrest (Figure 2E). Moreover, after 2 h in Colcemid, we note a further increase in H3.3S31ph staining intensity on chromosome arms and in the proportion of mitotic spreads (increased by 25%) with H3.3S31ph staining on chromosome arms (Figure 2F). These data suggest that a prolonged mitotic period may contribute to the increased intensity of H3.3S31ph on chromosome arms in these ALT cell lines; however, it is unlikely to be the primary cause for aberrant H3.3S31ph distribution in these cells. Consistent with this line of argumentation is the absence of high level of H3.3S31ph on chromosome arms in A549 cells even after 2 h of mitotic arrest (Figure 2D). Furthermore, the extensive H3.3S31ph signals observed on the arm regions in ALT cells even during early mitosis indicate that ALT cell mitotic chromosomes are subjected to rapid phosphorylation at H3.3S31 as soon as they enter mitosis. Thus, it is likely that other factor(s) contribute to the aberrant H3.3S31 phosphorylation pattern in these ALT cancer cells.
Up-regulated CHK1 kinase activity drives H3.3S31 phosphorylation in ALT cancer cells
A possible hypothesis is that an altered protein kinase activity could account for the aberrant H3.3S31ph level in ALT cancer cells. Unlike other canonical H3 residues, the kinase that promotes the phosphorylation of H3.3S31 remains unknown. The Checkpoint protein 1 (CHK1) is a serine/threonine kinase involved in DDR signaling. It has previously been shown to phosphorylate Serine10 and Threonine 11 on canonical H3 histone (30,31). Considering the high levels of DNA damage and the elevated DDR signaling in ALT cancer cells (6), CHK1 could be a candidate that drives the aberrant H3.3S31 phosphorylation pattern in ALT cancer cells. As predicted, we detected a higher level of activated CHK1 in ALT cells even in the absence of an external DNA damaging insult, as indicated by western blot analysis using an antibody against CHK1 Ser317 phosphorylation (Figure 3A). This result is in agreement with increased CHK2 Thr68 phosphorylation (a mark of DDR signaling) in ALT cells (Figure 3A; see also (6)). It is important to note that although ALT cells show significantly elevated CHK1 and CHK2 activation, likely as a response to persistent DNA damage and genomic instability (6)), these cell lines did not show elevated levels of total CHK1 and CHK2 compared to A549 cells (Figure 3B).
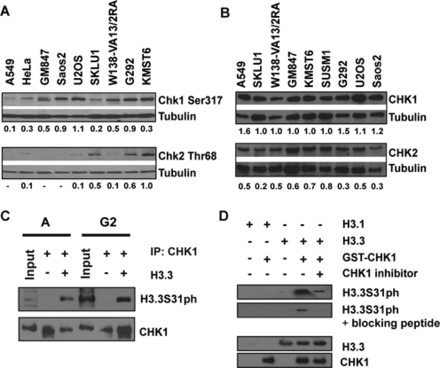
Phosphorylation of H3.3S31 in ALT cancer cell lines is mediated by CHK1 kinase. (A) Cell lysates were prepared from asynchronous populations from telomerase-positive and ALT-positive cell lines. In contrast to telomerase-positive HeLa and A549, the ALT cell lines GM847, Saos-2, U2OS, W138-VA13/2RA, G292 and KMST6 showed elevated levels of CHK1 Ser317 phosphorylation (CHK1 Ser317). A low level of CHK1 Ser317 is also detected in SKLU1 cells. In some of these ALT cell lines, CHK2 Thr68 phosphorylation (CHK2 Thr68) levels were also high (6). Tubulin protein levels were used as loading controls. Relative protein levels are shown as ratios of CHK1Ser317 and CHK2Thr68 against the tubulin loading controls. (B) ALT cancer cell lines did not show significantly difference in CHK1 and CHK2 levels, when compared to HeLa and A549 (6). Tubulin protein levels were used as loading controls. Relative protein levels are shown as ratios of CHK1 and CHK2 against the tubulin loading controls. (C) CHK1 protein was immunoprecipitated from either asynchronous or G2-enriched U2OS cells. The immunoprecipitated CHK1 was used in an in vitro kinase assay with recombinant H3.3 protein, followed by western blot analysis with an antibody against H3.3S31ph. A strong band corresponding to H3.3S31ph was detected when CHK1 was co-incubated with recombinant H3.3 protein. (D)In vitro kinase reaction was also performed using recombinant GST-tagged CHK1 and H3.1 and H3.3 proteins, followed by western blot analysis with anti-H3.3S31ph antibody. Lane 1: recombinant H3.1 protein alone; lane 2: 1 μg recombinant H3.1 with 0.5 μg GST-tagged CHK1; lane 3: recombinant H3.3 protein alone; lane 4: 1 μg recombinant H3.3 with 0.5 μg GST-tagged CHK1; lane 5: 1 μg recombinant H3.3 with 0.5 μg GST-tagged CHK1 and 1 μM CHK1 inhibitor SB218078. Western blot analyses were performed using antibodies against H3.3S31ph (with and without blocking peptide), CHK1 and H3.3, respectively.
To determine the involvement of CHK1 in the phosphorylation of H3.3S31, endogenous CHK1 protein was immunoprecipitated from both asynchronous and mitotic U2OS cell populations for in vitro kinase assays. The immunoprecipitated CHK1 was incubated with recombinant H3.3 under phosphorylation conditions, and H3.3S31ph was examined by western blotting using an antibody against H3.3S31ph. The kinase reaction yielded a clear H3.3Ser31ph product, indicating that CHK1 activity can phosphorylate H3.3S31 in vitro (Figure 3C). The in vitro kinase assay was also performed using recombinant GST-tagged CHK1 protein and a strong 17 kDa H3.3S31ph product was detected by western blot analysis. The intensity of the H3.3S31ph product was reduced when the immunoblot was co-incubated with an H3.3S31ph blocking peptide (Figure 3D), thus demonstrating the specificity of the antibody against phosphorylated H3.3S31. In addition, this 17 kDa product was also not observed when H3.1 recombinant protein was used in the assay or when the CHK1 inhibitor SB218078 was added to the in vitro kinase reaction. Together, these data show that CHK1 can phosphorylate H3.3S31 in human ALT cancer cell lines.
In addition to the in vitro kinase assays, we examined in cells the ability of CHK1 to phosphorylate H3.3S31. Western blot analysis was performed to assess the effect of the CHK1 inhibitor SB218078 on the level of H3.3S31ph. All cell lines examined showed an increase in H3.3S31ph after treatment with Nocodazole, a reversible microtubule toxin used to arrest cells in mitosis (Figure 4A). We find that the CHK1 inhibitor SB218078 leads to a reduction in H3.3S31ph in ALT cell lines including SKLU1, U2OS, Saos-2 and KMST6, but not in non-ALT, telomerase-positive A549 cells (Figure 4 and Supplementary Figure S7). Treatment of these ALT cells with a CHK2 inhibitor did not cause a change in H3.3S31ph level (Figure 4A), indicating that CHK2 is unlikely to be involved in driving H3.3S31 phosphorylation in ALT cells. Our results argue, therefore, for an in vivo role of CHK1 in phosphorylating H3.3S31 in ALT cells. To further investigate this, immunofluorescence analysis was performed on CHK1 inhibitor treated cells. Unlike A549 cells, CHK1 inhibition in U2OS ALT cells caused a significant reduction in H3.3S31ph staining on chromosome arms (Figure 4B), however, the presence of H3.3S31ph remains prominent at pericentric satellite DNA repeats. Similar data were also obtained when SUSM1 and KMST6 ALT cell lines were subjected to treatment with CHK1 inhibitor (Supplementary Figure S7). These data reveal a novel role of CHK1 as an H3.3S31 kinase and that in ALT cancer cells, the up-regulated CHK1 kinase activity not only increases the level of H3.3S31ph but also accounts for the aberrant distribution of H3.3S31ph across the entire chromosome.
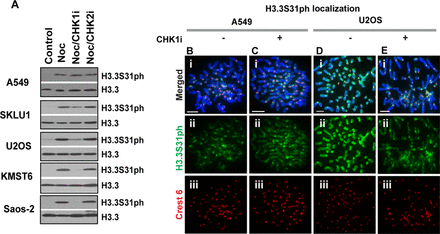
CHK1 inhibition reduces phosphorylation of H3.3S31 in ALT cancer cell lines. (A) Western blot analysis performed on cells arrested at mitosis for 3 h by Nocodazole treatment, either in the presence or absence of CHK1 inhibitor. In all lines, an increase in H3.3S31ph level was detected following Nocodazole treatment. Notably, when cells were treated with Nocodazole in the presence of 2.5 μM CHK1 inhibitor SB218078, there was a reduction in H3.3S31ph level in all ALT cancer cells. Incubation with 250 μM CHK2 inhibitor C3742 did not affect H3.3S31ph levels. Lane 1: lysates prepared from asynchronous population; Lane 2: lysates from mitotic cells; Lane 3: CHK1 inhibitor was co-incubated with mitotic cells; Lane 4: CHK2 inhibitor was co-incubated with mitotic cells. Similar observations were made with another CHK1 inhibitor UCN-01 at 1 μM (data not shown). (B–E) A549 (B–C) and U2OS cells (D–E) were arrested at G2 boundary using RO-3306. Cells were released either in the presence or absence of CHK1 inhibitor SB218078 for 20 min, followed by Colcemid treatment for 15 min. Immunofluorescence analysis was performed on these G2 released cells. Treatment with 2.5 μM CHK1 inhibitor SB218078 resulted in a significant reduction in the level of H3.3S31ph (green) in U2OS ALT cancer cells, but not in telomerase-positive A549 cells. Centromeres were stained with human CREST antiserum (red). Scale bar = 5 μm.
Besides the use of CHK1 inhibitor, siRNA depletion of CHK1 was also performed to examine the effect of CHK1 loss on H3.3S31ph. However, we found that the loss of CHK1 perturbed the cell-cycle distribution profile in both telomerase-positive and ALT cells (Supplementary Figure S8). This aligns with the role CHK1 plays in regulating cell-cycle progression (12). Furthermore, following siRNA knockdown of CHK1, we observed a significant increase in the level of the DNA damage chromatin mark γH2AX in interphase nuclei, in particular with the ALT cells (Supplementary Figure S8). As such, CHK1 inhibitors were used in subsequent experiments to determine the effect of the loss of CHK1 activity on H3.3S31ph in mitotic cells.
Loss of CHK1-mediated H3.3S31 phosphorylation on the chromosome arms affects the viability of ALT cancer cells by the activation of DDR during mitosis
Considering the importance of H3 phosphorylation in regulating chromatin remodeling during mitosis (39), we investigated the function of this CHK1-mediated H3.3S31 phosphorylation in ALT cancer cell lines. ALT cells were arrested in mitosis with Colcemid for 3 h with or without the CHK1 inhibitor SB218078, and distribution and level of the DNA damage chromatin mark γH2AX were examined. In telomerase-positive A549 cells, the low levels of γH2AX level remain unchanged with CHK1 inhibition (Figure 5A and B). In contrast, inhibition of CHK1 activity in mitotic cells led to the emergence of a high level of γH2AX on the chromosome arms and at some of the telomeres in a number of ALT cell lines including SUSM1, W138-VA13/2RA and GM847 (Figure 5C–H). In U2OS cells (Figure 5I–J), CHK1 inhibition did not induce a high level of γH2AX on chromosome arms but an increased number of telomeric foci with positive γH2AX signal were observed. We have also examined the impact of CHK1 inhibition on a synchronized cell population in order to validate that the increase in γH2AX on the chromosomes is not caused by Colcemid treatment. Cells were synchronized at the G2/M boundary with RO-3306 and released into mitosis either in the absence or presence of the CHK1 inhibitor SB218078 (Supplementary Figure S9). CHK1 inhibition did not alter the γH2AX level in A549 cells released from the G2/M boundary into mitosis; however, an increase in γH2AX was detected across the entire chromosome including some telomeric region in the GM847 ALT cells treated with the CHK1 inhibitor. These data suggest that the increase in γH2AX on chromosomes in ALT cells following CHK1 inhibition was not caused by Colcemid treatment, but a consequence of CHK1 activity inhibition.
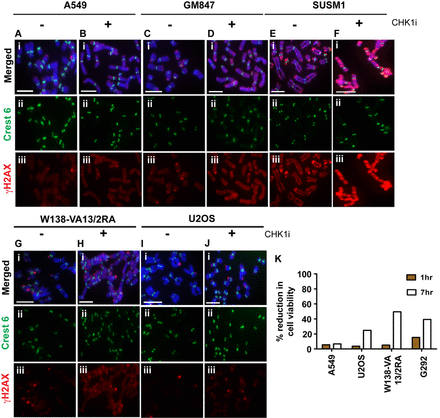
Inhibition of CHK1 in mitotic ALT cancer cell lines induces γH2AX and affects cell viability. A549, GM847, SUSM1, W138-VA13/2RA and U2OS cells were arrested at mitosis by 3 h treatment with Colcemid, either in the presence or absence of CHK1 inhibitor SB218078. Immunofluorescence analysis was then performed to determine the distribution and level of γH2AX (green). Centromeres were stained with human CREST antiserum (red). (A–B) In A549 cells, γH2AX was found only at low levels, with occasional staining at telomeric ends. Treatment with CHK1 inhibitor (CHK1i) did not cause a significant change to the distribution and level of γH2AX. (C–H) In ALT cell lines including GM847, SUSM1 and W138-VA13/2RA, there was a significant increase in γH2AX level on the chromosome arms upon the removal H3.3S31 phosphorylation by inhibiting CHK1 activity during mitosis. In U2OS cells, an increase in telomeric foci with positive γH2AX staining was detected following CHK1 inhibition. (I–J) Representative images of 50 chromosome spreads are shown. Scale bar = 5 μm. (K) A549 and ALT cancer cell lines including U2OS, W138-VA13/2RA and G292 were arrested at mitosis by overnight treatment with Nocodazole. Cells were released either in the presence or absence of CHK1 inhibitor SB218078 for either 1 h or 7 h, followed by thymidine treatment (at 2.5 mM) to prevent cells from progressing through G1/S. Cell viability was determined by staining of cells with 0.4% Trypan blue solution. The percentage of cell death was calculated as a percentage of difference between the untreated control and CHK1 inhibited cell populations. No significant change was observed in A549 at both time points. Similarly, there was no significant increase observed in ALT cell lines U2OS and W138-VA13/2RA after 1 h of release in the presence of CHK1 inhibitor. However, in G292 cells, there was a 15% increase in cell death. After 7 h of release in the presence of CHK1 inhibitor, all three ALT cancer cell lines showed increased cell death, ranging from 25 to 50%. An average of two independent experiments is presented in the graph.
Next, we examined the impact of CHK1 inhibition on cell viability of ALT cancer cell lines. Telomerase-positive A549 cells and ALT cancer cell lines including U2OS, G292 and W138-VA13/2RA were arrested in mitosis with Nocodazole for 16 h. They were then released into G1 phase either in the presence or absence of CHK1 inhibitor for 1 h and 7 h, respectively (Figure 5K). In A549, 1 h of CHK1 inhibition did not induce an increase in cell death. Likewise, in the ALT cancer cell lines, the overall cell viability was also unaffected after 1 h of CHK1 inhibition, except for G292 cells that showed a 15% increase in cell death. However, after 7 h of release in the presence of CHK1 inhibitor, there was a significant increase in the percentage of cell death with all three ALT cell lines, ranging from 25 to 50% when compared to the untreated cell population. These results demonstrate that maintenance of CHK1 activity during mitosis is essential for the survival of ALT cancer cells.
To further determine the importance of CHK1-mediated H3.3S31ph, we expressed myc-tagged H3.3 carrying a mutation in Serine 31 (substituted with an Alanine residue) in the ALT cell lines (Figure 6 and Supplementary Figure S10). In these cells, we examined the impact of H3.3 Serine 31 to Alanine mutation (S31A) on the staining pattern of H3.3S31ph on the mitotic chromosomes (Figure 6). In HeLa cells, the expression of mutant myc-H3.3S31A resulted in a reduced level of H3.3S31ph staining at pericentric DNA repeats (Figure 6A and B), and in W138-VA13/2RA cells, the expression of mycH3.3S31A resulted in a significant reduction of H3.3S31ph on the mitotic chromosomes (Figure 6C and D). The expression of H3.3S31A did not alter the number of ALT associated PML bodies in W138-VA13/2RA cells (data not shown).
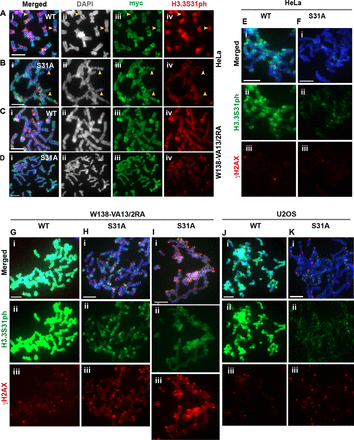
Substitution of the Serine 31 with an alanine residue alters the H3.3 S31ph staining pattern in ALT-positive cancer cells. (A–D) HeLa and W138-VA13/2RA cells were transfected with wild-type (WT) mycH3.3 and mutant mycH3.3S31A (S31A) DNA constructs, and immunofluorescence analysis was performed with antibodies against myc (green) and H3.3 S31ph (red) 96 h post-transfection. (A–B) In HeLa cells expressing WT myc-H3.3, H3.3S31ph was clearly detectable at the pericentric satellite DNA repeats on mitotic chromosomes. When the Serine 31 of H3.3 was substituted with an Alanine and over-expressed in cells, the level of H3.3S31ph at the pericentric DNA repeats was reduced. (C–D) Unlike the expression of WT myc-H3.3, over-expression of the H3.3S31A mutant in W138-VA13/2RA ALT cells led to a significant reduction in H3.3S31ph level at the pericentric DNA repeats and on chromosome arms. (E–K) Immunofluorescence analysis was performed with antibodies against H3.3S31ph (green) and γH2AX (red) on HeLa, W138-VA13/2RA and U2OS cells expressing either WT myc-H3.3 or mutant H3.3S31A. (E–F) The intensities of γH2AX on the mitotic chromosomes were low in HeLa cells expressing WT mycH3.3 and mycH3.3S31A mutant, respectively. (G–K) In W138-VA13/2RA cells, the over-expression of myc-H3.3S31A led to increases in γH2AX signals on the chromosomes arms (H) and at the telomeres (I). In U2OS cells, there was no significant increase in γH2AX staining on the chromosome arms; however, there was a significant increase in telomeric foci showing positive γH2AX signals (K). Representative images of 50 chromosome spreads are shown. Scale bar = 5 μm.
Next, we investigated the dynamics of the γH2A.X on mitotic chromosomes following the over-expression of mycH3.3S31A. In HeLa cells, the expression of mycH3.3S31A did not induce an increase in γH2AX level on the mitotic chromosomes (Figure 6E–F). In contrast, we observed a significant increase in γH2AX signal on chromosome arms in W138-VA13/2RA cells expressing mycH3.3S31A (Figure 6G–I). In U2OS cells, the expression of mycH3.3S31A did not induce a high level of γH2AX on chromosome arms but it induced an increase in the number of telomeric foci with positive γH2AX signals (Figure 6J–K). This data is consistent with the observation made with the inhibition of CHK1 activity in ALT cancer cells (Figure 5J).
Together, our data show that the presence of H3.3S31ph is important to maintain chromatin integrity in ALT cells. Inhibition of CHK1 activity and loss of H3.3S31 phosphorylation during mitosis lead to the presence of a DNA damage chromatin mark γH2AX in ALT cancer cells. Based on these findings, we hypothesize that CHK1-mediated H3.3S31 phosphorylation may act as a mark to regulate chromatin integrity by suppressing the high levels of γH2AX DNA in ALT cells. This then facilitates mitotic progression of ALT cells by preventing the induction of global DDR signaling in chromatin.
DISCUSSION
The deposition of histone variants by specific chaperones, and associated PTMs, can significantly impact chromatin structure and function. Recent studies have reported that human ALT cancers show a high frequency of mutations in H3.3 and in its chaperones ATRX and DAXX (6,8,9,12). Considering the importance of ATRX/DAXX/H3.3 in the maintenance of chromatin repression in heterochromatin (17,19), ALT cancer cells expectedly show severe telomere dysfunction and global genome instability. ATRX/DAXX mutations are also likely to affect the deposition pattern of H3.3 and its PTM profile in ALT cells. In this study, we report that despite the absence of H3.3 over-expression, ALT cancer cells show greatly elevated H3.3S31ph levels at pericentric satellite DNA repeats, along with aberrant H3.3S31ph localization on chromosome arms. We also show evidence of CHK1, a serine/threonine kinase linked to the DDR, as a novel H3.3S31 kinase. Furthermore, we demonstrate that in the ALT cancer cells, the elevated levels of activated CHK1 elicit high levels of H3.3S31ph and its mislocalization on chromosome arms. More importantly, inhibition of CHK1 activity during mitosis induces an increase in γH2AX levels on mitotic chromosome arms and compromises the viability of these ALT cancer cells. Lastly, over-expression of H3.3S31A mutant affects H3.3S31ph levels and induces γH2AX on the chromosome arms and at the telomeres in these cells. Altogether, our data suggests the importance of CHK1-mediated H3.3S31ph in chromatin maintenance and cell survival in ALT cancer cells.
All ALT cell lines tested in our study, except for the ATRX-positive SKLU1, show remarkable up-regulation in H3.3S31ph and mislocalization on chromosome arms. An up-regulated level of H3.3S31ph is also induced by siRNA knockdown of ATRX in telomerase-positive A549 and ALT-positive SKLU1 cells, although to a lesser extent than the aberrant H3.3S31ph pattern found in other ATRX-deficient ALT cancer cells. This suggests that ATRX loss is unlikely to be the sole factor that drives aberrant phosphorylation of H3.3S31ph in these cells. This aligns with the proposition that a singular dominant mutation in ATRX is insufficient to elicit an ALT phenotype (6,8), and it would likely involve unidentified cooperating genetic or epigenetic changes. A recent study suggested that beyond the initial activation of ALT activity induced by depletion of histone chaperone anti-silencing factor 1, ALT activity might be maintained via currently unidentified, cooperating epigenetic alterations (37). Here, we provide the first evidence of the existence of aberrant epigenetic histone modification profiles in ALT cells. It is currently unclear whether abnormal H3.3S31ph dynamics reported here is a consequence of ALT activation, or if it contributes to the ALT phenotype. Nevertheless, our findings suggest that telomere dysfunction and subsequent genome instability could promote a global change in chromatin status, due to elevated DDR signaling. In addition, we propose that aberrant H3.3S31ph labeling may constitute another marker of ALT activation in cancer cells. CHK1-mediated aberrant H3.3S31ph dynamics may be unique to ALT cells and represent a putative therapeutic target for treatment of ALT cancers.
Previous studies have identified the role of CHK1 as a histone kinase that mediates phosphorylation of Ser10 and Thr11 on histone H3, an interaction involved in transcription regulation (31,40). Our finding of a novel CHK1 function in phosphorylating H3.3S31 further implicates the regulatory role of CHK1 in chromatin metabolism. Although the H3.3S31ph PTM was first reported almost a decade ago (29), we report, to our knowledge, the first identification of an H3.3S31 kinase in vitro and in cells. It is important to note that CHK1 inhibition does not result in a complete loss of the H3.3S31ph signal, particularly at pericentric satellite DNA repeats. This suggests that CHK1 is unlikely to be the kinase that phosphorylates H3.3 at pericentric DNA repeats, and that other kinases of H3.3S31 remain to be discovered.
As shown above, the ALT cancer cells show significant up-regulation of activated CHK1, even though global CHK1 levels are unaltered compared to telomerase-positive cells. This finding is consistent with previous data of high basal levels of DNA damage and DDR signaling (as evidenced by high levels of γH2AX and activated CHK2) in these cells (6). Additionally, impaired G2/M checkpoint has been speculated to permit continued proliferation of ALT cancer cells despite a considerable burden of DNA damage both at telomeres and elsewhere in the genome (6). Considering the association of CHK1 with DDR signaling, and the protective role of H3.3S31ph against DNA damage that was recently reported (41), H3.3S31ph chromatin mark may be laid down as a response to chronic unresolved DNA damage. This would help ALT cells progress through mitosis without the induction of DDR, which can affect the maintenance of chromatin stability (42). Indeed, when CHK1 is inhibited or an H3.3S31A mutant is over-expressed in ALT cancer cells, the decrease in H3.3S31ph is accompanied by the appearance of increased levels of γH2AX on the chromosome arms and at telomeres, indicating a widespread increase in the level of DNA damage mark γH2AX across (mitotic) chromosomes. Inhibition of CHK1 activity in mitotic ALT cells also leads to increased cell death compared to untreated cells. These findings indicate that CHK1-mediated H3.3S31ph is pivotal for the maintenance of chromatin integrity and ALT cell survival. In these cells, CHK1-mediated H3.3S31ph may keep γH2AX low to potentially prevent spurious recruitment of the DDR machinery during mitosis. Further investigation is required to determine whether H3.3S31ph dynamics in ALT cells directly suppresses the formation of γH2AX foci during mitosis, or if the increase in γH2AX, when H3.3S31ph is lost, is indicative of further DNA damage during mitosis in ALT cells.
Collectively, our findings reveal CHK1 as a novel H3.3S31 kinase. We show that CHK1 is able to catalyze H3.3S31 phosphorylation in vitro, and more importantly, that elevated levels of activated CHK1 drive the aberrant H3.3S31ph dynamic in ALT cells. Previous studies have suggested CHK1 as a histone kinase mediating H3 Ser10 and Thr11 phosphorylation. Interestingly, recent evidence also indicates that CHK1 phosphorylation of H3 Ser10 and Thr11 influences the establishment of PTM profiles of adjacent residues on H3 (31,40). Furthermore, as the binding of an H3.3 Lysine 36 reader has been shown to be directly affected by the Serine 31 phosphorylation (43), it will be interesting to determine whether the aberrant H3.3S31ph influences PTMs on neighboring residues and affects the global PTM profile in ALT cells. For example, changes to the methylation/acetylation dynamics of the neighboring Lys27 and Lys36 may have drastic consequences for the cellular transcription profile. Our finding of H3.3S31 as a new substrate for CHK1 phosphorylation further implicates a regulatory role of CHK1 in chromatin metabolism, and shows for the first time how misregulation of CHK1 activity in a diseased state, such as in ALT cancers, may affect global chromatin regulation.
We thank Dr Jane Lin for critical reading of the manuscript.
FUNDING
National Health and Medical Research Council (NHMRC) of Australia; Postgraduate Scholarship from the Cancer Council of Victoria, Australia (to C.F.T.M.); Australia Research Council (ARC) Future Fellowship Award from the ARC, Australia (to L.H.W.). Funding for open access charge: National Health and Medical Research Council (NHMRC).
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments