





Abstract
Adenosine-to-inosine (A-to-I) RNA editing, catalyzed by Adenosine DeAminases acting on double-stranded RNA(dsRNA) (ADAR), occurs predominantly in the 3′ untranslated regions (3′UTRs) of spliced mRNA. Here we uncover an unanticipated link between ADARs (ADAR1 and ADAR2) and the expression of target genes undergoing extensive 3′UTR editing. Using METTL7A (Methyltransferase Like 7A), a novel tumor suppressor gene with multiple editing sites at its 3′UTR, we demonstrate that its expression could be repressed by ADARs beyond their RNA editing and double-stranded RNA (dsRNA) binding functions. ADARs interact with Dicer to augment the processing of pre-miR-27a to mature miR-27a. Consequently, mature miR-27a targets the METTL7A 3′UTR to repress its expression level. In sum, our study unveils that the extensive 3′UTR editing of METTL7A is merely a footprint of ADAR binding, and there are a subset of target genes that are equivalently regulated by ADAR1 and ADAR2 through their non-canonical RNA editing and dsRNA binding-independent functions, albeit maybe less common. The functional significance of ADARs is much more diverse than previously appreciated and this gene regulatory function of ADARs is most likely to be of high biological importance beyond the best-studied editing function. This non-editing side of ADARs opens another door to target cancer.
INTRODUCTION
Adenosine DeAminases acting on dsRNA (ADAR) are highly conserved family of enzymes catalysing adenosine to inosine deamination (A-to-I editing) (1,2). There are three ADAR proteins (ADAR1, ADAR2 and ADAR3) in human which all share a common modular structure characterized by two to three N-terminal dsRNA binding domains (dsRBDs) and a conserved C-terminal catalytic deaminase domain (3,4). Being the best-studied function associated with ADAR1 and ADAR2 (ADARs), A-to-I RNA editing contributes to multi-level gene regulation depending on where it occurs. ADAR3, which has no documented deaminase activity, is only reported in central nervous system (5). The Caenorhabditis elegans genome encodes 2 ADAR proteins, ADR-1 and ADR-2 (6), while Drosophila has a single Adar gene encoding a deaminase with two dsRBDs, similar to the mammalian ADAR2 (7). In coding regions, A-to-I RNA editing can lead to a codon change and the consequent alterations of protein-coding sequences since inosine is interpreted by the ribosome as guanosine (3). The differential editing frequencies of these recoding sites are found to impact on human diseases such as neurological disease and cancer (8–14). In non-coding regions, the vast majority of A-to-I RNA editing sites are in introns and repetitive Alu elements embedded in 3′ untranslated regions (3′UTRs) (15–17). The biological significance of editing within non-coding regions of RNA is still poorly understood. Previously described fates of mRNAs undergoing extensive A-to-I editing at their 3′UTRs are via RNA editing-dependent mechanisms including nuclear retention, nuclease-mediated degradation, and alteration of microRNA (miRNA) targeting (18–22), thereby influencing the expression of target genes.
ADARs have been found to be critical for normal development through and/or beyond A-to-I editing in different genetically modified animal models. Notably, the early post-natal lethality of the Adar2–/– mouse could be rescued by homozygous replacement of endogenous Gria2 (Glutamate Ionotropic Receptor AMPA Type Subunit 2) with a pre-edited allele, suggesting the editing activity of ADAR2 is essential for normal mouse development (23). Whether ADAR1 editing activity is similarly responsible for the embryonic lethality of Adar1–/– mouse is still being elucidated. Recent studies have shown that the embryonic lethality of Adar1–/– mouse might be resulted from the activation of innate immune responses (24–26). Mannion et al. have reported that dsRNAs containing multiple IU pairs (IU-dsRNAs), which resembled hyper-edited Alu dsRNAs, were found to inhibit the interferon pathway that is aberrantly activated in Adar1–/– mouse embryonic fibroblasts (25). Similarly, the embryos of editing-deficient knock-in mice (AdarE861A/E861A) died at ∼E13.5 due to the inability to hyper-edit long dsRNAs present in 3′UTRs of endogenous transcripts and the consequent activation of MAVS (Mitochondrial Antiviral Signalling Protein)-mediated interferon signalling, which might be relevant to Adar1–/– mouse phenotype (24). Besides these studies, a more recent study has identified MDA5-MAVS which is regulated by the p150 isoform of ADAR1, as the specific innate immune pathway responsible for both the dysregulation of interferon-stimulated genes (ISGs) and the embryonic lethality of Adar1–/– mice (26). However, in their study, whether these observed phenotypes are relevant to the editing or other non-editing activities of ADAR1 is still under investigation (26). Furthermore, the primary microRNA (pri-miRNA) cleavage by Drosha/DGCR8 complex was found to be inhibited by ADARs independent of their editing activities in both cell culture and the Drosophila models (27). Another very recent study by Anantharaman et al. has shown that the association of ADAR2 with RNA could stabilizes Cat2 transcribed nuclear RNA (Ctn RNA) by limiting the binding of HuR and PARN [Poly(A)-specific ribonuclease] in an editing-independent manner (28). Combining these editing-independent observations with the fact that an editing-incompetent ADAR family member is present in different species (e.g. ADAR3 in humans; Adar3 in mouse; ADR-1 in C. elegans) (6,29,30), it is most likely that ADARs can exert important functions as RNA binding proteins or through the formation of binding complexes with other proteins, beyond functioning as editing enzymes per se.
Hepatocellular carcinoma (HCC) is the most common type of liver malignancy, and accounts for approximately 700,000 cancer associated deaths per year worldwide (31). As reported previously, the deregulated RNA editing enzyme ADARs, as reflected by overexpression of ADAR1 and downregulation of ADAR2, occurs in >50% of patients with HCC (8), rendering a disrupted RNA editing balance in both coding and non-coding regions (8,9). Moreover, the recurrent editing of AZIN1 (antizyme inhibitor 1) which converts the codon 367 from serine to glycine has been demonstrated to predispose HCC development (9). However, most of A-to-I RNA editing occurs in the non-coding regions, and is enriched in 3′UTRs (32). The contributions of 3′UTR editing by ADARs to cancer development have not yet been fully illustrated. Moreover, whether major regulatory mechanisms of ADARs on the expression of target genes with promiscuously edited 3′ UTRs are independent or dependent of their RNA editing capability, remain to be further explored.
To this end, we carried out the first systematic analysis of A-to-I editing events within 3′UTRs using our previously published RNA-Sequencing (RNA-Seq) datasets of three matched pairs of primary hepatocellular carcinoma (HCC) tumors and their adjacent non-tumor (NT) liver specimens (8,9), followed by the evaluation of a direct link between RNA editing at 3′UTRs and the expression of target transcripts. Surprisingly, a majority of target pre-mRNA transcripts with extensive editing at their 3′UTRs were found to be regulated by ADARs independent of their deaminase and dsRNA binding functions, providing new insights that the multiple A-to-I editing at 3′UTRs might be merely a footprint of ADAR binding, which is dispensable for the regulation of target gene expression. As a target gene with multiple editing sites at its 3′UTR, METTL7A could be regulated through a non-canonical regulatory mechanism of ADARs in which ADARs act as a modulator of the RNAi machinery rather than RNA editing. We also found that the downregulation of METTL7A by ADARs has biological implication in cancer development.
MATERIALS AND METHODS
Cell lines
SNU-398, SNU-449, and HEK293T cell lines were purchased from the American Type Culture Collection (ATCC). Huh-7 was purchased from Japanese Collection of Research Bioresources Cell Bank (JCRB). SNU-398 and SNU-449 cells were cultured in Roswell Park Memorial Institute (RPMI) medium (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco BRL). Huh-7, SMMC-7721 and HEK293T cells were maintained in Dulbecco's Modified Eagle's medium (DMEM) supplemented with 10% FBS. All cell lines used in this study were regularly authenticated by morphological observation and tested for mycoplasma contamination (MycoAlert, Lonza Rockland, ME, USA). The cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Clinical samples
Total of 15 paired human primary HCC and adjacent NT liver tissues that were surgical removed and snap frozen in liquid nitrogen were obtained from the tissue repository of National University Hospital (NUH), Singapore. All patients gave written informed consent for the use of their clinical specimens for medical research. All samples used in this study were approved by the Institutional Review Board of National University of Singapore (NUS-IRB) (Reference Code: B-14-239E).
Publically available databases
Two publicly available datasets for survival data analysis of patients with HCC with respect to METTL7A expression: RNA-Seq datasets (including 370 HCC tumors and 50 adjacent NT liver samples) from TCGA (LIHC) (33) (https://tcga-data.nci.nih.gov/tcga/) and microarray gene expression datasets (including 81 HCC tumors and 80 adjacent NT liver samples) from GEO54236 (10) (http://www.ncbi.nlm.nih.gov/gds). Prior to the survival analysis, raw RNA-Seq counts were normalized using the total numbers of mappable reads across all samples, while the microarray data normalization was performed using the Cross-Correlation method (34). Normalized and log2 transformed METTL7A expression data was subjected to the survival analysis using Kaplan–Meier plots and log-rank tests. The median tumoral METTL7A expression was used to define METTL7A-high and METTL7A-low groups of patients with HCC for both datasets. The METTL7A expression in tumors and matched NT tissues were compared using the Mann–Whitney U test.
Luciferase reporter assay
The pmirGLO Dual-Luciferase expression vector was purchased from Promega (Promega Corporation, Madison, WI, USA). Full-length wild-type 3′UTRs of selected candidate genes (CCNYL1, TNFAIP8L1, MDMD2, METTL7A, MTDH and RBBP9) were amplified from normal human placental genomic DNA and cloned into pmirGLO immediately downstream of the firefly luciferase open reading frame. The third Alu-depleted mutant of METTL7A 3′UTR (ΔAlu) and the miR-27a seed mutant of METTL7A 3′UTR (Mut) were cloned from wild-type pmirGLO-METTL7A-3′UTR. Sequences of the cloning primers are listed in Supplementary Table S2.
The initial screening assay was performed in HEK293T cells by the co-transfection of pmirGLO-3′UTR reporter construct and ADAR1/2 expression construct generated by our previous study (9), at a mass ratio of 1:9 in 96-well plates. Luciferase activity was measured 48 h after transfection using the Dual-Glo® Luciferase Assay System (Promega, USA). Dose-dependent titration of the luciferase reporter assay was performed in HEK293T cells with 20 ng of pmirGLO-METTL7A-3′UTR and increasing amount of ADAR1/2 overexpression construct at mass ratios of 1:0.2, 1:0.4, 1:0.8, 1:1.6 and 1:3.2 forty-eight hours post-transfection, luciferase activity was measured as described above. Results were presented as relative firefly luciferase activity after normalization to internal control renilla luciferase.
Subgrouping of patients with HCC
We examined the protein expressions of ADARs in HCC tumor and their matched NT specimens by Western blot analysis. The ImageJ program (available at http://rsb.info.nih.gov/ij/) was used for densitometric analyses of western blots. The criteria for the classification of patients into two groups: ADAR1/2 high (n = 11) and ADAR1/2 low or normal (n = 4) are shown as follows:
Patients in the ADAR1/2 high group demonstrated higher protein expression levels (≥2-fold difference) of ADAR1 and ADAR2 in tumors than their matched NT samples (case nos. 1–3 and 6–10), or undetectable ADAR2 protein expression in both tumor and NT samples but higher level of ADAR1 in tumors than their matched NT samples (case nos. 4 and 11). Patients demonstrating lower (≥2-fold difference) or similar (<2-fold difference) protein expression levels of ADAR1 and ADAR2 in tumors than their matched NT samples (case nos. 12–14) or undetectable ADAR2 expression in both tumor and NT samples but lower or similar level of ADAR1 in tumors than their matched NT samples (case no. 15) were classified into ADAR1/2 low or normal group.
Cell fractionation assays
Nuclear and cytoplasmic fractions of HEK293T cells were isolated using Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's protocol. Briefly, 1 ml of cytoplasmic faction with approximately 5 mg of cytoplasmic proteins and 100 μl of nuclear fraction with ∼250 μg of nuclear proteins were obtained from HEK293T cells that were cultured in 15-cm dish with 80–90% confluence. To check the purity of each fraction, an equal amount (50 μg) of proteins were loaded for Western blot analysis using specific antibody against Fibrillarin (Abcam, ab4566), Lamin A/C (Abcam, ab40567), or α-tubulin (Santa Cruz Biotechnology, sc-5286).
Reciprocal pulldown assays
HEK293T cells that were cultured in 10-cm dish with 50–60% confluence were transfected with 8 μg of the FLAG-tagged wild-type or mutated ADAR1/2 plasmid, followed by the protein extraction 48 h post-transfection. For the treatment of RNase A prior to the immunoprecipitation, the total lysates were incubated with 1 μg/ml RNase A (20 mg/ml, Thermo Fisher Scientific, MA, USA) at 37 °C for 30 min. To pulldown FLAG-tagged protein, ∼5 mg of total cell lysates were immunoprecipitated with anti-FLAG M2 Magnetic Beads (Sigma-Aldrich) for 2 h at 4°C. In order to determine the binding of endogenous ADARs to Dicer, Huh-7 cells were analysed by reciprocal pulldown assays using the ADAR1, ADAR2 or Dicer-specific antibody. To pulldown Dicer, ADAR1 or ADAR2 protein from cell lysates of Huh-7 or pulldown Dicer from the transfected HEK293T cells, approximately 5 mg of total cell lysates were precleared with 5μg mouse IgG (Santa Cruz Biotechnology, sc-2025) and 40 μl Protein G Dynabeads (Invitrogen, CA, USA) on a rotating platform for 1 h at 4°C.
To conduct pulldown assays for different cell fractions, nuclear fractions, cytoplasmic fractions and total cell lysates were isolated from HEK293T cells, followed by reciprocal pulldown assays using the ADAR1, ADAR2 or Dicer-specific antibody. Approximately 5 mg of cytoplasmic proteins, 500 μg of nuclear proteins and 5 mg of total cell lysates were precleared as described above.
Precleared lysates were transferred to a new tube, and 5μg mouse IgG or the specific antibody against Dicer (Abcam, ab14601), ADAR1 (Abcam, ab88574) or ADAR2 (Sigma-Aldrich, SAB1405426) was added and incubated at 4°C for overnight, on a rotating platform. After an extensive washing in washing buffer (150 mM NaCl), the beads were boiled in 50 μl of 2 × Laemmli Sample Buffer (Bio-Rad, Hercules, CA, USA) and analyzed by Western blot analysis using antibodies to FLAG, Dicer, ADAR1 and ADAR2. We used 5% of the whole lysates (5% input) as a positive control.
Statistical analysis
The SPSS statistical package for Windows, version 16 (SPSS), and Microsoft Excel (Excel in Microsoft Office 2013 for Windows) were used for data analysis. The expression of METTL7A in tumors and matched NT tissues were compared using the Mann–Whitney U test. The relative editing frequencies of 10 representative editing sites at METTL7A 3′UTR in response to overexpression of the control or different ADARs in HEK293T cells were compared using the Mann–Whitney U test. Kaplan–Meier plots and log-rank tests were used for overall survival analysis. The unpaired, two-tailed Student's t test was used to compare the number of foci, colony formation, tumor volume and relative expressions of target genes, the luciferase activities between any two preselected groups. P < 0.05 was considered statistically significant.
RESULTS
Regulation by ADARs on the expression of target genes undergoing extensive A-To-I editing at 3′UTR sequences
Genes with extensive 3′UTR editing were selected based on our previously published RNA-sequencing (RNA-Seq) datasets (8), which have been generated from three paired primary HCC tumors and their adjacent NT liver specimens (Supplementary Data, Supplementary Figure S1). To investigate if there exists any functional interaction between ADARs and the edited 3′UTRs, we constructed a 3′UTR-luciferase reporter system (Luc-3′UTRs) containing full-length 3′UTR sequences of 6 selected genes (CCNYL1, TNFAIP8L1, MDM2, METTL7A, MTDH and RBBP9) (Supplementary Data, Supplementary Figures S1 and S2, and Supplementary Tables S1 and S2). Genetic structures of these six genes are illustrated in Figure 1A. Co-transfection of Luc-3′UTRs together with the ADAR1-p110 (the isoform responsible for pre-mRNA editing (15)) or ADAR2 expression constructs into HEK293T cells demonstrated that both ADAR1 and ADAR2 had suppressive effects on the luciferase activities linked to 3′UTRs of TNFAIP8L1 and METTL7A gene, and the luciferase activities associated with CCNYL1, MDM2 or RBBP9 3′ UTRs were only inhibited upon ADAR2 overexpression; however, neither ADAR1 nor ADAR2 demonstrated any effect on MTDH 3′UTR (Figure 1B and Supplementary Figure S3).
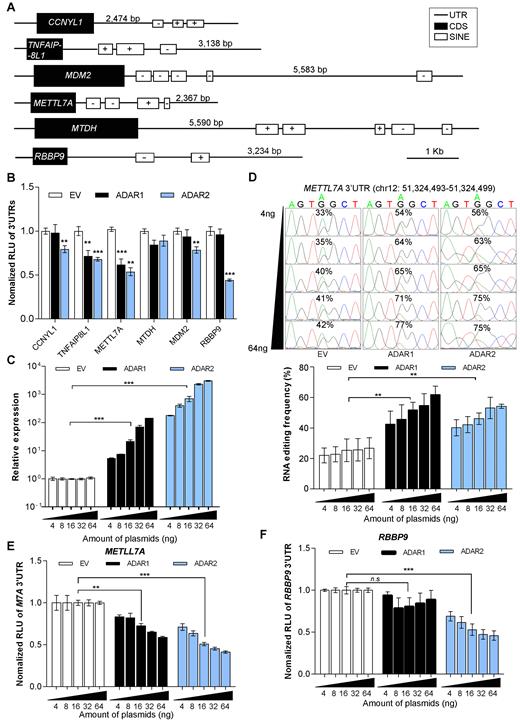
Regulatory effects of ADARs on target genes undergoing extensive 3′UTR editing. (A) Genetic structures of 6 selected genes. Black boxes represent the coding sequences (CDS), open boxes represent short interspersed elements (SINEs), and lines indicate untranslated regions (UTRs), using the RepeatMasker track of the UCSC genome browser (64). ‘+’ and ‘–’ signs indicate the strand specificity (sense or antisense, respectively) of SINEs. (B) Luciferase activities of pmirGLO-3′UTRs for the indicated genes were measured at 48 h post co-transfection with the indicated expression constructs into HEK293T cells. RLU, relative luminescence unit. EV, empty vector (C) Quantitative real-time PCR (qRT-PCR) analyses of ADARs expression 48 hours post co-transfection of pmirGLO-METTL7A-3′UTR construct with increasing amounts of ADARs expression constructs into HEK293T cells. (D) Sequence chromatograms of one editing site in METTL7A 3′UTR in response to the co-transfection as described in (C). From top to bottom, the amount of ADARs expression construct was gradually increased from 4 to 64 ng. Percentages denote the editing frequencies of the corresponding editing sites (top panel). The bar chart represents the average editing frequency of 10 editing sites in METTL7A 3′UTR (identified by our RNA-Seq (8)) in each group of cells (bottom panel). (E, F), Luciferase activity of pmirGLO-METTL7A-3′UTR (E) or pmirGLO-RBBP9–3′UTR (F) measured 48 h post co-transfection with increasing amounts of ADAR expression constructs into HEK293T cells, as described in (A). Data are presented as the mean ± S.E.M. of six replicates from a single experiment and representative of three independent experiments (B–F). Statistical significance is determined by unpaired, two-tailed Student's t test (B–F) (**P < 0.01; ***P<0.001; n.s., not significant).
Given the significantly reduced luciferase activities by ADAR1 and/or ADAR2, METTL7A and RBBP9 were selected for further investigation whether they are bona fide editing targets and more importantly, the suppressive effects of ADARs on their expression. In the light of the fact that a double stranded RNA (dsRNA) structure is essential for ADARs binding and A-to-I RNA editing (15), 3′UTR sequences of both METTL7A and RBBP9 were predicted to form long dsRNA secondary structures using CentroidFold (35) (Supplementary Figure S4). As expected, in HEK293T cells transfected with increasing amounts of either ADAR1 or ADAR2 expression construct, a dose-dependent reduction in luciferase activity of pmirGLO-METTL7A-3′UTR correlated negatively with the increase in the average editing frequency of 10 editing sites in the METTL7A 3′UTR (Figure 1C–E and Supplementary Figure S5A). As for RBBP9, we did observe a negative correlation between the luciferase activity of pmirGLO-RBBP9–3′UTR and the expression level of ADAR2 but not with ADAR1, in a dose-dependent manner (Figure 1F). Unexpectedly, none of 6 edited sites within the RBBP9 3′UTR demonstrated any increase in editing levels (Supplementary Figure S5A and B). Altogether, we have shown that the expression of METTL7A and RBBP9 could be regulated by ADAR1 and/or ADAR2; however whether this regulation is through an RNA editing-dependent or independent mechanism requires further investigation. Although we observed the significantly reduced 3′UTR-associated luciferase expression of CCNYL1 and TNFAIP8L1 upon ADAR1 or/and ADAR2 overexpression (Figure 1B), there was no obvious change in the editing frequencies of their 3′UTR editing sites (Supplementary Figure S5C). All these data suggest ADARs are most likely to regulate the expression of target genes undergoing extensive 3′UTR editing beyond their catalytic functions.
RNA editing/dsRNA binding-independent suppression of ADARs on target genes
To this end, we generated different ADARs mutants devoid of either the enzymatic activity (DeAD mutants) (36) or the dsRNA binding capability (EAA mutants) (37), which are both required for A-to-I RNA editing (Supplementary Materials and Methods, Supplementary Figure S6). Editing frequencies of 10 editing sites within 3′UTR of both endogenous and exogenous METLL7A were examined in cells transfected with either wild-type or mutant form of ADARs (Figure 2A and B; Supplementary Figure S7). Consistent with a previous report (37), ADARs DeAD mutants functioned as dominant negative forms, as they could compete for homodimerization with endogenous wild-type ADARs, rendering the wild-type partners inactive. As for ADARs EAA mutants, there was no significant difference in the basal editing level of METTL7A 3′UTR when compared to the control (ADAR1 EAA: P = 0.14; ADAR2 EAA: P = 0.47) (Figure 2A and B), which might be attributed to the loss of dimerization ability of EAA mutants (38). Consistent with the data described above (Figure 1B), luciferase activities of pmirGLO-METTL7A-3′UTR and pmirGLO-TNFAIP8L1–3′UTR could be suppressed by both wild-type and mutant forms of ADARs (Figure 2C and E). Luciferase activities linked to 3′UTRs of CCNYL1 and RBBP9 were only suppressed by different forms of ADAR2 in HEK293T cells (Figure 2D and F). Similar effects of different ADAR forms on luciferase activities of pmirGLO-METTL7A-3′UTR and pmirGLO-RBBP9–3′UTR were also observed in the HCC cell line SNU-398 (Supplementary Figure S8). To exclude the possibility that 3′UTR editing may lead to gene expression change, we deleted the third Alu element of METTL7A 3′UTR which is required for the formation of secondary structure (Supplementary Figures S4 and S9). Luciferase activities of wild-type METTL7A 3′UTR and the Alu-depleted (ΔAlu) mutant were suppressed by the overexpression of ADARs to a similar extent (Supplementary Figure S10). All these data strongly suggested a non-canonical regulatory function of ADARs on gene expression through 3′UTRs independent of their RNA editing and dsRNA binding capabilities.
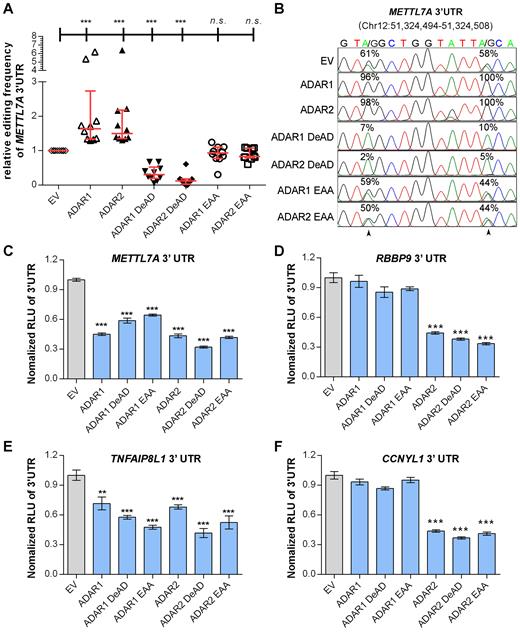
RNA editing/dsRNA binding-independent suppression of target genes by ADARs. (A) Scatter plots showing the relative editing frequencies of 10 editing sites at METTL7A 3′UTR in response to overexpression of different ADARs (wild-type: ADAR1 or ADAR2; DeAD mutant: ADAR1 DeAD or ADAR2 DeAD; dsRNA-binding mutant: ADAR1 EAA or ADAR2 EAA) in HEK293T cells. The data are presented with median (horizontal line) and interquartile range (error bar) for each group (Mann–Whitney U test; ***P < 0.001; n.s., not significant). (B) Sequence chromatograms of two editing sites within METTL7A 3′UTR. Percentages denote the editing frequencies of the corresponding editing sites indicated by arrowheads. (C–F) Luciferase activity of pmirGLO-METTL7A-3′UTR (C), pmirGLO-RBBP9–3′UTR (D), pmirGLO-TNFAIP8L1–3′UTR (E), or pmirGLO-CCNYL1–3′UTR (F) 48 h post co-transfection with the control empty vector (EV) or different ADARs constructs (wild-type and mutants) in HEK293T cells. Data are presented as the mean ± S.E.M. of six replicates from a single experiment and representative of three independent experiments (**P < 0.01; ***P < 0.001). Statistical significance is determined by unpaired, two-tailed Student's t test (C–F).
Suppression by ADARs on METTL7A expression in HCC cells and primary tumors
To demonstrate that our observations have biological implications in cancer development, beyond exogenous luciferase reporter assays, the METTL7A gene was selected for further investigation of ADARs-mediated effects on endogenous gene expression. Both wild-type, DeAD and EAA mutants inhibited endogenous METTL7A expression at both mRNA and protein levels in the HCC cell line Huh-7, which has the highest endogenous METTL7A expression among 9 HCC cell lines (Figure 3A and B; Supplementary Figure S11A). Conversely, specific shRNAs against ADAR1 or ADAR2 increased METTL7A expression in the HCC cell line SMMC7721, expressing relatively high levels of ADARs among nine cell lines (Figure 3C; Supplementary Figure S11B and C). Altogether, ADARs could exert RNA editing and dsRNA binding-independent suppression on METTL7A expression in cancer cells.
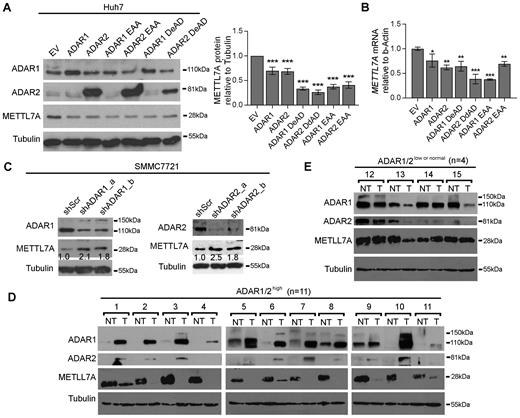
RNA editing/dsRNA binding-independent suppression by ADARs on METTL7A expression in HCC cells and primary tumors. (A) Western blot analyses of the indicated proteins in Huh-7 cells 72 h after transfection with the wild-type, DeAD or EAA mutants of ADARs (left panel). The bar chart represents the normalized densitometry unit of METTL7A protein on Western blot (right panel). (B) qRT-PCR analysis of METTL7A mRNA level measured in the same samples as described in (A). (C) Western blot analyses of the indicated proteins SMMC7721 cells 72 h post-transfection with shRNAs against either ADAR1 or ADAR2. Value indicates the normalized densitometry unit of METTL7A protein on western blot. (D and E) Western blot analyses of METTL7A, ADAR1 and ADAR2 proteins in two groups of HCC cases including ADAR1/2 high (D) and ADAR1/2low or normal (E). Tubulin is the loading control. T, primary HCC tumor; NT, matched non-tumor liver. Data are presented as the mean ± S.E.M. of six replicates from a single experiment and representative of three independent experiments (A, B). Statistical significance is determined by unpaired, two-tailed Student's t test (A, B). (**P < 0.01; ***P < 0.001).
To study if this non-canonical regulatory mechanism is of high biological importance during cancer development, we went on to examine ADARs-mediated suppression on METTL7A expression in 15 matched pairs of primary HCC tumor and their matched NT liver specimens. Based on the protein expression levels of ADAR1 and ADAR2 as detected by western blot analysis, patients with HCC were divided into 2 groups: ADAR1/2 high (n = 11) and ADAR1/2 low or normal (Materials and Methods). When compared to their matched NT specimens, all of ADAR1/2 high HCCs demonstrated either lower or absent expression of METTL7A (Figure 3D); while three out of four (75%) ADAR1/2 low or normal HCCs demonstrated an increase or no change in METTL7A expression (Figure 3E). This suggested the ADARs-mediated suppression may be a major mechanism of METTL7A downregulation in HCCs.
MiR-27a as a mediator between ADARs and METTL7A
In light of the fact that 3′UTR is the major miRNA targeting region, miRNAs are most likely to mediate this non-canonical regulation of ADARs on METTL7A expression. Editing-independent effects of ADARs on RNA interference (RNAi) and miRNA processing, which were first delineated in Drosophila (27), have been recently observed in mammalian systems (39).
In order to identify miRNAs targeting METTL7A, the METTL7A 3′UTR sequence was subjected to miRNA prediction using miRWalk across multiple algorithms (miRanda, miRDB, miRWalk, RNA22 and Targetscan) (40). Only miRNAs that can be predicted by at least two algorithms were selected for further investigation. As seen in Figure 4A, the introduction of miR-27a into HEK293T cells significantly repressed the luciferase activity of pmirGLO-METTL7A-3′UTR, indicating that miR-27a might truly target the METTL7A 3′UTR. Further, the luciferase activity of pmirGLO-METTL7A-3′UTR was found to be repressed by miR-27a, in a dose-dependent manner (Figure 4B). Subsequently, the pmirGLO-METTL7A-3′UTR mutant was constructed by mutating the seed region (indicated by box; Figure 4C) in the pmirGLO-METTL7A-3′UTR construct. Transfection of the pmirGLO-METTL7A-3′UTR mutant into HEK293T cells completely abolished the inhibition by miR-27a of the luciferase activity (Figure 4C), indicating a direct interaction between miR-27a and the METTL7A 3′UTR. Moreover, the expression of endogenous METTL7A protein was also found to be inhibited by miR-27a in Huh-7 cells (Figure 4D).
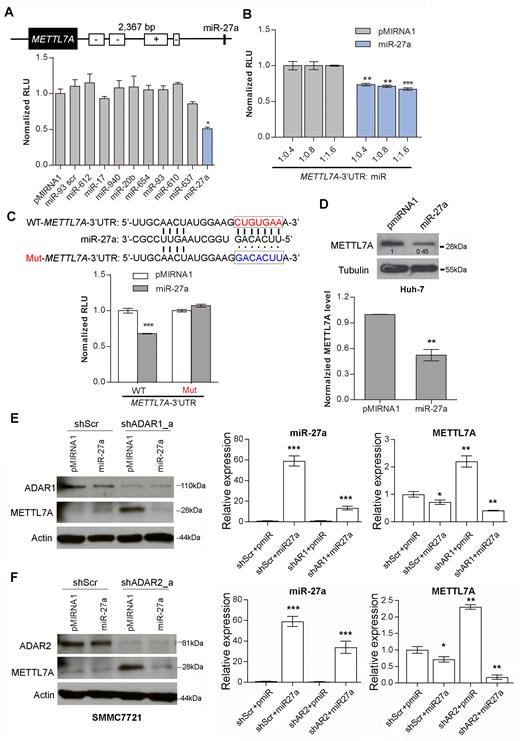
MiR-27a as a mediator between ADARs and METTL7A. (A) Schematic diagram demonstrates the predicted miR-27a targeting region on the METTL7A 3′UTR. The bar chart presents the luciferase activity of pmirGLO-METTL7A-3′UTR 48 h after co-transfection with the indicated miRNAs at a mass ratio of 1:9 in HEK293T cells. pMIRNA1, empty vector; miR-93 scr, scrambled control (scrambled mature miR-93 seed sequence). (B) Luciferase activity of pmirGLO-METTL7A-3′UTR measured 48 h post co-transfection with the increasing amount of miR-27a in HEK293T cells (**P < 0.01; ***P<0.001). (C) Schematic diagram showing the mutations being introduced to the predicted miR-27a target site of the METTL7A 3′UTR (top panel). WT, wild-type; Mut, mutant. The bar chart presents the luciferase activity associated with the wild-type or mutant pmirGLO-METTL7A 3′UTR 48 h after co-transfection with miR-27a in HEK293T cells (bottom panel). (D) Western blot analysis of METTL7A protein in Huh-7 cells stably overexpressing miR-27a. Value indicates the normalized densitometry unit of METTL7A protein on western blot. The bar chart presents the densitometry unit of METTL7A from 3 independent experiments. Tubulin was the loading control. (E) Left panel: Western blot analyses of the indicated proteins in SMMC7721 cells that were co-transfected with shRNA against ADAR1 (shADAR1_a) or scrambled shRNA (shScr) and pMIRNA1-miR-27a (miR-27a) or pMIRNA1 only (pmiR). Right panel: The qRT-PCR measurement of mature miR-27a and METTL7A in the same groups of cells described in left panel. (F) Left panel: Western blot analyses of the indicates proteins in SMMC7721 cells that are co-transfected with shRNA against ADAR2 (shADAR2_a) or shScr and miR-27a or pMIRNA1. Right panel: The qRT-PCR measurement of mature miR-27a and METTL7A in the same groups of cells described in left panel. Data are presented as the mean ± S.E.M. of six replicates from a single experiment and representative of three independent experiments (A-C, E: right panel; F: right panel). Statistical significance is determined by unpaired, two-tailed Student's t test (A–F) (*P < 0.05; **P < 0.01; ***P < 0.001).
If miR-27a is a key mediator for ADARs-mediated suppression of METTL7A, we assumed that the upregulation of METTL7A caused by the depletion of ADARs could be suppressed by the introduction of miR-27a into cells. To this end, shRNA against ADAR1 or ADAR2 together with pMIRNA1-miR-27a were co-transfected into SMMC7721 cells. As expected, the elevated mRNA and protein expression of METTL7A in ADAR1/2-knockdown cells was found to be obviously repressed by miR-27a, but not the pMIRNA1 control (Figure 4E and F; Supplementary Figure S12A), suggesting that the ADARs-mediated suppression of METTL7A is most likely to be mediated by miR-27a. Besides, editing in 3′UTR of METLL7A was dramatically repressed in ADAR1/2-knockdown cells (Supplementary Figure S12B and C), which did not affect the suppression of METTL7A expression by miR-27a, indicating that editing of the 3′UTR of METLL7A was also not required for its silencing by miR-27a.
Moreover, we further confirmed that there was no A-to-I editing occurring in mature miR-27a, primary miR-27a (pri-miR-27a) and its targeted METTL7A 3′UTR sequences, ruling out the possibility that the impact of ADARs on METTL7A expression was via the enhancement of miR-27a:METTL7A mRNA interaction arising from the A-to-I editing of miR-27a and/or its target METTL7A 3′UTR sequence (Supplementary Figure S13).
ADARs interact with dicer to promote miR-27a expression independent of RNA editing/dsRNA binding activities
To elucidate the mechanism by which miR-27a functions as a mediator of ADARs-mediated suppression of METTL7A, we investigated whether the expression of miR-27a could be affected by different forms of ADARs. Indeed, mature miR-27a was found to be upregulated upon overexpression of wild-type, DeAD or EAA mutants of ADARs in Huh-7 cells (Figure 5A). We went on to study which stage of miR-27a biosynthesis could be targeted by ADARs. There was no obvious increase in the expression of miR-27a precursors, pri-miR-27a, and pre-miR-27a, upon the overexpression of wild-type, DeAD or EAA mutant (Figure 5B and C). Further, Northern blot analysis for miR-27a confirmed that the introduction of both wild-type and EAA forms of ADARs could process pre-miR-27a to mature miR-27a more efficiently than the control (Supplementary Figure S14). All these observations indicated that processing of pre-miR-27a to mature miR-27a is likely to be regulated by ADARs independent of RNA editing and dsRNA binding.
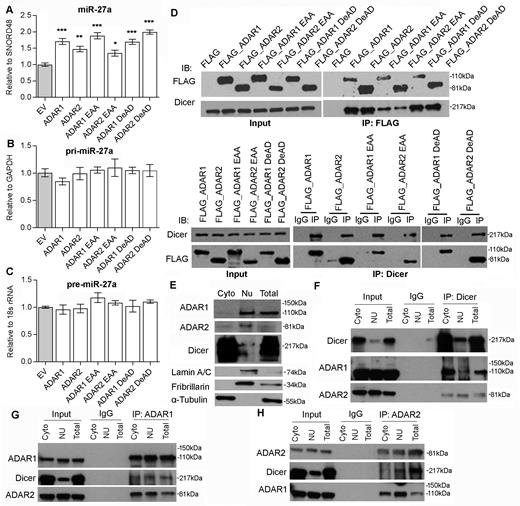
ADARs interact with Dicer to promote miR-27a expression independent of RNA editing/dsRNA binding activities. (A–C) The qRT-PCR measurement of mature miR-27a (A), pri-miR-27a (B), and pre-miR-27a (C) in Huh-7 cells upon overexpression of empty vector (EV) or different ADARs expression constructs including the wild-type, DeAD and EAA mutants. Data are presented as the mean ± s.e.m. of triplicates from a single experiment and representative of 3 independent experiments (*P < 0.05; **P < 0.01; ***P < 0.001). Statistical significance is determined by unpaired, two-tailed Student's t test. (D) Reciprocal immunoprecipitation (IP) of ADARs and Dicer is performed in HEK293T cells overexpressing FLAG only or FLAG-tagged wild-type and mutant form of ADARs, using anti-FLAG antibody conjugated magnetic beads (IP: FLAG; top panel), or Dicer-specific antibody (IP: Dicer; bottom panel) or mouse IgG (IP: IgG; bottom panel). Western blot analyses of the indicated proteins in FLAG, Dicer or mouse IgG-IPed products are conducted. (E) The purity of cytoplasmic (Cyto) and nuclear (Nu) fractions of HEK293T is verified by western blot analyses of ADAR1, ADAR2, Dicer, Lamin A/C, Fibrillarin and α-tubulin. (F–H) Reciprocal IP of endogenous ADARs and Dicer is conducted in the cytoplasmic (Cyto) and nuclear (Nu) fractions of HEK293T cells using an Dicer (F), ADAR1 (G), ADAR2 (H)-specific antibody (IP: ADAR1, ADAR2 or Dicer) or mouse IgG (IP: IgG). Western blot analyses of the indicated proteins in ADAR1, ADAR2, Dicer or mouse IgG-IPed products are conducted. Input indicates 5% of each fraction or total lysates from HEK293T cells. Total lysates (Total) are included in the experiments describe in (E–H) as positive controls.
Processing of pre-miRNAs to miRNAs is catalysed by Dicer in complex with TRBP (Tar RNA binding protein) (41). Also, it has been recently reported that ADAR1 promote pre-miRNA processing via binding to Dicer (39). Therefore, we hypothesized that both ADARs promote miR-27a processing and expression through interacting with Dicer. As expected, endogenous Dicer was detected in FLAG-tagged wild-type, DeAD or EAA mutant pull-down products (Figure 5D). Reciprocally, FLAG-tagged wild-type ADARs, DeAD or EAA mutants could also be detected in anti-Dicer immunoprecipitates (Figure 5D). Despite that the EAA mutants were found to lose significant binding affinity to Dicer, they could still demonstrate the similar effect to the wild-type and DeAD forms of ADARs on the expression of miR-27a (Figure 5A). To further confirm the less binding affinities of EAA mutants to Dicer have no effect on the expression of miR-27a, the wild-type, DeAD, or EAA forms of ADAR1/2 was transfected into cells in a dose-dependent manner, the expression of mature miR-27a was found to be gradually increased dose-dependently (Supplementary Figure S15), suggesting that although the dsRNA interaction may be involved in the binding of ADARs to Dicer, it is most likely to be indispensable for augmenting miR-27 processing by ADARs. Further, the reciprocal pull-down assays indicated endogenous Dicer could interact with both endogenous ADAR1 and ADAR2 in Huh-7 cells (Supplementary Figure S16). As pre-miRNAs are exported to the cytoplasm and processed by Dicer to mature miRNAs, we further investigated whether the interaction of Dicer to ADAR1/2 indeed occurs in the cytoplasm. As expected, the binding of Dicer to ADAR1/2 was detected in both cytoplasmic and nuclear fractions (Figure 5E–H). The observed interaction between ADAR1 and ADAR2, known to form heterodimer in human cells (42,43), serves as positive controls.
Taken together, ADARs were found to augment the processing of pre-miR-27a to mature miR-27a via binding to Dicer, in turn targeting the METTL7A 3′UTR and decreasing METTL7A expression.
METTL7A as a novel tumor suppressor in HCC
Having identified a non-canonical function of ADARs beyond their catalytic and dsRNA-binding capabilities, we sought to demonstrate that the altered expressions of target genes has a biological impact on cancer development. METTL7A expression was analyzed using RNA-Seq datasets from the Cancer Genome Atlas (TCGA) project (33) and a microarray gene expression dataset from the NCBI Gene Expression Omnibus (GEO) database (GEO542361) (10). Both datasets confirmed significantly decreased expression of METTL7A in HCC tumors compared to adjacent NT tissues, and also predicted shorter overall survival for patients with HCC (Figure 6A, B). We further analysed the expression level of each of two ADAR genes individually in METTL7A-high and METTL7A-low groups of patients using the TCGA dataset. The METTL7A-low patients demonstrated the significantly higher expression level of ADAR1 (P < 0.0001; Supplementary Figure S17). Although there was no statistical significance, we could still observe the METTL7A-low patients had the higher expression level of ADAR2 (P = 0.1353; Supplementary Figure S17), which might be due to the fact that the METTL7A expression was inhibited to a greater extent by ADAR1 than ADAR2 which was much less abundantly expressed than ADAR1 in patient samples.
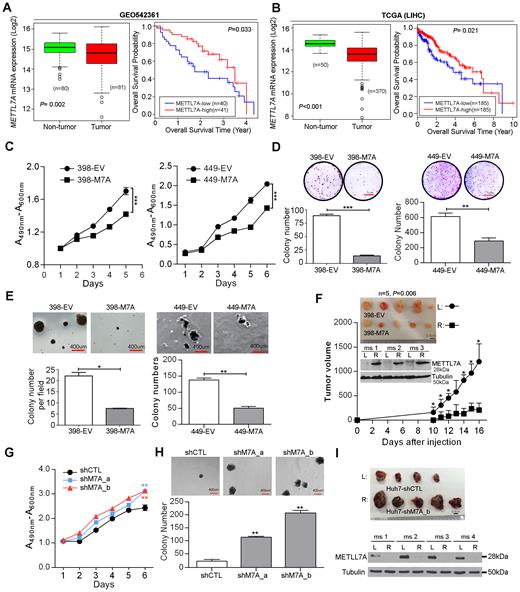
METTL7A is a novel tumor suppressor in HCC. (A, B) Box plots showing METTL7A expression in HCC and matched NT liver specimens, as retrieved from the GEO54236 microarray gene expression datasets in (A), from TCGA (Liver Hepatocellular carcinoma, LIHC) RNA-Seq datasets in (B). The data are presented as box plots with median (horizontal line), 25–75% (box) and 5–95% (error bar) percentiles for each group and the open dots indicate the outliers (Mann–Whitney U test). Kaplan–Meier plots for the overall survival rate of patients with HCC in the group with high (METTL7A-high: n = 41 for GEO54236; n = 185 for LIHC) or low METTL7A expression (METTL7A-low: n = 40 for GEO54236; n = 185 for LIHC) which was categorized based on the median expression of METTL7A in HCCs. The P value is calculated by log rank test. (C) 2,3-bis-(2-Methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay showing growth rates of the indicated stable cell lines. A490nm and A600nm, absorbance at 490 and 600 nm, respectively (***P < 0.001). (D) Quantification of foci formation induced by the indicated stable cell lines (**P < 0.01; ***P < 0.001). Scale bar, 1 cm. (E) Quantification of soft agar colonies induced stable cell lines (*P < 0.05; **P < 0.01). Scale bar, 400 μm. (F) Tumors derived from the indicated cell lines 3 weeks after subcutaneous injection (n = 5 mice per group, top panel). Scale bar, 0.5 cm. Western blot analysis of METTL7A protein in tumors developed in three representative mice at end point (middle panel). Growth curves of tumors derived from the indicated stable cell lines over the observation period (bottom panel). Data are presented as the mean ± S.E.M. *P < 0.05, determined by unpaired, two-tailed Student's t test. (G) XTT assay showing growth rates of the stably knockdown cell lines. A490nm and A600nm, absorbance at 490 and 600 nm, respectively (**P < 0.01). (H) Quantification of soft agar colonies induced stably knockdown cell lines (**P < 0.01). Scale bar, 400 μm. (I) Top panel shows the xenograft tumors derived from the indicated stable cell lines 4 weeks post subcutaneous injection (n = 5 mice per group). Scale bar, 0.5 cm. Western blot analysis of METTL7A protein in tumors developed in four representative mice at end point (bottom panel). Tubulin was the loading control. All data are shown as the mean ± S.E.M. of triplicate wells with the same experiment and representative of three independent experiments (C, D, E, G, H), and statistical significance is determined by unpaired, two-tailed Student's t test (C–H).
To further investigate the role of METLL7A in HCC development, three HCC cell lines (SNU-398, SNU-449 and Huh-7) were selected for establishing overexpression and knockdown cell models and the subsequent functional studies (Supplementary Figure S18A and B). As detected by cell culture assays predictive of tumorigenicity, overexpression of METTL7A in both SNU-449 and SNU-398 cells (398-M7A and 449-M7A) significantly reduced the cell viability, frequency of focus formation, and the number of colonies formed in soft agar when compared to cells transduced with empty vector lentiviruses (398-EV and 449-EV) (Figure 6C–E). Moreover, xenograft tumors derived from 398-M7A cells, with overexpression of METTL7A, grew much slower than tumors derived from 398-EV cells during a 3-week observation period (Figure 6F). Conversely, specific shRNAs-mediated silencing of METTL7A augmented the tumorigenicity of Huh-7 cells, as indicated by the increased cell viability and higher frequency of colony formation in soft agar (Figure 6G, H and Supplementary Figure S18C). As seen in Figure 6I, xenograft tumors derived from stably knockdown cells (Huh7-shM7A_b) grew much more aggressively than tumors derived from control cells (Huh7-shCTL), as a consequence of the persistent silencing of METTL7A during a 4-week observation period (Figure 6I). All these data suggest that METTL7A downregulation, possibly mainly due to the RNA editing/binding-independent suppression by ADARs, is closely linked to HCC development.
DISCUSSION
Extensive A-to-I editing at 3′UTR sequences has been associated with different fates of the edited pre-mRNA transcripts. Due to the distinct base pairing preference between adenosine and inosine, RNA editing at 3′UTRs can regulate the stability of target transcripts by creating or eliminating miRNA targeting sites (44,45). Alternatively, the promiscuously edited 3′UTRs can recruit inosine-specific nucleases, e.g. the RNA-induced silencing complex (RISC) subunit Tudor-SN and human endonuclease V, leading to mRNA degradation (19,20). Certain transcripts with edited 3′UTRs were shown to be retained in the nucleus by an inosine-specific nuclear protein complex composed of P54nrb, splicing factor PSF, and matrin-3 (3,46). All of the above reported fates of inosine-containing RNAs highlighted the importance of inosine in either changing the base-pairing properties or recruiting inosine-specific protein complexes, thereby affecting the translational efficiency of the edited mRNAs. In contrast, a previous study in C. elegans reported no significant difference in expression of mRNAs with structured and edited 3′UTRs across different adr strains (47). Furthermore, mRNAs with edited 3′UTRs have been demonstrated to associate with translating polyribosomes in both C. elegans and human HeLa cells, indicating that not all the edited mRNAs are retained in the nucleus (47). Thus, a relevant question is whether RNA editing is necessary to induce the observed effect of ADARs on 3′UTRs?
Surprisingly, using different ADAR mutants which are devoid of either RNA editing or dsRNA binding capability, we found the promiscuous A-to-I editing within 3′UTRs of transcripts is most likely to be a footprint of ADARs binding, which is not required for the manipulation of target gene expression, at least for a subset of targets. Relatively few protein variants produced by RNA editing have been demonstrated to be causative of biological phenotypes observed in animal models with genetically modified ADARs suggesting that editing-independent functions of ADARs might also be of importance in the observed phenotypes (48). Furthermore, a recent study suggested that the ADAR1 binding to non-Alu regions could affect 3′UTR usage through competing with 3′UTR-binding factors, such as cleavage and polyadenylation-relevant proteins (49), thereby influencing the expression of target genes. More importantly, the impact of ADAR1 on 3′UTR usage is only dependent on RNA editing for certain 3′UTRs, but others could be affected by ADAR1 in an editing-independent manner. In this study, five out of the six selected genes carrying multiple editing sites at 3′UTRs were found to be regulated by ADAR1 and/or ADAR2 beyond their deaminase and dsRNA binding functions. Therefore, the functional significance of ADARs is much more diverse than previously appreciated and this gene regulatory function of ADARs may be of biological importance beyond their better-studied editing function.
Until now, gene regulatory mechanisms of ADARs have not been clearly understood. Given the fact that ADAR-mediated A-to-I RNA editing competes for shared dsRNA substrates with RNAi machinery, interactions between ADARs and RNAi pathway have gained intensive attention during the past decade (50). Effects of hyper-editing in protecting dsRNA from entering RNAi were first confirmed by biochemical assays, and further observed in C. elegans with adr-1/adr-2 double mutant models (51,52). Editing of primary miRNAs has been reported to inhibit miRNA biosynthesis at multiple stages, e.g. Drosha and Dicer cleavage, the loading to RISC, or redirecting silencing targets (18,53–56). In contrast to antagonistic effects of ADARs on the RNAi pathway, a recent study on the impact of ADAR1 on pri-miRNA processing in the nucleus suggested that ADAR1 might enhance pri-mRNA processing via its interaction with the pri-mRNAs prior to Drosha/DGCR8 binding, which may not rely on RNA editing (49). This finding complements the previous report that the interaction between ADAR1 and RISC component proteins revealed an editing-independent agonistic effect of ADAR1 on miRNA processing, by forming a complex with Dicer in the cytoplasm (39). MiR-27a is known to be an oncogenic microRNA and is highly expressed in numerous types of cancer, including breast (57), ovarian (58), liver (59) and gastric (60). However, mechanisms that may plausibly account for altered expression of miR-27a in tumors are not well understood. At genomic DNA level, the amplification was only found to upregulate miR-27a expression in gastric cancer cells (61). At epigenetics level, SNPs (single nucleotide polymorphisms) and methylation have been reported to be associated with the regulation of miR-27a expression. In HCC, He et al. found that hypomethylation contributes to aberrant miR-27a expression (59). Besides, the A→G change of rs895819 could shorten the stem-loop structure and affect the processing of miR-27a and increase the gastric cancer risk (62). While the other functional SNP rs11671784 G→A variation in miR-27a interrupts the expression of miR-27a, which decreases chemo-sensitivity of bladder cancer and increase the target RUNX-1 expression (63). Here, our mechanistic studies, adding a novel mechanism in which miR-27a is upregulated in cancer cells, indicated that the interaction of either ADAR1 or ADAR2 with Dicer augments the processing from pre-miR-27a to mature miR-27a and subsequent miR-27a targeting of the METTL7A 3′UTR, leading to reduced expression of METTL7A.
Thus, our study represents the first report that both ADAR1 and ADAR2 can augment miRNA processing from pre-miRNAs to mature miRNAs through interacting with Dicer, independent of RNA editing and dsRNA binding capabilities of ADARs. Nevertheless, the interaction between Dicer and ADARs may be mediated by RNA molecules, as RNase A treatment of cell lysates prior to immunoprecipitation resulted in the dramatically decreased binding of Dicer to ADAR1/2 (Supplementary Figure S19). As reported by our previous studies (9), ADAR1 and ADAR2 were significantly upregulated and downregulated in HCC, respectively. Besides, the expression of ADAR1 was ∼30-fold higher than that of ADAR2 in tumors. All these findings could explain although ADAR1 and ADAR2 have opposing expression patterns in HCC and have the same inhibitory effect on METTL7A expression via enhancing miR-27a processing, the expression of METTL7A was still dramatically decreased in tumors. Therefore, it is likely that although maybe less common, there are a subset of genes that are regulated by ADARs via non-canonical editing/dsRNA binding-independent mechanism(s), such as CCNYL1, MDM2, TNFAIP8L1 and RBBP9, which have been identified in this study. However, for other target genes, the precise regulatory mechanism(s) of ADARs will need to be examined on a case-by-case basis. Moreover, as the 3′UTR sequences often carry binding sites for specific proteins that affect the export, localization, stability and translation of mRNAs, further investigation will be required to determine whether the editing of 3′UTRs may be substantially involved in these processes that are important for regulating genes in normal cells or in other conditions.
The next question is whether this mechanism may have important functional relevance in cancer development. As a target gene undergoing 3′UTR editing, METTL7A was identified to be a tumor suppressor in HCC. Moreover, the survival analysis of two individual HCC cohorts indicated the tumoral downregulation of METTL7A predicted poor prognosis of patients with HCC. Unexpectedly, even with multiple editing sites in the 3′UTR, the expression of METTL7A was found to be regulated by ADARs through miRNA expression modulation in an RNA editing and RNA binding-independent manner. Also, we found that the tumoral downregulation of METTL7A was not associated with the genomic loss of METLL7A gene which is located at chromosome 12q13.12 (Supplementary Figure S20). Given the widespread effects of miRNA targeting, METTL7A will likely not be the only target influenced by ADARs under this mechanism. Recent studies reported the expression of miRNAs is globally inhibited in adar1−/− mouse embryos, in turn altering the expression of miRNA target genes (39). Also, in human U87MG cells, a gene otology (GO) analysis of target genes of ADAR1-affected miRNA yields a number of genes functionally enriched in pathways related to cell proliferation, growth, apoptosis and cellular response to stimuli or DNA damage (49).
Our study revealed that the functional essentiality/importance of ADARs is likely to stem from its involvement in processes other than RNA editing and dsRNA binding alone, at least for a subset of target genes relevant to cancer development. As we and other have demonstrated, the enhancement of miRNA processing via the interaction between ADARs and Dicer (39) or Drosha (49) may function as a major mechanism responsible for gene regulatory function of ADARs beyond their deaminase and RNA binding activities. Disruption of the interaction between ADARs and Dicer or Drosha might represent a targetable approach for rescuing expression of tumor suppressors such as METTL7A as a novel therapeutic modality in HCC and other cancers.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank and acknowledge the patients for tumor tissue donation and Dr J. Yan (Cancer Science Institute of Singapore, National University of Singapore, Singapore) for providing the pMIRNA1 lenti-miR expression vector.
Author contributions: L.Q., Y.S., L.C. and D.G.T. initiated the study. L.Q., Y.S. and L.C. wrote the manuscript with inputs from T.H.M.C., H.Q., J.J.P.M., D.G.T., and J.S.L., L.Q., Y.S. and L.C. designed the experiments. L.Q., Y.S. and L.C. performed all experiments with assistance from T.H.M.C., H.Q., L.C., J.S.L., D.J.T.T., S.J.T, X.X.H and V.N. C.H.L., K.T.T. and H.Y. performed all bioinformatics analyses of RNA Sequencing datasets. H.Y., L.Q., Y.S. and L.C. contributed to data acquisition, analysis and interpretation. L.C. and D.G.T. supervised the project.
FUNDING
National Research Foundation Singapore; Singapore Ministry of Education under its Research Centers of Excellence initiative; NMRC Clinician Scientist—Individual Research Grant New Investigator Grant (CS-IRG NIG) [NMRC/CNIG/1117/2014]; NMRC Clinician Scientist-Individual Research Grant (CS-IRG) [NMRC/CIRG/1412/2014]; NUS Young Investigator Award (NUS YIA) [NUSYIA_FY14_P22]; Yong Siew Yoon Research Grant (Tier 2; National University Cancer Institute, Singapore), NUS Start-up fund [NUHSRO/2015/095/SU/01]; Singapore Ministry of Health's National Medical Research Council under its Singapore Translational Research (STaR) Investigator Award; RNA Biology Center at the Cancer Science Institute of Singapore, NUS, as part of funding under the Singapore Ministry of Education's Tier 3 grants [MOE2014-T3-1-006]. Funding for open access charge: Singapore Ministry of Education’s Tier 3 grant [MOE2014-T3-1-006] and NMRC Clinician Scientist-Individual Research Grant New Investigator Grant (CS-IRG NIG) [NMRC/CNIG/1117/2014].
Conflict of interest statement. None declared.
REFERENCES
Author notes
These authors contributed equally to this work as first authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments