





Abstract
MicroRNAs (miRNAs) and heterogeneous nuclear ribonucleoproteins (hnRNPs) are families of sequence-specific, posttranscriptional modulators of gene expression. Despite extensive mechanistic and functional studies on both regulatory classes, the interactions and crosstalk between them are largely unexplored. We have reported that competition between miR-297 and hnRNP L to bind a 3΄UTR-localized CA-rich element (CARE) of VEGFA mRNA regulates its translation. Here, we show that translation of VEGFA mRNA in human myeloid cells is dictated by a bi-directional interaction between miR-574-3p, a CA-rich microRNA, and hnRNP L. In normoxia, miR-574-3p, acting as a decoy, binds cytoplasmic hnRNP L and prevents its binding to the CARE and stimulation of VEGFA mRNA translation, simultaneously permitting miR-297-mediated translational silencing. However, in hypoxia, cytoplasmic accumulation of Tyr359-phosphorylated hnRNP L sequesters miR-574-3p, overcoming its decoy activity and seed sequence-dependent gene silencing activity. Ectopically expressed miR-574-3p binds multiple RNA recognition motif (RRM) domains of hnRNP L, synergizes with miR-297, reduces VEGFA mRNA translation, and triggers apoptosis, thereby suppressing tumorigenesis. Our studies establish a novel condition-dependent interplay between a miRNA and an hnRNP that regulates their functions in a bidirectional manner.
INTRODUCTION
Gene expression is temporally and spatially regulated at multiple synthetic and degradative steps including transcription, pre-mRNA splicing, mRNA transport, mRNA stability, translation, protein stability and post-translational modification. Translational control of gene expression offers the benefits of rapid response, reversibility, conservation of resources, fine control and coordinate regulation of transcript families (1). Translational control can be subdivided into two types on the basis of regulatory mechanisms and extent of the target mRNA group, i.e. global and transcript-selective translational control. The latter is generally mediated by the interaction of mRNA-binding proteins (2–4) or microRNAs (miRNAs) (5,6) with structural or linear cis-elements in non-coding regions of target mRNAs. The 3΄-untranslated region (UTR) is the principal binding site for trans-acting proteins and miRNAs that generally repress, rather than activate, translation of an ensemble of mRNAs (7). miRNAs are endogenous, ∼22-nt RNAs that regulate expression of mRNA targets (6,8). Mature miRNAs associate with members of the argonaute (Ago) family of proteins in the RNA-induced silencing complex (RISC), and target mRNAs by binding regions complementary to ‘seed’ sequences at miRNA 5΄-termini (nt 2–8), resulting in transcript degradation or translation repression, or both. miRNAs contribute to the pathogenesis of cardiovascular disease and cancer, among other human ailments (9,10).
Compound, protein-directed RNA switches have been recently shown to regulate translation in vertebrate cells (11–13), adding another layer of complexity to gene regulation. These RNA switches feature mutually exclusive interactions of positive and negative trans-acting RBPs or ribonucleoprotein (RNP) complexes with the same or vicinal regulatory RNA elements, by direct competition (14) or by structural rearrangement (15). These complex interactions can integrate signals from disparate input conditions to markedly influence transcript-specific gene expression and cellular function. RNA switches can be linear or structural, each with their own class of regulators. Linear switches are directed by primary sequence-specific interacting factors, including miRNAs and heterogeneous nuclear ribonucleoproteins (hnRNPs) (12,14), whereas structural switches involve secondary or tertiary structure-specific binding factors, such as the gamma-interferon activated inhibitor of translation (GAIT) complex and double-stranded RNA-binding proteins (11,15). RNA switches can reversibly inhibit or activate transcript-specific translation, depending on the environmental state. However, the regulation of vertebrate RNA switches and the pathophysiological consequences of their ablation remain largely unexplored.
The hnRNP L-mediated 3΄UTR RNA switch controls translation of vascular endothelial growth factor-A (VEGFA) mRNA in myeloid cells, and responds differentially to hypoxic and inflammatory signals (11,14,15). VEGF-A is an essential regulator of physiological and pathological angiogenesis (17,18). Myeloid-derived VEGF-A plays a critical role in the ‘angiogenic switch’ during tumor vascularization (19). VEGF-A protein expression in myeloid cells is fine-tuned by hnRNP L at the post-transcriptional level (14,15,20). hnRNP L contains four RNA recognition motif (RRM) domains and is a CA-rich element (CARE)-binding protein that binds RNA sequences bearing multiple nearby CACA or ACAC motifs (21). It can shuttle between the nucleus and cytoplasm, participating in nuclear pre-mRNA splicing and cytoplasmic mRNA switching, respectively (11,22–24). Under normoxic conditions, hnRNP L primarily localizes in the nucleus with a minor component in the cytoplasm (14). However, under hypoxic stress substantial hnRNP L translocates out of the nucleus to interact with two other proteins to form the cytoplasmic HILDA (hypoxia-induced hnRNP L-DRBP76-hnRNP A2/B1) complex (11). The HILDA complex binds a 21-nt CARE in the VEGFA mRNA 3΄UTR and blocks miR-297-RISC binding to an overlapping CA-rich sequence in the 126-nt hypoxia stability region (HSR) and prevents GAIT complex binding to the HSR, thereby stimulating VEGFA translation by two distinct mechanisms (Supplementary Figure S1A) (11,14). In addition, miR-297 and hypoxia-activated hnRNP L control translation of a second oncogenic target, DGK-α (diacylglycerol kinase-alpha) mRNA, in human glioma cells, thereby contributing to glioblastoma progression (25). Thus, hnRNP L-directed RNA switches might regulate expression of an ensemble of target mRNAs in multiple cell types.
Emerging evidence indicates that miRNAs, via seed sequence-independent interaction with RNA-binding proteins (RBPs), can act as sequence-specific decoys and modulate target RBP function (26,27). Conceivably, RBP–miRNA interactions could work in the reverse direction, namely, RBP-mediated inhibition of target miRNA function. However, biological systems taking advantage of this converse mechanism have not been reported. Here, we report a novel, dual miRNA-mediated mechanism that regulates the VEGFA mRNA switch under pathophysiological conditions. Individual transcripts are often regulated by multiple miRNAs that target distinct sites, primarily in the 3΄UTR. We show potentiation by two distinct miRNAs, miR-297 and miR-574-3p, where the first targets a 3΄UTR RNA element, and the other targets the cognate RNA element-binding protein. miR-574-3p is a tumor suppressor RNA of unknown mechanism, and is down-regulated in multiple cancer tissues (28–32). We now report that miR-574-3p, via its CA-rich sequence, binds hnRNP L thereby preventing its CARE-mediated activation of the VEGFA mRNA switch that drives VEGF-A expression, and simultaneously permitting inhibition by CARE-targeting miR-297. We also show a converse regulatory activity, namely, hypoxia-inducible accumulation of cytoplasmic hnRNP L overcomes the decoy and RISC silencing activity of miR-574-3p. In a potential therapeutic approach we show that overexpression of miR-574-3p in hypoxia decoys hnRNP L, reverses the VEGFA mRNA switch, and inhibits tumor cell growth in vitro, as well as tumorigenesis in a mouse model. Together, these findings establish condition-dependent, multi-modal regulation of translation mediated by miRNA–hnRNP interaction, and provide a rationale for miRNA-based, hnRNP-targeted therapeutic strategies against cancer.
MATERIALS AND METHODS
Reagents
Phospho-safe extraction buffer was from Novagen (Madison, WI, USA). Dual luciferase reporter system was from Promega (Madison, WI). Human monocyte nucleofector kit was from Lonza (Switzerland). Reagents for protein purification and immunoanalysis were from Pierce (Rockford, IL, USA). Primers, dNTP mix, TRIzol LS reagent, one-step RT-PCR system and competent cells were from Invitrogen (Carlsbad, CA, USA). Mouse monoclonal anti-hnRNP L antibody (NB120-6106) was from Novus (Littleton, CO, USA). Anti-DRBP76 antibody (orb40012) was from Biorbyt (Cambridge, UK). Antibodies against VEGF-A (sc-152), EP300 (sc-585), hnRNP A2/B1 (sc-53531), Dicer (sc-30226) and phosphotyrosine (sc-7020), and Protein A/G beads were purchased from Santa Cruz (Dallas, TX, USA). Anti-Ago2 (#2897) was obtained from Cell Signaling (Danvers, MA, USA). Anti-β-actin antibody (3662-100) was from Biovision (Mountain View, CA, USA). Anti-rabbit IgG and anti-mouse IgG were from GE healthcare (UK). hnRNP L siRNA was obtained from Origene (SR302174). Pre-miR™ miRNA precursors and anti-miR inhibitors were from Life Technologies (Cat. #AM17110 for negative control miR, AM17100 for miR-574-3p, AM17010 for negative control anti-miR inhibitor and AM17000 for anti-miR-574-3p; Carlsbad, CA). [α-32P]UTP was from PerkinElmer (Boston, MA).
Cell culture and transfection
Human U937 monocytic cells (ATCC, Rockville, MD) were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum (FBS), 2 mM glutamine and 100 U/ml of penicillin and streptomycin at 37°C and 5% CO2. For preparation of cytosolic extracts, cells were treated under either normoxic (21% O2) or hypoxic (1% O2) conditions for 8, 24 or 48 h. Cell lysates were prepared in Phosphosafe extraction buffer containing protease inhibitor cocktail. To silence endogenous genes, U937 cells (5 × 106 cells) were transfected with gene-specific siRNA or a scrambled control siRNA (100–200 nM) using human monocyte Nucleofector Kit (Lonza). To determine miRNA function, U937 cells (5 × 106 cells) were transfected with oligomers including pre-miR miRNA precursor mimics, miRNA-negative control, anti-miR inhibitors, anti-miR-negative control (50–200 nM, Ambion) and plasmid DNA (0.5–2.0 μg) using the Nucleofector Kit. Relatively high levels of miR mimics and anti-miR inhibitors are used due to the low transfection efficiency of U937 cells and the need to achieve high miRNA levels to be effective decoys. The sequences of anti-miR-574-3p inhibitor: 5΄ ugugggugugugcaugagcgug 3΄.
Plasmids, site-directed mutagenesis, and recombinant protein expression
pET28-hnRNPL-RRM12, pET28-hnRNPL-RRM12-H105A and pET28-hnRNPL-RRM34 plasmids were gifts from Dr Xu Li (33). His-tagged wild-type hnRNP L, Y359F mutant, RRM1,2, RRM1,2-H105A and RRM3,4 were expressed in Escherichia coli BL21(DE3) with IPTG induction, and purified with Ni-NTA resin (Qiagen). Expression of GST-tagged hnRNP L was expressed and purified with B-PER GST purification kit (Thermo Fisher) (11). pcDNA3-c-Myc-hnRNP L His105-to-Ala mutant was prepared using GeneArt Site-Directed Mutagenesis System (Thermo Fisher).
RNA analysis by RT-qPCR
Total small RNA was extracted with miRVana miRNA isolation kit (Thermo Fisher), and quality and quantity determined by NanoDrop spectrophotometer. miRNA was assessed by real-time PCR using TaqMan probe (Thermo Fisher) and primer sets using Applied Biosystems Real Time PCR StepOne Plus. Briefly, total small RNA (10 ng) was reverse-transcribed using Taqman MicroRNA Reverse Transcription Kit and amplified using TaqMan 2x Universal PCR Master Mix, No AmpErase UNG (Thermo Fisher). To determine VEGFA, ACTB and EP300 mRNAs, one-step reverse transcription coupled with real-time PCR was performed with 2x VeriQuest Probe One-Step qRT-PCR Master Mix (Affymetrix) using total RNA (0.5 μg) extracted with Trizol in an Applied Biosystems Real Time PCR StepOne Plus machine. RT-qPCR probes for the Taqman Gene Expression Assay, i.e. VEGFA (Hs00900055_m1), ACTB (Hs99999903_m1) and EP300 (Hs00914223_m1) were from Thermo Fisher (Cat. #4331182): The primers for semi-quantitative RT-PCR were: RT_ACTB-f: 5΄-ATGGATGATGATATCGCCGCG-3΄; RT_ACTB-r: 5΄-CTAGAAGCATTTGCGGTGGAC-3΄; RT_VEGFA-f: 5΄-ACAGAACGATCGATACAGAA-3΄; RT_VEGFA-r: 5΄-AAAGATCATGCCAGAGTCTC-3΄.
Immunoprecipitation
Most IP experiments were done using Co-Immunoprecipitation kit (Pierce) following the manufacturer's instruction to eliminate antibody contamination of IP products. In several IP experiments, more standard methods was used. Cells were lysed in Phospho-safe extraction buffer, and 500 μl of lysate was combined with 50 μl protein A/G agarose beads (50% bead slurry), and pre-cleared at 4°C for 60 min. The samples were centrifuged at 13 000 rpm for 10 min at 4°C and the supernatant added to 50 μl of protein A/G beads with 2 μg of antibody, and rotated for 4 h at 4°C. The beads were washed five times with 1 ml detergent-containing wash buffer (50 mM Tris–HCl [pH 7.6], 150 mM NaCl and 0.1% Triton X-100). Protein gel-loading dye (100 μl) was added, and the samples boiled and loaded onto the gel.
RIP-RT-qPCR
RIP was performed as described (11). Protein A/G beads (50 μl) were incubated with 500 μl of cell lysate (4 mg protein for IP) for 1 h at 4°C with rotation to pre-clear. The cell lysate was centrifuged and the supernatant collected. Mouse anti-hnRNP L or rabbit anti-Ago2 antibody (2 μg) was added (mouse or rabbit pre-immune IgG was used as negative control) and the mixture rotated at 4°C for 1 h. Protein A/G beads (50 μl) were added and incubated at 4°C for 4 h. The beads were washed five times with 1 ml of wash buffer (as in IP) with rotation at 4°C. Total immunoprecipitated RNA and total input RNA from 50 μl of lysate were extracted with Trizol. Immunoprecipitated RNA (5 μl) and 200 ng of total RNA were used in miRNA RT-qPCR with Taqman assays.
Isolation of translationally active and inactive mRNA pools by sucrose gradient fractionation
Polysome profiling was performed as described (11). Cycloheximide (CHX, 100 μg/ml, Sigma) was added to cells for 15 min before lysis. 107 cells were suspended in 350 ml TMK lysis buffer, and lysates centrifuged at 12 000 rpm for 10 min and the supernatants collected. RNase inhibitor (2 μl, 40 U/μl) and CHX (50 μl, 100 μg/μl) were added to 50 ml of 10% and 50% sucrose gradient solutions before use. Cytosolic lysates were loaded onto the sucrose solutions, centrifuged at 29 000 rpm for 4 h, and 12 fractions of ∼0.5–1 ml were collected, containing light RNP, 40S, 60S and 80S, and heavy polysome fractions. Total RNA was isolated from individual fractions by extraction with Trizol and purified by RNeasy minikit (Qiagen). RNA was quantitated and used for real-time PCR analyses. Polysome cushion assay was performed as described (34).
RNA electrophoretic mobility shift assay (EMSA)
Interactions between hnRNP L and miRNAs were evaluated by RNA EMSA. Internally [α-32P]UTP-labeled wild-type and mutant miR-574-3p and miR-574-5p RNAs were prepared by oligonucleotide-directed transcription using MEGAshortscript T7 Transcription Kit (Thermo Fisher) and synthesized DNA template (Life Technologies). RNA EMSA was done as described with some modifications (35). [α-32P]UTP-labeled miR (50 fmol) was incubated for 30 min at 4°C with purified recombinant His-tagged hnRNP L (25, 50, 100, 200 and 400 nM) in 20 μl of reaction buffer containing 50 mM Tris–HCl (pH 7.6), 150 mM NaCl, 0.25 mM dithiothreitol, 40 U of RNase inhibitor and 10% glycerol. RNA–protein complexes were resolved by 5% native PAGE gel electrophoresis at 4°C. The gel was dried, and the shifted probe visualized by autoradiography.
Dual luciferase reporter assay
U937 cells (5 × 106 cells) were transiently transfected with miR mimics (200 nM), anti-miR inhibitors (200 nM), or pcDNA3-based hnRNP L-bearing plasmid DNA (2 μg) with pCD-FLuc-VEGFA HSR (11) using human monocyte Nucleofector Kit. pRL-SV40 (1 μg) RLuc-expressing vector was co-transfected to normalize transfection efficiency. After 12 h, transfected cells were incubated under normoxia or hypoxia for up to 48 h, lysed and luciferase activities measured using a dual luciferase assay kit (Promega).
Surface plasmon resonance
Protein binding to VEGFA CARE, miR-574-3p, and VEGFA GAIT element RNA was determined by SPR in a Biacore 3000 system. Biotinylated RNAs were immobilized on a streptavidin sensor chip in buffer containing 10 mM Hepes (pH 7.4), 150 mM NaCl, 3 mM EDTA and 0.005% (v/v) surfactant P20. The analyte flow rate was 5 μl/min in the same buffer for immobilization; purified protein was applied at 20 μl/min. Flow cells were regenerated with three rinses of 1 M NaCl at 30 μl/min. Dissociation constants were calculated for a range of protein concentrations using Biaevaluation software (Biacore).
Cell proliferation assay
U937 cells were seeded into 24-well plates at a density of 30 000 cells/well and cultured for 2 days in RPMI medium with 10% FBS. Cells were incubated with methylthiazolyldiphenyl-tetrazolium bromide (MTT, 1 mM, Sigma) for 2 h at 37°C. After adding 100 μl of 10% SDS in 10 mM HCl, absorbance was measured at 570 nm with a reference at 620 nm in a Spectramax 190 spectrophotometer (Molecular Devices).
Caspase 3/7 activity assay
Cells in 96-well plates were equilibrated at room temperature. Caspase-Glo® 3/7 Reagent (100 μl, Promega) was added to each well of a white-walled 96-well plate containing 100 μl of blank, negative control cells, or treated cells in culture medium, and gently mixed at 300–500 rpm for 30 s and incubated at room temperature for 1 h. Luminescence was measured in a plate-reading luminometer.
Anchorage-independent colony formation assay
U937 cell suspension (20 000 cells in 0.1 ml) was mixed gently with 3 ml of 20% FCS in 2× RPMI and 3 ml of 0.7% agarose (all pre-warmed to 40°C). A 1.5 ml aliquot was added to 35-mm cell culture dishes containing a base of 1% agar. Plates were re-fed with cell culture media twice a week at 37°C in humidified incubator for 15 days, and stained with 0.5 ml of 0.005% crystal violet for 1 h and colonies counted using a dissecting microscope.
Myeloid cell-derived xenograft tumor mouse model
BALB/c nude mice (4–6 week males) were from the Case Western Reserve University Athymic Animal and Xenograft Core. Genetecin-selected U937 cells stably expressing pcDNA3-c-Myc-hnRNP L were transfected with miR-574-3p or control miR (200 nM miR for 5 × 106 cells). Transfected U937 cells at 1 × 107 were implanted s.c. into both mouse flanks. Tumor volume was measured every other day for 2–3 weeks and volume calculated as 0.525 × (X2Y), where X = tumor width and Y = tumor length. Mice with tumor volume larger than 1500 mm3 were sacrificed based on requirements from the IACUC protocol of the Tumor Core. Statistical analysis was done with two-way ANOVA with repeated measures.
RESULTS
CA-rich miR-574-3p Inhibits VEGFA mRNA Translation in Normoxia
hnRNP L is a nucleocytoplasmic shuttling protein predominantly localized in the nucleus of monocytic cells, but ∼10% is in the cytoplasm (Supplementary Figure S1B) (11,14). We showed that hypoxia induces Tyr359 phosphorylation of hnRNP L leading to marked accumulation in the cytoplasm where, together with hnRNP A2/B1 and DRBP76, it forms the HILDA complex which binds the 3΄UTR CARE of VEGFA mRNA and stimulates its translation (11) (Supplementary Figure S1A). We and others have observed that under normoxic conditions, cytoplasmic hnRNP L fails to interact with VEGFA mRNA (36) and does not stimulate VEGFA mRNA translation (14). However, the underlying molecular mechanism remains unclear. We considered the possibility that a CA-rich miRNA acts as a molecular decoy by binding the RRM domain of hnRNP L that recognizes the CA-rich region, i.e., CARE, in the 3΄UTR of VEGFA mRNA, blocking their interaction. We interrogated human miRNAs in miRBASE using a 24-nt, 12-CA repeat sequence as query. Three CARE-bearing miRNAs, miR-466, miR-574-3p and miR-299-5p were identified (Figure 1A, top). miR-466 and miR-299-5p expression was lowly expressed or below the detection limit in U937 monocytic leukemia cells, but expression of miR-574-3p was 15.6-fold higher than miR-466 after normalizing with RNU6B (Figure 1A, bottom). Interestingly, the CA-rich sequence ‘CACACACCCACA’ in the 3΄-terminus of miR-574-3p does not overlap the 5΄-terminus seed sequence, possibly indicating distinct functions for the two regions, for example, decoy of CARE-binding proteins and silencing of RISC target gene expression, respectively (Figure 1B).
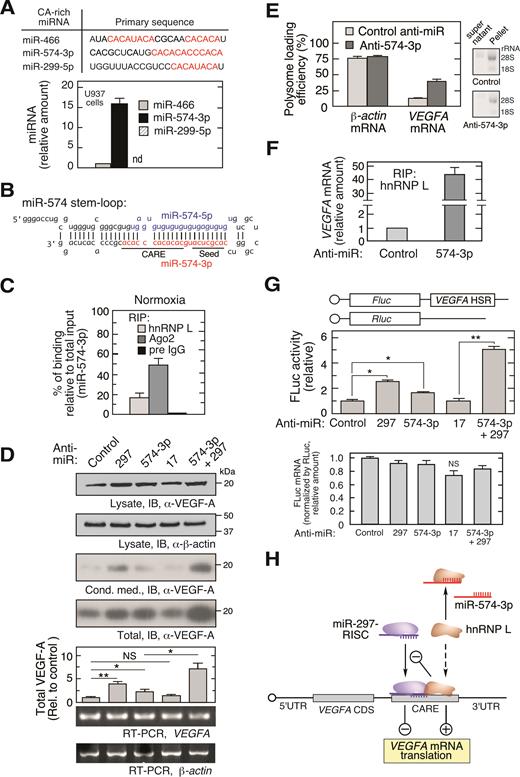
miR-574-3p represses hnRNP L binding to VEGFA mRNA and translational activation in normoxia. (A) Bioinformatic identification of CA-rich miRNAs and their expression in U937 cells. qRT-PCR was performed and expression normalized to RNU6B. (B) Sequence of precursor miR-574 stem–loop. Guide strand miR-574-5p (blue) and passenger strand miR-574-3p (red) are highlighted; seed and CARE regions are underlined. (C) Interaction of miR-574-3p with hnRNP L and RISC shown by percentage of binding relative to total input. U937 cells were cultured under normoxic condition for 24 h. Lysates with the same amount of total protein were subjected to IP with anti-hnRNP L, -Ago2 or pre-immune IgG antibodies, and then qRT-PCR using miR-574-3p-specific probe. Input RNA from the same amount of lysates was used as normalizer. (D) Endogenous miR-574-3p inhibits VEGF-A expression. U937 cells were transfected with negative control anti-miR, anti-miR-574-3p, -297 (200 nM), or both (100 nM/each) for 48 h, and lysates immunoblotted with anti-VEGF-A and -β-actin antibodies. VEGFA and β-actin mRNA was determined by semi-quantitative RT-PCR (equal total RNA for RT; 25 and 15 cycles for PCR of VEGFA and actin β-mRNA, respectively). (E) Inactivation of miR-574-3p increases VEGFA mRNA translation. U937 cells were transfected with anti-miR-574-3p or control anti-miR (200 nM) for 48 h. Ribosome-free and -bound mRNA were fractionated on a polysome cushion (rRNA was shown), and total RNA extracts were subjected to qRT-PCR using VEGFA- and β-actin-specific probes. (F) Endogenous miR-574-3p reduces the interaction between hnRNP L and VEGFA mRNA. U937 cells were transfected with anti-miR-574-3p or control (200 nM) for 24 h. Lysates were subjected to RIP with anti-hnRNP L antibody coupled with qRT-PCR using VEGFA-specific probe. (G) Endogenous miR-574-3p inhibits expression of HSR-bearing reporter. FLuc reporter bearing the VEGFA HSR was co-transfected into U937 cells with anti-miR-574-3p or anti-miR-297, or both, for 48 h in normoxic condition. FLuc activity was normalized by RLuc and expressed as percentage of control. FLuc mRNA level was shown by normalization with RLuc mRNA. (H) Schematic showing effects of CA-rich miR-574-3p and hnRNP L on normoxic VEGF-A expression. VEGFA mRNA translation is inhibited by miR-297-RISC binding to VEGFA 3΄UTR CARE, and by miR-574-3p binding to hnRNP L as RNA decoy in normoxia. In (A, C–G), data are presented as mean ± SD (n = 3; *P ≤ 0.05, **P ≤ 0.01, Student's t-test).
The CA-rich region of miR-574-3p resembles the top-ranking hnRNP L-interacting motif as defined by SELEX (Systematic Evolution of Ligands by EXponential enrichment) (21) and CLIP-Seq (cross-linking immunoprecipitation sequencing) analyses (23,24), consistent with miR-574-3p as an hnRNP L-binding miRNA. RNA ribonucleoprotein immunoprecipitation (RIP) coupled with miRNA-specific quantitative RT-PCR (qRT-PCR) revealed relatively specific binding; the ratio of miR-574-3p binding to hnRNP L compared to binding to the RISC constituent Ago2 was ∼0.3, whereas that ratio for a stable and abundant non-CA-rich miRNA miR-17 (37,38) was <0.01, suggesting its tight binding with Ago2 (Supplementary Figure S1C). Absolute quantitation confirmed that the percentage of miR-574-3p binding to hnRNP L and Ago2 versus total input is 17.4% and 47.8%, respectively (Figure 1C). Northern blot for hnRNP L RIP suggested that only the mature miR-574-3p but not the pre-miR-574 was bound by hnRNP L (Supplementary Figure S1D).
To determine whether endogenous miR-574-3p modulates VEGFA mRNA translation, chemically-stabilized anti-miR-574-3p, was transfected into U937 cells and found to increase VEGF-A protein expression by ∼2-fold (determined as sum of intra- and extracellular protein) without altering VEGFA mRNA, consistent with translational repression by miR-574-3p, but less than the nearly 4-fold stimulation by anti-miR-297 previously shown to increase VEGF-A expression (Figure 1D) (14). Combined treatment of anti-miR-574-3p and anti-miR-297 (at a half-dose of each) synergistically increased VEGF-A expression by ∼7-fold. Inactivation of miR-574-3p by anti-miR increased VEGFA mRNA loading into polysomes, confirming a translational control mechanism (Figure 1E), and increased the interaction of hnRNP L with VEGFA mRNA by ∼40-fold (Figure 1F). Consistent with this result, anti-miR-574-3p markedly reduced the interaction of hnRNP L with miR-574-3p (Supplementary Figure S1E). To interrogate the VEGFA mRNA region responsible for the observed translational control, cells were co-transfected with anti-miRs and a firefly luciferase reporter bearing the VEGFA HSR (3΄UTR nt 332–456) that includes the CARE (15). Anti-miR-574-3p diminished translational repression without altering mRNA stability indicating the VEGFA HSR contains the regulatory region, and combined treatment of anti-miR-574-3p and anti-miR-297 (at half-dose of each) exhibited marked synergistic activity (Figure 1G). The expression of FLuc reporter containing the VEGFA HSR with mutant CARE (CA dinucleotides changed into GU) did not respond to anti-miR-574-3p and anti-miR-297 inhibitors (Supplementary Figure S1F). These results are consistent with a dual inhibitory mechanism in which miR-574-3p binds hnRNP L as a decoy, thereby preventing its translational stimulatory activity, and simultaneously facilitating miR-297-mediated translational repression of VEGFA mRNA (Figure 1H).
Hypoxia-induced, hnRNP L-directed capture of miR-574-3p inhibits canonical RISC activity
Hypoxia induces phosphorylation of hnRNP L at Tyr359 and consequent translocation of ∼50% of the protein to the cytoplasm (Supplementary Figure S1B) (11,14). Hypoxia induced a dramatic shift of miR-574-3p binding from Ago2 to hnRNP L, compared to that observed in normoxia (Figures 1C and 2A, Supplemental Figure S1C). The cytoplasmic level of miR-574-3p was not substantially altered by hypoxic treatment (Supplementary Figure S2A). Transfected, Myc-tagged phospho-mimetic (Tyr359Asp, Y359D) (11) and wild-type hnRNP L bound essentially identical amounts of miR-574-3p (Supplementary Figure S2B). These results suggest that hypoxia drives trapping of miR-574-3p by hnRNP L primarily by cytoplasmic re-localization of hnRNP L, and not by induction or phosphorylation state.
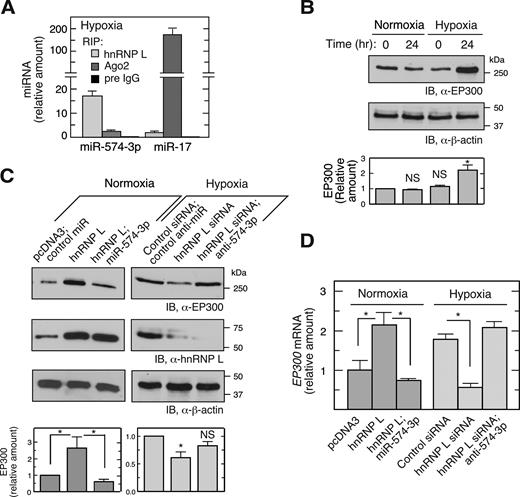
Hypoxia induces release of miR-574-3p from Ago2 suppressing RISC-mediated activity. (A) Hypoxia switches binding of miR-574-3p from Ago2 to hnRNP L. U937 cells were cultured in hypoxia for 24 h. Lysates with the same quantity of total protein were subjected to IP with anti-hnRNP L, -Ago2 or pre-immune IgG antibodies, and then to qRT-PCR using probes against miR-574-3p or miR-17. Signal of hnRNP L RIP-RT-qPCR for miR-17 under normoxia (Figure S1C) was used as normalizer. (B) Hypoxia induces EP300 expression. U937 cells were cultured in normoxia or hypoxia for 24 h and lysates immunoblotted using anti-EP300 antibody. (C) hnRNP L inhibits canonical RISC activity of miR-574-3p and stimulates EP300 expression. U937 cells were transfected with pcDNA3-Myc-hnRNP L (1 μg) or hnRNP L siRNA (with or without miR-574-3p or anti-miR-574-3p) (200 nM) in normoxia or hypoxia as indicated, and EP300 expression measured by immunoblot. Transfections with pcDNA3 vector or scrambled siRNA were used as negative controls. (D) hnRNP L induces EP300 mRNA by antagonizing miR-574-3p. U937 cells were treated as in (C) and total RNA subjected to qRT-PCR using EP300-specific probe and normalized to β-actin. Cond. Med.: conditioned medium. In (A–D), data are presented as mean ± SD (n = 3, Student's t-test).
To begin to explore the influence of hnRNP L binding to miR-574-3p on its RISC-driven tumor-suppressive activity (28,29,39), we showed that hypoxia induces expression of EP300, an established downstream target of miR-574-3p (Figure 2B) (30). EP300 is a transcriptional co-activator of NF-κB, a key transcription factor regulating inflammatory gene expression (40), and HIF1A (hypoxia-inducible factor 1, α-subunit), a transcription factor that stimulates expression of hypoxia-inducible genes including VEGFA (41). Moreover, overexpression of hnRNP L increased EP300 expression in normoxia, and the basal level was restored by co-introduction of pre-miR precursor mimetics (chemically modified, double-stranded RNAs that mimic endogenous mature miRNAs) of miR-574-3p (Figure 2C, left). Likewise, hnRNP L knockdown reduced EP300 expression in hypoxia, and co-introduction of anti-miR-574-3p inhibitor restored the level (Figure 2C, right). Changes in EP300 mRNA levels nearly paralleled changes in protein, suggesting that regulation by miR-574-3p-RISC and hnRNP L is primarily at the level of RNA stability rather than translation (Figure 2D). These results are consistent with a mechanism in which binding of miR-574-3p by hnRNP L prevents its interaction with RISC, thereby overcoming its suppressive effect on gene expression. To investigate whether miR-574-3p bound to hnRNP L binds RISC, the interaction between hnRNP L and Ago2, a major RISC constituent, was determined by co-immunoprecipitation (IP) and immunoblot analysis. hnRNP L did not interact with Ago2 or Dicer in either hypoxia (Supplementary Figure S2C, left panel) or normoxia (not shown). The reverse pull-down confirmed that hnRNP L does not interact with RISC (Supplementary Figure S2C, right panel). Thus, hypoxia-activated capture of miR-574-3p by hnRNP L inhibits its canonical RISC-mediated gene silencing activity, thereby inducing EP300 expression.
Ectopic miR-574-3p decoys hnRNP L and reverses the VEGFA RNA switch
miR-574-3p functions as an endogenous hnRNP L decoy and inhibits VEGFA mRNA translation in normoxia. We interrogated the effect of exogenously introduced miR-574-3p on VEGF-A expression under hypoxic conditions as a potential therapeutic for anti-angiogenic and anti-tumor applications. Transfection of U937 cells with pre-miR precursor mimetics of miR-574-3p for 48 h in hypoxia reduced VEGF-A protein expression by about 40% (Figure 3A, inset). Polysome profiling coupled with qRT-PCR showed that VEGFA mRNA (but not β-actin mRNA, not shown) was markedly shifted from polysome to non-polysome fractions, suggesting translational repression by miR-574-3p (Figure 3A). None of the miRNA target prediction programs, i.e., TargetScan, miRanda, miRWalk, PITA, miRDB, and DIANAmT, predicted miR-574-3p binding sites in VEGFA mRNA suggesting that repression might not be due to direct targeting of VEGFA mRNA, but rather by decoying hnRNP L and facilitating miR-297-mediated targeting of VEGFA mRNA.
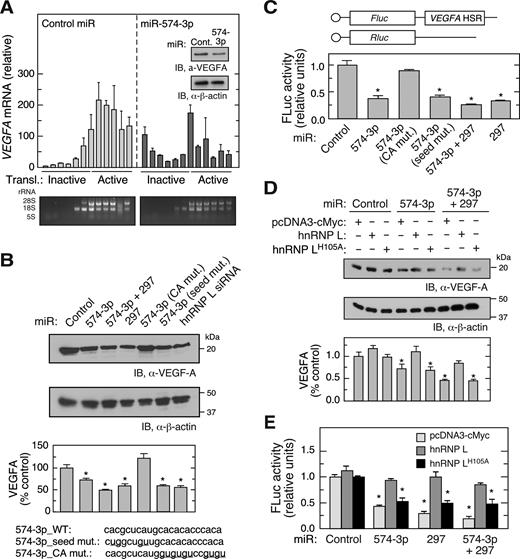
CARE-dependent inhibition of hnRNP L by miR-574-3p reduces VEGF-A expression in hypoxia. (A) Overexpression of miR-574-3p represses polysome loading of VEGFA mRNA in hypoxia. U937 cells were transfected with miR-574-3p or control miR (200 nM) for 48 h in hypoxia, and lysates subjected to sucrose density gradient fractionation. RNA isolated from each fraction was subjected to qRT-PCR to determine VEGFA mRNA distribution. Inset: VEGF-A expression was measured by immunoblot. (B) CARE is required for miR-574-3p-mediated inhibition of VEGF-A in hypoxia. U937 cells were transfected with wild-type and mutant miR-574-3p, miR-297, or hnRNP L siRNA (200 nM) as indicated. After 48 h, cell lysates were subjected to immunoblot analysis with anti-VEGF-A or -β-actin antibody, and quantitated by densitometry. (C) Overexpression of miR-574-3p inhibits translation of HSR-bearing reporter in hypoxia. FLuc reporter bearing VEGFA HSR was co-transfected into U937 cells with RLuc reporter and wild-type and mutant miR-574-3p, miR-297 or both for 48 h. FLuc activity was normalized by RLuc expression. (D) Overexpression of hnRNP L overcomes miR-574-3p-mediated repression of VEGF-A in hypoxia. U937 cells were co-transfected with pcDNA3-Myc-hnRNPL or the H105A mutant and with miR-574-3p or miR-297, or both, for 48 h in Hpx. Lysates were subjected to immunoblot analysis with anti-VEGF-A or -β-actin antibodies. (E) Overexpressed hnRNP L reverses miR-574-3p-mediated repression of HSR-bearing reporter in hypoxia. FLuc reporter bearing VEGFA HSR was co-transfected into U937 cells with miR-574-3p, miR-297, or both (or control miR) in the presence of pcDNA3-Myc-hnRNPL or its H105A mutant for 48 h in Hpx. FLuc was normalized by RLuc expression. In (A–E), data are presented as mean ± SD (n = 3; * P ≤ 0.05, Student's t-test).
To validate the decoy hypothesis and determine the sequence element within miR-574-3p that confers decoy activity, cells were transfected with pre-miR precursor mimetics of miR-574-3p, miR-574-3p mutants or miR-297, and VEGF-A was determined after 48 h in hypoxia. VEGF-A expression was significantly reduced by miR-574-3p (by ∼27%) or miR-297 (by ∼38%), and inhibited to a greater extent by combined transfection of both miRNAs (by ∼52% at half-dose of each) (Figure 3B). The decoy activity of miR-574-3p was abolished by several CA to GU mutations within the CARE; however, seed sequence mutation was ineffective. As a positive control, siRNA-mediated knockdown of hnRNP L reduced VEGF-A protein expression by a comparable amount. To establish the requirement for the VEGFA HSR, U937 cells were co-transfected with FLuc reporter bearing the VEGFA HSR, and with wild-type and mutant miR-574-3p. FLuc expression was repressed by at least 50% by wild-type and seed-mutant miR-574-3p, as well as by miR-297, but not by CA-mutant miR-574-3p (Figure 3C). Co-transfection of anti-miR-297 prevented miR-574-3p-directed translation repression of VEGFA mRNA (Supplementary Figure S3A) and HSR-bearing FLuc reporter mRNA (Supplementary Figure S3B). The expression of FLuc reporter containing the VEGFA HSR with mutant CARE did not respond to miR-574-3p and miR-297 (Supplementary Figure S3C). Consistent with loss-of-function assay in normoxia (Figure 1F), overexpression of miR-574-3p reduced hnRNP L binding of VEGFA mRNA in hypoxia (Supplementary Figure S3D). These data suggest that miR-574-3p renders CARE available for miR-297 binding and facilitates miR-297-RISC-mediated translational repression of VEGFA mRNA. As a cis-acting element, CARE is required for synergistic interaction between miR-574-3p and miR-297.
His105 in the hnRNP L RRM1 domain has been identified by crystal structure and mutagenesis analyses as a critical amino acid residue for binding to the CARE (33). Overexpression of wild-type hnRNP L antagonized the inhibitory effect of miR-574-3p, alone and in combination with miR-297, on VEGF-A expression in hypoxia, but the His105Ala mutant defective in CARE-binding was ineffective (Figure 3D). Similar results were observed in dual luciferase reporter assays, confirming that the VEGFA HSR contains the cis-acting element necessary and sufficient for control of VEGF-A expression by miR-574-3p and hnRNP L (Figure 3E).
miR-574-3p targets multiple hnRNP L RRMs and blocks VEGFA mRNA binding and HILDA complex assembly
We investigated the direct binding of miR-574-3p to hnRNP L by RNA electrophoresis mobility shift assay (EMSA). Recombinant hnRNP L formed stable complexes with radiolabeled miR-574-3p but not with GU-rich miR-574-5p (Figure 1B) or CARE mutant miR-574-3p (Figure 4A). By contrast, single site mutations within binding pockets of RRM2 (M232A) (42) and RRM3 (K410A) (33) both reduced the binding affinity of hnRNP L with miR-574-3p suggesting these two domains contribute to binding miR-574-3p (Figure 4A and D). An unrelated recombinant protein R1R2 from EPRS linker region (16) neither bound miR-574-3p directly nor competed with hnRNP L for binding (Supplementary Figure S4A). To determine which of the vicinal RRM pairs of hnRNP L are involved in miR-574-3p binding, we measured the affinity of recombinant domains with 5΄-terminal biotinylated miR-574-3p and VEGFA CARE using surface plasmon resonance (SPR). miR-574-3p bound both RRM 1,2 and RRM 3,4 with high affinity comparable to that of full-length hnRNP L (Table 1, Figure 4D). However, VEGFA CARE bound RRM 1,2, but not RRM 3,4, with high affinity, consistent with selective binding of VEGFA CARE by RRM 1,2 whereas RRM 3,4 serves as a docking site for DRBP76 to form the HILDA complex, consistent with a previous report (11). RRM 1,2 bearing the H105A mutation showed diminished binding to VEGFA CARE as well as miR-574-3p. Also, RRM 1 or RRM 2 by themselves failed to form a stable complex with miR-574-3p by EMSA showing a requirement for the vicinal pair (data not shown). As a negative control, binding of a 29-nt VEGFA GAIT element RNA to hnRNP L and its subdomains was undetectable.
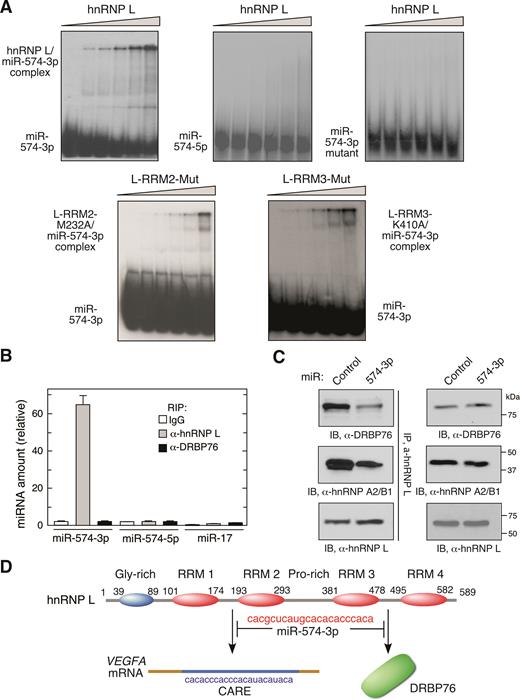
miR-574-3p binds multiple hnRNP L domains and inhibits binding to mRNA targets and HILDA complex formation. (A) hnRNP L and miR-574-3p form stable complexes. 32P-labeled miR-574-3p, miR-574-5p, or CARE mutant miR-574-3p were incubated with increasing amounts of recombinant hnRNP L or variants (0, 20, 50, 100, 200 and 500 nM), and RNA-protein complexes resolved by electrophoresis on a nondenaturing 5% polyacrylamide gel. (B) miR-574-3p-associated hnRNP L does not bind DRBP76 in hypoxia. U937 cells were cultured in hypoxia for 24 h. Lysates were IPed using anti-hnRNP L and -DRBP76 antibodies, and qRT-PCR was done with probes against miR-574-3p, miR-574-5p and miR-17. Data are presented as mean ± SD (n = 3, Student's t-test). (C) miR-574-3p blocks HILDA complex assembly. U937 cells were transfected with miR-574-3p or control miR, and lysates IPed with anti-hnRNP L antibody and immunoblotted using anti-DRBP76, -hnRNP A2/B1 and -hnRNP L antibodies. (D) Schematic depicting interaction of hnRNP L and miR-574-3p and its downstream consequences. miR-574-3p CARE interacts with RRM1,2 and RRM3,4, and prevents binding to VEGFA mRNA CARE and DRBP76, respectively.
KD for binding of hnRNP L and RRM domains to VEGF-A CARE and miR-574-3p
| Protein (or domain) | VEGF-A CARE (wild-type) KD (M) | miR-574-3p KD (M) | VEGF-A GAIT element KD (M) |
|---|---|---|---|
| hnRNP L | 8.5 ± 1.5 × 10−9 | 11.7 ± 3.6 × 10−9 | N.D. |
| RRM 1,2 | 7.6 ± 2.1 × 10−9 | 21.4 ± 9.3 × 10−9 | N.D. |
| RRM 1,2-H105A | 61.2 ± 18.6 × 10−9 | 91.0 ± 26.6 × 10−9 | N.D. |
| RRM 3,4 | 47.9 ± 8.7 × 10−9 | 18.1 ± 8.6 × 10−9 | N.D. |
| Protein (or domain) | VEGF-A CARE (wild-type) KD (M) | miR-574-3p KD (M) | VEGF-A GAIT element KD (M) |
|---|---|---|---|
| hnRNP L | 8.5 ± 1.5 × 10−9 | 11.7 ± 3.6 × 10−9 | N.D. |
| RRM 1,2 | 7.6 ± 2.1 × 10−9 | 21.4 ± 9.3 × 10−9 | N.D. |
| RRM 1,2-H105A | 61.2 ± 18.6 × 10−9 | 91.0 ± 26.6 × 10−9 | N.D. |
| RRM 3,4 | 47.9 ± 8.7 × 10−9 | 18.1 ± 8.6 × 10−9 | N.D. |
Biotinylated VEGF-A CARE, miR-574-3p and GAIT element RNA were immobilized on a streptavidin chip for SPR determination of binding of recombinant analyte protein. The KD was calculated using Biaevaluation software. N.D., not detected.
| Protein (or domain) | VEGF-A CARE (wild-type) KD (M) | miR-574-3p KD (M) | VEGF-A GAIT element KD (M) |
|---|---|---|---|
| hnRNP L | 8.5 ± 1.5 × 10−9 | 11.7 ± 3.6 × 10−9 | N.D. |
| RRM 1,2 | 7.6 ± 2.1 × 10−9 | 21.4 ± 9.3 × 10−9 | N.D. |
| RRM 1,2-H105A | 61.2 ± 18.6 × 10−9 | 91.0 ± 26.6 × 10−9 | N.D. |
| RRM 3,4 | 47.9 ± 8.7 × 10−9 | 18.1 ± 8.6 × 10−9 | N.D. |
| Protein (or domain) | VEGF-A CARE (wild-type) KD (M) | miR-574-3p KD (M) | VEGF-A GAIT element KD (M) |
|---|---|---|---|
| hnRNP L | 8.5 ± 1.5 × 10−9 | 11.7 ± 3.6 × 10−9 | N.D. |
| RRM 1,2 | 7.6 ± 2.1 × 10−9 | 21.4 ± 9.3 × 10−9 | N.D. |
| RRM 1,2-H105A | 61.2 ± 18.6 × 10−9 | 91.0 ± 26.6 × 10−9 | N.D. |
| RRM 3,4 | 47.9 ± 8.7 × 10−9 | 18.1 ± 8.6 × 10−9 | N.D. |
Biotinylated VEGF-A CARE, miR-574-3p and GAIT element RNA were immobilized on a streptavidin chip for SPR determination of binding of recombinant analyte protein. The KD was calculated using Biaevaluation software. N.D., not detected.
hnRNP L is joined by DRBP76 and hnRNP A2/B1 to form the HILDA complex that also binds the VEGFA mRNA HSR (Supplementary Figure S1A). We investigated whether DRBP76 binds the hnRNP L-miR-574-3p complex in vivo. A RIP experiment showed that miR-574-3p binds hnRNP L, but not DRBP76, suggesting that hnRNP L interaction with miR-574-3p does not require the HILDA complex (Figure 4B). We investigated whether miR-574-3p, by binding the hnRNP L component of the HILDA complex, inhibits formation of the complex. U937 cells were transfected with miR-574-3p mimics under hypoxic condition and subjected to co-IP and immunoblot analysis. Overexpression of miR-574-3p substantially reduced the interaction of hnRNP L with DRBP76 and hnRNP A2/B1 (Figure 4C). These results indicate that miR-574-3p binding to hnRNP L at both RRM pairs prevents both HILDA complex formation and interaction with VEGFA mRNA (Figure 4D).
Overexpression of miR-574-3p and miR-297 Inhibits myeloid cell proliferation and tumorigenesis
To explore the pathophysiological roles of miR-574-3p and hnRNP L, U937 monocytic leukemia cells were used as a model cell system to determine the effect of inactivation of hnRNP L by miR-574-3p. U937 cell growth was dramatically reduced by transfection of miR-574-3p mimics, and restored by co-transfection of hnRNP L-bearing expression plasmid (Figure 5A). Likewise cell proliferation, measured by MTT assay, was repressed by miR-574-3p and miR-297, and greater repression observed with both miRNAs in combination (at half-dose of each) (Figure 5B, upper panel). Wild-type hnRNP L reversed the miR-mediated inhibition of proliferation, but the hnRNP L mutant was less effective. Superimposed on their inhibitory effect on proliferation, cell apoptosis was enhanced by miR-574-3p and miR-297, and by both combined as indicated by caspase 3 activation (Figure 5B, lower panel; Supplementary Figure S4C). Similarly, anchorage-independent colony formation of hypoxia-treated U937 was reduced by miR-574-3p, miR-297 or combined miRNAs, and restored by co-expression of hnRNP L (Figure 5C). To test the influence of endogenous miR-574-3p on normoxic cell growth, U937 cells were incubated with anti-miR-574-3p and anti-miR-297, alone or in combination. Cell proliferation was moderately increased by inactivation of miR-574-3p, and increased to a greater extent by inactivation of both miRs (Supplementary Figure S4D). These experiments suggest that miR-574-3p might exhibit anti-tumorigenic activity against myeloid-derived tumors such as those generated following administration of U937 cells (43,44). U937 cells expressing miR-574-3p, with or without hnRNP L, were injected into both flanks of immunodeficient, nude mice, and tumor volume determined with external calipers. Compared to the control miR group, tumor growth was dramatically reduced by miR-574-3p overexpression; while co-expression of hnRNP L partially restored the tumor growth (Figure 5D). Overexpression of hnRNP L by itself did not significantly accelerate tumor formation. Together, these results suggest that miR-574-3p and hnRNP L antagonistically control tumor growth. Moreover, CA-rich miR-574-3p, possibly in combination with miR-297, might represent a potent therapeutic approach to repress myeloid cell-derived tumorigenesis.
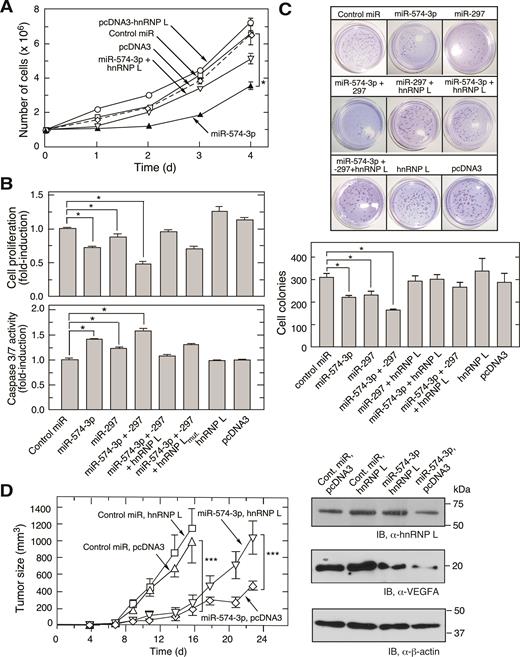
miR-574-3p inhibits proliferation and promotes apoptosis of U937 leukemia cells and suppresses tumorigenesis. (A) U937 cell proliferation is repressed by miR-574-3p and rescued by hnRNP L. U937 cells (1 × 106 cells) were transfected with miR-574-3p (or control miR, 50 nM), pcDNA3-Myc-hnRNP L (or empty vector, 500 ng), or both, for up to 4 days and cell number determined by hemocytometer. (B) Effects of miR-574-3p, miR-297, and hnRNP L on U937 cell proliferation and apoptosis. (Top) U937 cells (1 × 106 cells) were transfected with miRNAs (50 nM) as indicated in the presence or absence of hnRNP L or its mutant (500 ng). Cell proliferation rate was determined by MTT assay after 48 h transfection. (Bottom) Caspase 3/7 activity was measured using luminescent assay after 48 h. (C) miR-574-3p reduces anchor-independent U937 cell colony formation. U937 cells were transfected as in Figure 5B and seeded (5 × 103 cells) in 0.35% soft agarose gel containing RPMI with 10% serum. Cells were incubated for 2 weeks and stained with crystal violet and cell colonies imaged (above) and quantified (below). Data information: In (A–C), data are presented as mean ± SD (n = 3; *P ≤ 0.05, Student's t-test). (D) miR-574-3p represses tumor growth in mouse xenograft tumor model. miR-574-3p was transfected into U937 cells stably expressing hnRNP L (or pcDNA3 vector) for 48 h. Transfected cells (1 × 107 cells) were injected into flanks of nude mice, and tumor growth monitored for 23 days. Data are presented as mean ± SD (n = 20; *** P ≤ 0.001, two-way ANOVA).
DISCUSSION
Our results indicate that interplay between hnRNP L and miR-574-3p directs a dual regulatory mechanism that modulates both of their activities under physiological, pathological and therapeutic conditions determined by their functional stoichiometry (Figure 6). Under physiological, i.e. normoxic conditions, the bulk of hnRNP L is localized in the nucleus; thus, less abundant cytoplasmic hnRNP L is prevented by miR-574-3p from binding target VEGFA mRNA. We quantified miR-574-3p in the cytoplasm of U937 cells by RT-qPCR with synthetic miRNA as copy number standard, and hnRNP L by western blot with recombinant protein as standard. About 8000 copies of miR-574-3p (Supplementary Figure S1G) and 100 000 copies of hnRNP L (∼10% of 1.1 × 106 copies per cell) were determined in the cytoplasm per cell (Supplementary Figure S1H). The lower level of miR-574-3p and its subpopulation in hnRNP L-binding compared to that of entire cytoplasmic hnRNP L suggests a small sizable fraction of hnRNP L in binding of VEGFA mRNA besides its various binding partners and multiple functions in cytoplasm (45–47). Based on our results (Figures 1 and 2, Supplementary Figure S1), we assume that the quantity of miR-574-3p (and combined with the amount of miR-297) is comparable to the fraction of available cytoplasmic hnRNP L for binding VEGFA mRNA in normoxia. This allows the decoy activity of miR-574-3p to antagonize interaction of hnRNP L with VEGFA mRNA. Under pathological conditions, such as hypoxia, the cytoplasmic increase in phosphorylated hnRNP L overwhelms miR-574-3p, and impairs its canonical RISC-dependent gene silencing function. Moreover, binding of phospho-hnRNP L to the VEGFA CARE prevents binding of the negative regulator, miR-297, to the CARE, further amplifying VEGFA mRNA translation (14). In a potential therapeutic modality, ectopic introduction of an excess of miR-574-3p outcompetes CARE-containing VEGFA mRNA and interacts with hnRNP L, thereby permitting binding of miR-297 to the VEGFA CARE, preventing hnRNP L-directed VEGFA mRNA translation, and reducing tumorigenesis.
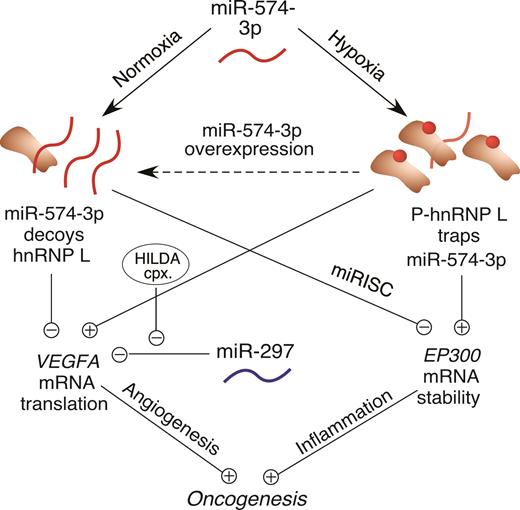
Schematic of interplay between hnRNP L and miR-574-3p and their cellular and oncogenic consequences.
miR-574-3p acts as an hnRNP L Decoy to suppress VEGFA mRNA translation
MicroRNAs, hnRNPs and target mRNAs are interconnected regulatory components forming a triangular network controlling mRNA translation. On one hand, hnRNPs compete with miRNAs for binding to mRNAs, thereby modulating the effect of the latter on translation or mRNA stability (14). On the other hand, miRNAs can directly bind hnRNPs by mimicking local primary sequence of target mRNAs, and acting as decoys to suppress their activity (26). miRNAs have been characterized as molecular ‘rheostats’ that tune the rate of tumor growth as either oncogenes or tumor suppressors (8). Under-expression of tumor suppressive miRNAs, or aberrant processing of their primary miRNAs, can increase cellular transformation and tumorigenesis (9,26,48). As one example in myeloid progenitor cells, highly expressed, C-rich miR-328 decoys the poly(rC)-binding protein hnRNP E2, releasing hnRNP E2 from the 5΄UTR C-rich element in the target, CEBPA, thereby inducing its translation and promoting myeloid cell differentiation (26). Importantly, loss of miR-328 in blast crisis chronic myelogenous leukemia (CML-BC) prevents myeloid cell differentiation. In this case, the altered expression of miR-328 can switch the translation inhibitory function of hnRNP E2 on or off. Here, we show that miR-574-3p exerts a two-pronged anti-tumoral activity by: (i) acting as an hnRNP L decoy, suppressing its binding to the 3΄UTR CA-rich element in VEGFA mRNA thereby reducing its translation, and (ii) canonical function in which the miRNA destabilizes oncogenic target mRNAs via the RISC pathway (e.g. EP300 and EGFR (30)) (Figure 6). Given the abundance of mRNAs containing 3΄UTR CA-rich regions, other oncogenic transcripts might be identified (in addition to VEGFA (14) and DGK-α (25)) that are activated by cytoplasmic hnRNP L-mediated RNA switch pathway and form a posttranscriptional regulon subject to translational co-regulation via hnRNP L-miR-574-3p interaction.
One unresolved issue in our proposed decoy model is the mechanism of the shuttling mechanism of miR-574-3p between Ago2 and hnRNP L. It is possible that hnRNP L binding with the 3΄-tail of RISC-associated miR-574-3p would displace the miRNA from RISC and this process is likely to be fast and transient as we did not observe stable interaction between hnRNP L and Ago2. Alternatively, TDP-43 (TAR DNA binding protein 43) has been reported to bind to the terminal loop of pre-miR-574 (49) and is a candidate factor that may recruit hnRNP L to interact with and protect miR-574-3p. A future investigation using an in vitro reconstitution cell lysate system with titrated factors (e.g., hnRNP L, TDP-43, etc.) will pursue the mechanism by which miR-574-3p is transferred to hnRNP L from RISC. The TaqMan miRNA RT-qPCR probe can sufficiently distinguish pre-miRNA and mature miRNA because the RT primer is in a stem-loop structure that inhibits hybridization of the RT primer to miRNA precursors and other long RNAs (50). Our northern blot data show that hnRNP L binds to single-stranded, mature miR-574-3p but not the double-stranded precursor miR-574, which is consistent with the sequence-specific binding property of hnRNP L for CA-rich single strand sequences. Though CARE is specific for hnRNP L in this work, we also tested the possibility that other RBPs with specificity for CAREs can also bind to miR-574-3p. As an example, the paralog protein of hnRNP L, hnRNP L-like, can bind to miR-574-3p (Supplementary Figure S4B) but the in vivo functional significance warrants further study depending on the cellular localization of hnRNP LL. By contrast, hnRNP L-binding protein in HILDA complex, hnRNP A2/B1 did not bind to miR-574-3p (Supplementary Figure S4B).
Recent studies resolving the crystal structures of hnRNP L RRM1 and RRM34 domains proposed a looping model for the interaction of hnRNP L with CA-rich splicing elements in pre-mRNAs (33). However, structural information addressing the interaction of full-length hnRNP L with its CARE target is lacking as is the interaction of CARE with other components of the HILDA complex. Our experiments show miR-574-3p interacts with two pairs of RNA-recognition domains, RRM1,2 and RRM3,4, but not with single RRM domains. Whereas RRM1,2 binds to the VEGFA CARE, RRM3,4 binds DRBP76 for assembly of the HILDA complex (11). miR-574-3p binding to RRM1,2 blocks hnRNP L binding to VEGFA CARE, and miR-574-3p binding to RRM3,4 prevents DRBP76 recruitment and HILDA complex assembly. The CA-rich domain in miR-574-3p is critical for its interaction with specific RRM domains of hnRNP L. Interestingly, two single nucleotide polymorphisms (SNPs) are present in the sequence of mature human miR-574-3p (Ensembl Genome Browser). The SNPs alter the CACA CARE motif in the 3΄-end of miR-574-3p to UACA or CAUA, possibly reducing the affinity of binding to hnRNP L thereby suppressing decoy activity (21). In summary, miRNAs can modulate both protein-RNA and protein-protein interactions through binding to multiple RBP domains. Thus, an important advantage of small miRNAs for decoying RBPs is their ability to interact with multiple RNA binding pockets, such as RRM domains, that enables muting multiple activities, notably RNA- and protein-binding capacities.
Inactivation of tumor suppressor miRNAs via trapping by RBPs
These studies reveal an unexpected, stress-responsive function of cytoplasmic hnRNP L in hypoxia, namely, trapping of CA-rich miR-574-3p. This interaction leads to inactivation of canonical RISC-dependent gene silencing activity, but also restoration of translation of downstream target mRNAs, such as EP300. Due to their reciprocal activities, the stoichiometric balance between cytoplasmic hnRNP L and miR-574-3p determines the net target mRNA translation. Increased cytoplasmic hnRNP L antagonizes miR-574-3p, suggesting a novel regulatory mechanism in which miRNA activity is suppressed, not by reduced expression, but rather by RBP-mediated sequestration. miR-574-3p is significantly down-regulated in colon, prostate, gastric, bladder and esophageal cancers compared to healthy tissues (28–32). In contrast, miR-574-3p is up-regulated in the serum of tumor patients with prostate, liver, and brain cancers (51–53). These observations suggest that altered expression of miR-574-3p might not be a major tumorigenic mechanism, but rather hnRNP L-mediated capture of miR-574-3p might be more significant. Our finding that hypoxia does not significantly alter the cytoplasmic level of miR-574-3p in U937 cells is consistent with this proposed mechanism. Anti-miR-574-3p treatment of U937 cells in normoxia triggered hnRNP L binding of VEGFA mRNA but the same treatment in hypoxia did not increase binding possibly due to saturated interaction of VEGFA mRNA by hnRNP L (Supplementary Figure S3E).
Tumor-suppressive activity of RBP-targeted miRNA
Consistent with its reported tumor suppressive activity, overexpression of miR-574-3p inhibits tumor cell proliferation, migration, and invasion, and promotes apoptosis in multiple cancers (28–30,32,39). Similarly, we showed that miR-574-3p repressed proliferation and induced apoptosis of U937 leukemia cells. We also showed that miR-574-3p reduced anchorage-independent colony formation of U937 cells and tumorigenesis in a U937 cell-dependent mouse xenograft model. Our findings are consistent with reports showing that agents that alter endogenous miR-574-3p levels in tumor cells regulate oncogenesis. For example, genistein, an anti-angiogenic and anti-tumoral phytoestrogen extracted from beans and coffee up-regulates miR-574-3p, represses VEGF-A expression and inhibits tumor progression in prostate (30). In contrast, TGF-β1 down-regulates miR-574-3p, induces VEGF-A expression, and promotes gastric cancer cell invasion and metastasis (32). These reports support our hypothesis of a potent negative regulatory activity of miR-574-3p reducing VEGFA mRNA translation, and consequent tumorigenesis. In addition, the anti-tumor activity of miR-574-3p relies on both the decoy activity of the 3΄ CA-rich element and the RISC silencing activity of the 5΄ seed region. Multiple downstream targets of hnRNP L and miR-574-3p seed region combined (in addition to VEGFA) are likely to contribute to overall pathogenic activity (Figure 6).
We have identified hnRNP L as a non-canonical protein target of miR-574-3p. Previous studies have suggested an oncogenic activity of the RNA-binding function of hnRNP L, and investigated the potential therapeutic efficacy of targeting hnRNP L. siRNA-mediated knockdown of hnRNP L reduced alternative splicing of caspase 9 pre-mRNA in non-small cell lung cancer cells, and inhibited lung cancer formation in an xenograft mouse model (54). However, dysfunctional splicing activity following T lymphocyte-specific conditional knockout of hnRNP L caused defective proliferation and chemotaxis of thymic pre-T cells (55). Thus, systemic depletion of hnRNP L, by inactivation of the nuclear splicing activity of hnRNP L, might compromise T-cell-mediated adaptive immunity. Our studies reveal that cytoplasmic hnRNP L, by targeting oncogenic mRNA switches, exhibits an alternate, nucleus-independent tumorigenic activity, and suggest that agents specifically targeting the cytoplasmic form might offer therapeutic efficacy with minimal adverse side-effects. miR-574-3p is one such agent that specifically targets cytoplasmic hnRNP L, providing RISC-dependent gene silencing and CARE-dependent decoy activities that synergistically contribute to anti-tumoral activity. Moreover, down-regulation of multiple oncogenic genes (e.g. VEGFA and DGKA, etc.) at the posttranscriptional level likely contributes to the tumor-suppressive activity of miR-574-3p. Current available small RNA therapeutics primarily block the expression of target mRNAs encoding pathogenic proteins (56). Our data reveal a new therapeutic activity of miRNAs, namely, targeting disease-causing RBPs. Our studies provide the founding example of a miRNA that, acting as an RNA decoy, regulates mRNA translation by synergistically promoting the RISC activity of another miRNA. This synergistic interplay is based on simultaneous targeting of CARE-binding proteins (i.e., hnRNP L) and CARE-bearing mRNAs (e.g. VEGFA mRNA) by miR-574-3p and miR-297, respectively. Combinatorial use of both hnRNP L-targeted miR-574-3p and CARE-targeted miR-297 maximizes repression of VEGFA (and possibly other oncogenic targets), providing a new avenue for achieving robust regulation of gene expression using multiple miRNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
Studies were supported by the Case Comprehensive Cancer Center Athymic Animal and Xenograft Core and NCI core grant P30 CA043703-23. We are grateful to Donna Driscoll and Paul DiCorleto for helpful discussions and Dalia Halawani for critical reading of the manuscript. We appreciate the assistance from Qiuqing Wang in statistical analysis. We acknowledge Xu Li for the generous gift of plasmids of pET28-hnRNPL-RRM12, pET28-hnRNPL-RRM12-H105A, and pET28-hnRNPL-RRM34. None of the authors have any financial conflict of interest with the information in this manuscript.
Author contributions: P.Y. designed and performed experiments, analyzed the data, wrote the initial draft of the manuscript, and contributed to all subsequent revisions. D.L. performed the tumor xenograft experiment. J.W. performed part of the experiments and helped read and edit the late drafts of the manuscript. P.L.F. supervised the project and contributed to manuscript and figure preparation.
FUNDING
National Institutes of Health [P01 HL029582, P01 HL076491, R01 GM086430, R01 GM115476 to P.L.F.] (in part); Scientist Development Grant from the American Heart Association (National Center) (to P.Y.); National Institutes of Health [R56 HL132899-01] (to P.Y.); start-up funds from Aab Cardiovascular Research Institute of University of Rochester Medical Center (to P.Y.). Funding for open access charge: American Heart Association Scientist Development Grant.
Conflict of interest statement. None declared.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments