





Abstract
Proper chromosome segregation is crucial for preserving genomic integrity, and errors in this process cause chromosome mis-segregation, which may contribute to cancer development. Sister chromatid separation is triggered by Separase, an evolutionary conserved protease that cleaves the cohesin complex, allowing the dissolution of sister chromatid cohesion. Here we provide evidence that Separase participates in genomic stability maintenance by controlling replication fork speed. We found that Separase interacted with the replication licensing factors MCM2–7, and genome-wide data showed that Separase co-localized with MCM complex and cohesin. Unexpectedly, the depletion of Separase increased the fork velocity about 1.5-fold and caused a strong acetylation of cohesin's SMC3 subunit and altered checkpoint response. Notably, Separase silencing triggered genomic instability in both HeLa and human primary fibroblast cells. Our results show a novel mechanism for fork progression mediated by Separase and thus the basis for genomic instability associated with tumorigenesis.
INTRODUCTION
Cohesion between sister chromatids is essential for ensuring that chromosomes are distributed correctly to daughter cells. In eukaryotes, sister chromatid cohesion is mediated by an evolutionarily conserved complex called cohesin whose SMC1A and SMC3, belonging to the Structural Maintenance of Chromosomes protein family and RAD21 (a single α kleisin) form a tripartite ring structure topologically encircling sister chromatids, according to an embrace model. Finally, the fourth subunit, STAG, associates with RAD21 (1). Cohesin loading, mediated by NIPBL, occurs in G1 in yeast or at the end of telophase of the previous cell cycle in mammalian cells. The presence of ESCO factors allows cohesin establishment in the S phase whereas PDS5 proteins ensure its maintenance. Once loaded into the cohesin ring, the interaction of DNA with SMC3 head domain stimulates ATPase activity at the SMC3-SMC1A interface, causing the opening of the cohesin ring. This process permits WAPL to open the SMC3-RAD21 interface, fully releasing the DNA (2). On the other hand, lysine acetylation of the SMC subunits is critical for proper SMC1A and SMC3 heads’ engagement and for cohesin association with the chromosomes thus preventing ring opening (3). The removal of cohesin from chromosomes is closely regulated by a set of cohesin interactors. The bulk of cohesin is removed from chromosome arms following the phosphorylation of RAD21 and STAG subunits by Polo-like kinase 1 during prophase and prometaphase (4,5). Only small amounts of cohesin remain on chromosomes, preferentially at centromeres. Centromeric-cohesin is indeed protected by specific proteins, such as the SGO1–PP2A complex, which keeps cohesin in a hypophosphorylated state and maintains centromeric cohesion (6–9). At the metaphase–anaphase transition, the remaining cohesion is dissolved by the endopeptidase Separase, which cleaves the cohesin's RAD21 subunit. This cleavage permits opening of the cohesin ring, causing it to dissociate from chromosomes (10). Separase is activated by the proteolysis of its inhibitory molecular partner Securin and the simultaneous degradation of CDK1’s subunit cyclin B. This process is mediated by a ubiquitin protein ligase called Anaphase Promoting Complex (APC) and its cofactor Cdc20 (11,12).
Beyond sister chromatid cohesion, the cohesin pathway is an emerging player in many biological processes. Cohesin is a major contributor to interphase genome organization through the formation of chromatin loops. Cohesin regulates gene expression through long-range interactions with regulatory elements associated with CTCF (13–16) or with enhancers and promoters (17–19), it organizes DNA replication factories, and facilitates gene recombination (20,21). Mutations in core cohesin and cohesin-regulatory genes are responsible for rare human diseases, collectively called cohesinopathies (22). It is worth noting that cohesinopathy cells display many markers of genome instability, as well as aneuploidy, chromosome aberrations, micronucleus formation, and sensitivity to genotoxic drug treatments (23,24). Mutations in genes that regulate sister chromatid cohesion have also been identified in human cancers, including colorectal carcinoma and myeloid neoplasms, characterized by genome destabilization (25–29). These observations further support the notion that the cohesin pathway plays a role in preserving genome stability.
Given the importance of the metaphase–anaphase transition, maintenance of genomic stability is ensured through the concerted action of many cohesin proteins, and in this regard Separase plays a key role. Separase knockout results in embryonic lethality (30,31) whereas its depletion by small interfering RNA (siRNA) causes chromosome missegregation and genomic instability in fission yeast, mouse and human cells (30,32–36). Moreover, Separase dysregulation has been shown to cause mitotic spindle defects, premature sister chromatid separation and lagging chromosomes (37–39). Here, we describe a novel role for Separase, which appears to be engaged in the regulation of replication fork speed and in maintaining genome integrity. We provide evidence that Separase works together with MCM proteins and cohesin. Down-regulation of Separase results in speeding up fork replication progression. In addition, Separase silencing leads to chromosome missegregation and unexpectedly, to structural aberrations in both primary human fibroblasts and HeLa cells. Our data show a novel mechanism for fork progression, mediated by Separase engagement. Loss of this regulatory arrangement leads to increased DNA damage and may contribute to genomic instability found in cancer cells.
MATERIALS AND METHODS
Cell culture
Both HeLa and normal human primary fibroblast cells (established as previously described (40), were grown in Dulbecco's minimal essential medium supplemented with 10% fetal calf serum and antibiotics in a humidified 5% CO2 atmosphere.
siRNA treatment
Smart pool siRNA and four different siRNAs against Separase, and mock siRNA were purchased from Dharmacon. The target sequences were siRNA1 CCGAGGAUCACUUGAAAUA, siRNA2 GGAGAAGGCUCACAGUUAC, siRNA3 GAUCGUUUCCUAUACAGUA, siRNA4 GGAACGAAUUCUCUUUGUC. Both HeLa cells and human fibroblasts (at 40–60% confluence) were transfected with 20 nM Separase siRNA by using Oligofectamine Reagent. After 24 h in the presence of siRNA, cells were harvested and processed for further analyses (Supplementary Figure S1A and B).
Chk1 transfection
HeLa cells (at 40–60% confluence) were transfected with a vector overexpressing Chk1 by means of TurboFect transfection kit (Thermo Scientific) according to the manufacturer's instructions.
Aphidicolin treatment
HeLa cells were treated with 0.1 μM aphidicolin for 15 h (Supplementary Figure S1C and D).
Cytogenetic analysis
Metaphase spreads were prepared under standard conditions. Colcemid was added to the cultures for 60 min, followed by a 30-min incubation in 0.075 M KCl hypotonic solution at 37°C and multiple changes of Carnoy's fixative. One hundred metaphases were analyzed. Chromosome aneuploidy, micronuclei and aberrations were visualized by staining slides in Giemsa stain and detected by direct visualization using the Leica DM2500 microscope.
Antibodies
Antibodies used in this study are as follows: Actin (Bethyl), Chk1 (Bethyl), pS345-Chk1 (Abcam), Chk2 (Bethyl), MCM2 (Bethyl), MCM4 (Bethyl), MCM6 (Bethyl), MCM7 (Bethyl), RPA2 (Bethyl), Separase (Bethyl), SMC1A (Bethyl), acetylated-SMC3 (MBL) and SMC3(Bethyl).
Immunoprecipitation
To perform immunoprecipitation experiments, a volume containing 800 μg of total protein extracts from HeLa cells was dissolved in 1 mL of incubation buffer (5 M NaCl, 50 mM Tris–HCl pH 8, 0.5 M EDTA, protease inhibitor cocktail). The solution were precleared with 20 μl Dynabeads protein A (Invitrogen) for 1 h. The supernatants were then incubated overnight at 4°C with 5 μg of Separase or MCM2 antibody coupled to the 40 μl Dynabeads protein A. Samples were boiled in sample buffer and separated by SDS-PAGE.
Western blotting
Whole protein extracts were resuspended with lysis buffer and protein concentration was estimated by the Bradford Protein Assay (Thermo Scientific). Proteins, 50 μg per lane, were separated by SDS-PAGE. Proteins were transferred to nitrocellulose membranes (Amersham) and incubated with the primary antibody, Chk1, pS345-Chk1, Chk2, MCM2, MCM4, MCM6, MCM7, RPA2, Separase, SMC1A, acetylated-SMC3 and SMC3. Membranes were incubated with secondary antibody-peroxidase conjugate (Sigma), processed for detection by chemiluminescence (Amersham) and imaged on Biomax film (Kodak). Actin antibody was used as loading control.
Flow cytometry
Control, aphidicolin- and siRNA-treated cells were fixed in 70% ethanol at 4°C, then treated with 1μg/ml RNaseA (Sigma) at 37°C for 20 min, stained with 5 μg/ml propidium iodide (Sigma) and analyzed for DNA content (25 000 cells/sample) with a FACScalibur flow cytometer (Becton Dickinson).
Genome-wide localization of Separase, MCM2 and SMC1A-cohesin by ChIP-sequencing
ChIP was performed in HeLa cells with antibodies against Separase, MCM2 and SMC1A as previously described (41). At first, cells were crosslinked with 1% formaldehyde for 15 min and quenched with 125 mM glycine. Pellets of cells were incubated with lysis buffer 1 (50 mM HEPES–KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40 and 0.25% Triton X-100), then lysis buffer 2 (10 mM Tris–HCl, 100 mM NaCl and 1 mM EDTA) and sonicated in lysis buffer 3 (10 mM Tris–HCl, 100 mM NaCl, 1mM EDTA, 0.1% sodium deoxycolate and 0.5 sarkosyl). Each sample was incubated with Dynabeads protein A (Invitrogen) previously bound with 10 μg of Separase, MCM2, SMC1A antibody or negative control rabbit IgG (Sigma). Beads were washed with low salt buffer (20 mM Tris–HCl, pH 8, 150 mM NaCl, 0.5 mM EDTA, 0.1% SDS, 1% Triton X-100), high salt buffer (20 mM Tris–HCl, pH 8, 500 mM NaCl, 0.5 mM EDTA, 0.1% SDS, 1% Triton X-100) and RIPA buffer (50 mM HEPES–KOH, pH 7.5, 0.5 mM EDTA, 10% NP-40, 0.7% sodium deoxycolate and 0.5 M LiCl) and eluted overnight at 65°C. The eluates were incubated with proteinase K and the purificate with QIAquick Purification Kit (Qiagen).
Library preparation and sequencing
DNA was then processed as previously described (41,42). DNA libraries were validated and processed with Illumina cBot for cluster generation on the flowcell, following the manufacturer's instructions. The libraries were sequenced on single-end 50 bp mode on HiSeq2500 (Illumina) at a depth of ∼30–50 million sequences per sample. CASAVA 1.8.2 version (Illumina pipeline) was used to processed raw data for both format conversion and de-multiplexing.
ChIP-seq bioinformatics analysis
ChIP-seq bioinformatics analysis was performed as previously described (41,42). Briefly, FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used for the quality control of raw sequence files. The quality-checked reads were mapped to the human reference genome GRCh37/hg19. Only uniquely mapping reads were used for the peak calling by MACS2 (43) with 0.05 FDR used as a cut-off value. MACS2 was also used for all comparisons between input track as control and each one of the data sets as treatment. Custom UNIX shell scripting was used to obtain binding site overlaps between different sample sets. Genome-wide analysis of enrichment of chromosomal features was determined using CEAS package (44). A co-localization was defined by an overlap of the regions by at least one base. ChIP-seq data were validated by qPCR. Each sample was run in duplicate and repeated at least three times. Corresponding primers of the selected regions are described in Supplementary Table S1. Fold enrichment of MCM2, Separase and SMC1A was calculated relative to Input in four genomic regions on chromosomes 3, 4, 8 and 9 in which MCM2, Separase and SMC1A co-localize. All NGS raw files have been deposited into NCBI Sequence Read Archive under accession number SRP078978.
Cohesin ChIP-qPCR in unsynchronized and synchronized cells
Briefly, 107–108 unsynchronized and synchronized in S-phase cells were used to perform cohesin-ChIP according to a previously described protocol (41). At least three independent ChIP assays were carried out and analyzed by qPCR. Each sample was run in duplicate and repeated at least three times. Corresponding primers of the selected genes are described in Supplementary Table S2.
Proteomic analysis
Proteins co-purified with Separase or control IgG were separated on a 1D-gel NuPAGE 4–12% (Novex, Invitrogen), run in morpholinepropanesulfoninic acid (MOPS) buffer, and stained with the Colloidal Blue Staining kit (Invitrogen). Whole gel lanes were cut into 10 sequential slices. Each band was subjected to cysteine reduction with DTT and alkylation by iodoacetamide, and finally digested with trypsin as described (45). Peptide mixtures were analyzed by nanoflow reversed-phase liquid chromatography tandem mass spectrometry, using an Ultimate 3000 HPLC (DIONEX) connected on line with a linear Ion Trap (LTQ, Thermo). After desalting in a trap column (Acclaim PepMap 100 C18, DIONEX), the peptides were separated in a 10-cm-long silica capillary (Silica Tips FS 360–75-8, New Objective), packed in-house with 5 μm, 200 Å pore size C18 resin (Michrom BioResources). Peptide elution was obtained with a 40 min linear gradient from 5% to 60% acetonitrile, in the presence of 0.1% formic acid. Analyses were performed in positive ion mode (1.7–1.8 kV HV) and data-dependent acquisition allowed fragmentation of the five most abundant ions. Tandem mass spectra were searched against the Swiss-Prot human database (20 121 sequences, 11 321 258 residues) using Proteome Discoverer software (version 1.4, Thermo Electron): peptide spectral matches (PSM) were filtered using Percolator based on q-values, at a 0.01 FDR (high confidence). Searches allowed for specific trypsin hydrolysis, with two possible miss cleavages. Proteins identified with at least two PSMs were taken into account. Network and KEGG pathway enrichment (FDR < 0.05) analyses were performed by the STRING tool (version 2.0), using experimental and co-expression data as active interaction sources and adopting the high-confidence interaction score. Cluster analysis was visualized with Cytoscape (version 2.8.2), using the MCL plugin.
Molecular combing
Exponentially growing HeLa cells were labelled with 50 μM 5-Iodo-2′-deoxyuridine (IdU, Sigma Aldrich) and 100 μM 5-chloro-2′-deoxyuridine (CldU, Sigma Aldrich) by sequential pulses of 30 min each. In order to investigate the effect of Separase depletion on replication fork speed, cells were treated with siRNA against Separase for 24 h and labelled during the last hour of siRNA treatment (Supplementary Figure S1B). Cells were harvested and immobilized into agarose plugs (150 000 cells/plug), incubated overnight at 53°C with 2 mg/ml proteinase K solution (1% N-laurosylsarcosine, 0.1 M EDTA pH 8, 0.01 M Tris–HCl pH 8, 0.02 M NaCl) and stored at +4°C in EDTA 0.5 M pH 8. To isolate genomic DNA at high molecular weight two plugs were digested with 3 U of β-agarase I (New England Biolabs) in 0.1 M MES, pH 6.1 at 42°C. DNA stretching was performed on silanized surfaces (Genomic Vision S.A.) according to our validated protocol (46,47). Preparations were denatured (15 min in 1 M NaCl, 50 mM NaOH), rinsed in 0.01 M Tris–HCl pH 7.6 and replication tracks were immunodetected by a 30 min incubation step with a mix of anti-BrdU antibodies from Becton Dickinson (developed in mouse, 2:7) and from Abcam (developed in rat, 1:40) which crossreact with IdU and CldU respectively. Slides were rinsed with PBS pH 7.4 (3 × 5 min) before being incubated for 30 min with a mix of AlexaFluor 488-conjugated anti-mouse IgG and AlexaFluor 594-conjugated anti-rat IgG (Life Technologies, 1:50). After washing at the above conditions, counterstaining of the stretched DNA molecules was performed by three successive incubation/washing steps: mouse IgG2A anti-ssDNA (Millipore, Clone 3034, 1:25 dilution, 90 min), AlexaFluor 350-conjugated anti-mouse IgG2A (developed in goat, Life Technologies, 1:50 dilution, 45 min), AlexaFluor 350-conjugated anti goat IgG (developed in donkey, Life Technologies, 1:50 dilution, 30 min). A summary of the procedure for labelling and immunodetection is shown in Figure 3B. Preparations were scored under a motorized fluorescence microscope (Zeiss Axio Imager.M1) equipped with a CCD camera (Photometrix, Coolsnap HQ2) and a 40× oil immersion objective (N.A. = 1.30). Images were acquired from adjacent microscope fields at the different wavelentgths, merged and aligned using Adobe Photoshop CS2. Analysis of fluorescent patterns were done using the Metavue Research Imaging System (Molecular Devices), following the stringent criteria described in details elsewhere (47).
Statistical analysis
Molecular combing data were analyzed by the Kruskal-Wallis test, a non-parametric method used for comparing independent samples of equal or different sample sizes. All other data were analyzed by Student's t-test or the chi-squared test. P-values of <0.05 were considered statistically significant.
RESULTS
Separase interacts with replisome and cohesin
To date securine and cohesin-RAD21 are the only well-established interactors for Separase. However, the observations that Separase is involved in centriole disengagement and telomere stability (39,48) suggest that it might play novel roles, possibly through interaction with additional proteins. We therefore sought to identify molecular partners of Separase using immunoprecipitation followed by mass spectrometry (MS) analyses. Eighty proteins were identified as co-precipitating with Separase (FDR < 0.01, Supplementary Table S3). All identified proteins were grouped according to their prevalent biological function, as described in the KEGG pathway database (Figure 1A). Though proteomic data suggest that Separase may be involved in several pathways, here we focused on processes related to its interaction with DNA. Strikingly, proteins belonging to replisome assembly such as MCM (Mini Chromosome Maintenance) 4, MCM6 and MCM7, SMC1A, RPA1 and RPA2 figured in this pathway (Figure 1B and C, Supplementary Table S3). The interactions between Separase, MCM proteins, SMC1A and RPA2 were confirmed by immunoprecipitation and immunoblot assays (Figure 2A and B). Time course analysis showed that 24 h of siRNA treatment was effective in silencing Separase expression (Supplementary Figure S2A). It is worth noting that these interactions disappeared following Separase depletion (Supplementary Figure S2B), further supporting the notion that Separase interacts with replisome proteins.
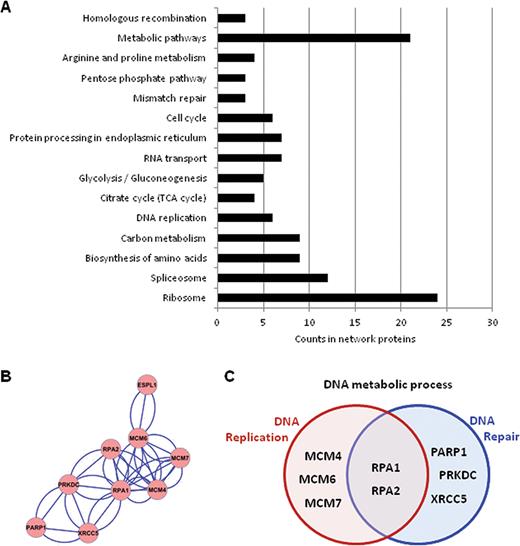
Analysis of proteins co-purified with Separase identified by MS in two replicated experiments. (A) KEGG pathway enrichment (FDR < 0.05) of protein network analyzed by STRING showed enrichment for proteins important for metabolic pathways, ribosome, RNA splicing. (B) Protein–protein interaction clustered by Cytoscape MCL plugin. (C) KEGG pathway enrichment of proteins shown in the cluster in panel (B).
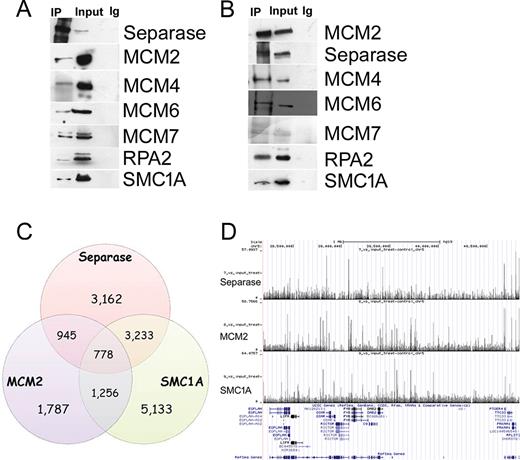
Separase physically interacts with MCM2–7 complex and co-localizes with cohesin and MCM2. (A) Separase immunoprecipitate (IP) tested by western blot for MCM2–7 proteins, SMC1A and RPA2. (B) MCM2 IP tested by western blot for MCM4–7 proteins, SMC1A and RPA2. No signal was detected in the IPs using IgG-coated beads. Note that of the antibodies available for the MCM proteins, only that for MCM2 worked properly in co-IP experiments (data not shown). (C) Venn diagram showing the overlap between Separase, MCM2 and SMC1A as determined by ChIP-seq. (D) Genomic binding of Separase subunit at a selected region of chromosome 5. The cohesin's SMC1A subunit and MCM2 binding profiles are also shown. This region was chosen to illustrate the typical binding patterns throughout the human genome of Separase, MCM2 and cohesin.
Next, we aimed to determine the genome-wide distribution of Separase, MCM2 and cohesin in HeLa cells. For this, we used chromatin immunoprecipitation (ChIP) followed by massively parallel DNA sequencing (ChIP-seq) with antibodies against Separase, MCM2 and the cohesin's SMC1A subunit. Genome-wide analysis of the sequenced tags defined 8118, 4776 and 10 400 occupied regions for Separase, MCM2 and SMC1A (P-value of 10−3 and FDR ≤ 0.05) respectively. A total of 778 Separase sites (9.5%) overlap MCM2 and cohesin (Figure 2C and D). Their association with transcriptional elements was verified by the cis-regulatory element annotation system (CEAS). Data showed that all three proteins bound preferentially to the promoter regions (Supplementary Figure S3A–C). ChIP-seq data validation was performed by RT-PCR (Supplementary Figure S3D). We looked next at their genome-wide distribution in cells synchronized in S-phase by aphidicolin treatment for 15 h (Supplementary Figure S1C). We found 2169, 20 704 and 23 426 sites for Separase, MCM2 and SMC1A (Supplementary Figure S4, P-value of 10−3 and FDR ≤ 0.05) respectively and again, all three proteins bound preferentially to the promoters (Supplementary Figure S5). Separase shares 650 (29.9%) interaction sites with both MCM2 and SMC1A (Supplementary Figure S4). Furthermore, at variance with unsynchronized cells, the remaining Separase sites were engaged with either SMC1A or MCM2. Unsynchronized and synchronized cells shared 9% of Separase, MCM2 and SMC1A sites and most of them bound promoter regions (data not shown). Together, these observations indicate that Separase co-localizes with MCM2–7 complex and cohesin preferentially at gene promoter regions.
Separase depletion speeds up replication fork involving SMC3 acetylation and checkpoint response
The finding that Separase interacts with replisome and cohesin prompted us to attain a more comprehensive understanding of the replication role of Separase. The effect of Separase silencing on DNA replication was analyzed by molecular combing in HeLa cells. Fork rate, inter-origin distances, organization and size of the replication clusters, as well as the proportion of unidirectional, paused/arrested and asynchronous forks, were evaluated 24 h after the start of siRNA treatment (Supplementary Figure S1B, Figure 3A, B, C and E). Fork velocity was significantly higher in Separase-depleted than in control cells, 0.90 ± 0.035 kb/min and 0.57 ± 0.037 kb/ min respectively (Figure 3C, Supplementary Table S4, P < 0.001, Kruskal-Wallis test) as well as the inter-origin distance, 98.7 ± 9.26 kb versus 65.2 ± 6.10 kb (Figure 3D, Supplementary Table S4, P < 0.001, Kruskal–Wallis test). In addition, we found a slight decrease in the proportion of unidirectional forks in siRNA-treated when compared to control cells (Supplementary Table S4, P < 0.05, chi-square test) whereas, the size of the replication clusters, paused/arrested and asynchronous forks were not affected by Separase inhibition (Supplementary Table S4). To gain further insight into the molecular mechanism underlying S-phase velocity control mediated by Separase, we investigated the progression of the cell cycle in control and Separase-silenced cells stained with propidium iodide and analyzed by flow cytometry. A significant decrease (P < 0.05) in the number of cells in S-phase was found 24 h after the start of siRNA treatment in Separase-depleted cells (Supplementary Figure S6). This effect is associated with a significant increase (P < 0.05) in the proportion of G1-phase.
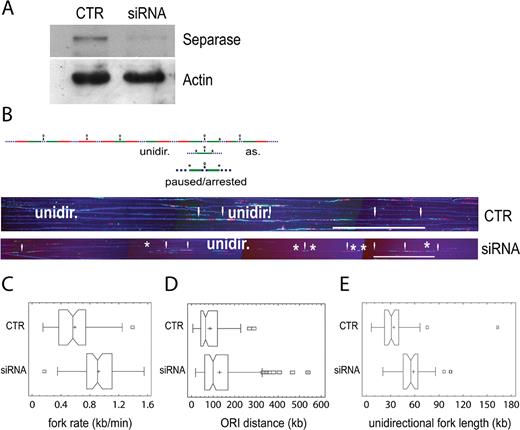
Separase controls global replication dynamics and its silencing causes a quick replication fork progression. (A) Western blotting showing the down-regulation of Separase in HeLa cells treated with 20 nM of smart pool siRNA. Actin was used as loading control. (B) Single-molecule analysis of replication fork dynamics in Separase-depleted cells. Cells were labelled with IdU for 30 min (first pulse, green), then washed and pulsed with CldU for a further 30 min (second pulse, red). DNA immunostaining with an anti-ssDNA antibody was used to verify the integrity of the molecule (blue dotted line). Bidirectional origins (O) may be mapped in the middle of the distance between the two divergent arms or in the middle of a green- or a red-only track. Paused/arrested forks may involve one or both arms (*). As = asynchronous fork. Unidir = unidirectional fork. Representative images of DNA molecules analyzed by combing are shown under the scheme. White arrows indicate bidirectional origins. (C) Fork velocities were measured in more than 50 tracks per sample. (D) Inter-origin distances were measured in around 100 tracks per sample. (E) Unidirectional fork length distribution. Median bar is reported in each box-plot and the mean value is indicated by a cross. Scale bar, 200 kb.
According to results described above, we reasoned that Separase loss could influence the restart of fork progression in response to replication stress. To test this hypothesis, we treated cells with 0.1 μM of aphidicolin for 15 h (Supplementary Figure S1C), because it is well-known that low concentrations of aphidicolin slow elongation during DNA replication process (49). As a consequence of delayed fork progression, we observed, as expected, the accumulation of cells in S-phase leading to an effect of synchronization of the cell population. However, Separase down-regulation in aphidicolin-treated cells (Supplementary Figure S1D) led to a significant decrease (P < 0.001) in cells in S-phase (T0, Figure 4A) and, at the same time, to an increased number of cells in G1 in comparison with non silenced cells. This difference is lost progressively during the time course (T1-T3).
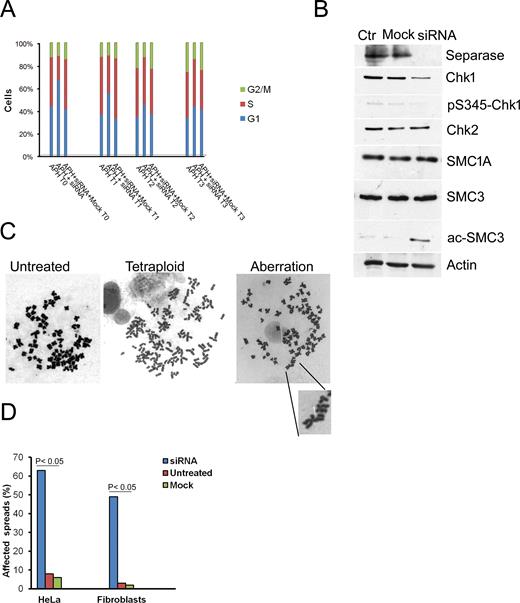
Effects of Separase depletion on cell cycle and genome stability. (A) HeLa cells were treated with 0.1 μM aphidicolin (APH) for 15 h then washed and transfected with siRNA against Separase for 24 h. Cells were analyzed by flow cytometry immediately after the end of siRNA treatment (T0), after 2 h (T1), 4 h (T2) and 6 h (T3). The values are the mean of three independent experiments. (B) Separase down-regulation caused the reduction of both Chk1 and pS345-Chk1. Furthermore, a strong band corresponding to acetylated SMC3 was detected in Separase-depleted cells. The cohesin's subunits, SMC1A and SMC3, did not change their level of expression. Actin is shown as loading control. (C) Karyotypic analysis of 100 Giemsa staining metaphase spreads revealed the presence of near tetraploid HeLa cells and structural chromosome aberrations. A chromosome break is highlighted. (D) Collectively, the frequency of affected metaphases was higher in siRNA-treated than control cells.
Next, we asked whether in Separase-depleted cells the progression of S-phase is mediated by checkpoint response. Chk1 and Chk2 are the main effector kinases respectively activated by ATR and ATM in response to DNA damage. The levels of Chk1 and its active-phosphorylated form (pS345-Chk1) showed a decrease in Separase-treated cells (Figure 4B, Supplementary Figure S7A and B) while no difference occurred for Chk2 (Figure 4B).
We then investigated the effect of Separase expression on cohesin. The levels of SMC1A and SMC3 were similar in control and Separase-depleted cells whereas a stronger band corresponding to acetylated-SMC3 was found in the latter (Figure 4B). It is well-established that SMC3 and Chk1 play different roles during DNA replication. Indeed, SMC3 acetylation speeds the replication forks and promotes sister chromatid cohesion in association with the replication machinery (50,51), whereas Chk1 plays a part in controlling both the initiation of DNA replication and the progression of S-phase (52,53). Therefore, to gain insight into the mechanism of SMC3 acetylation, we overexpressed Chk1 in HeLa cells (Supplementary Figure S7C). We found that the overexpression of Chk1 caused a slight increase in acetylated-SMC3, whereas its level was higher following siRNA against Separase and combined treatment (siRNA plus Chk1 overexpression, Supplementary Figure S7D). We also evaluated whether SMC3 acetylation could be explained by increased cohesin loading. To this aim, we measured chromatin-bound cohesin by ChIP-qPCR at four sites previously identified by ChIP-seq (41). Results showed the cohesin level increased significantly in FOXM, RASA1 and UBE2I genes following synchronization whereas it was comparable to the control in RALY gene (Supplementary Figure S8). Together, these results suggest that Separase is a component of the replication complex and regulates the replication fork speed. The depletion of Separase may cause the acceleration of S-phase; at the same time, failure of the intra-S phase checkpoint allows cells to proceed to the next G1.
Separase depletion induces genomic instability
Since an unstable replication fork is a major contributor to DNA damage in eukaryotes (54) our findings that Separase controls fork progression prompted us to investigate its role in genomic stability. For this, asynchronous HeLa and human primary fibroblast cell lines were transiently transfected with siRNA. In order to avoid off-target effects of siRNA treatment, we assayed four siRNAs against four different nucleotide targets on Separase (Supplementary Figure S9). Separase expression levels were strongly reduced 24 h after transfection, whereas mock transfection had no effect on its levels (Figure 4B, Supplementary Figure S10A). Microscopic analyses revealed that siRNA-Separase transfected cells showed a heterogeneous population of nuclear morphology. Besides morphologically normal nuclei, we found lobed ones and nuclei 1.5–3 times larger than normal (data not shown). Similar results were found in Separase-depleted cells analyzed at the first mitosis following aphidicolin treatment (data not shown). In addition, micronuclei were observed in 25% and 12% of HeLa cells and fibroblasts respectively (Supplementary Figure S10B). To determine whether these abnormal morphologies were the consequence of chromosome missegregation, we scored the chromosome number in mitotic cells. Chromosome spreads of primary fibroblasts ranged from near diploid to hyperdiploid; even though the HeLa genome is altered per se, we observed an increased chromosome number (data not shown). In addition, karyotypic analyses revealed structural aberrations such as chromatid gaps and breaks (Figure 4C, Supplementary Figure S10C). We obtained similar results using the siRNA4, one of the four above-described siRNAs (see Material and Methods, data not shown). The frequency of affected spreads varied from 49% to 63% in fibroblast and HeLa cells respectively (Figure 4D). Altogether, these data indicate that Separase is important for the maintenance of genomic stability, and its depletion causes both mis-segregation and a high degree of chromosome aberrations.
DISCUSSION
Based on the results presented in this study, we show that Separase interacts with MCM complex and cohesin and we provide new and unexpected insights into the relationship between Separase and replication fork speed. Though several genetic studies have pointed out the proteins contributing to DNA replication machinery, this is a matter for discussion. Previous reports showed the physical interaction between MCM complex and cohesin (20,55). Here, both proteomic and genome-wide data indicate that Separase is a novel component of the replication apparatus since it was found to have been co-immunoprecipitated with MCM2, MCM4, MCM6, MCM7, SMC1A and RPA2 proteins. Genome-wide analysis identified 8118, 4766 and 10400 occupied regions for Separase, MCM2 and SMC1A respectively. The number of cohesin sites is consistent with previous published data (41,56), whereas to our knowledge, this is the first time that genome-wide distribution of MCM2 in asynchronous cells and Separase has been carried out. Furthermore, we found that during replication stress conditions induced by aphidicolin, cohesin and MCM2 regions were higher than under an unperturbed S-phase. On the contrary, the Separase sites decreased but, the overlapping among MCM2, Separase and cohesin sites rose from about 9% to about 30% in cells synchronized in S-phase with a few regions in common between control and aphidicolin-treated cells. Furthermore, at variance with asynchronous cells, the remaining 70% of Separase sites co-localized with either MCM2 or SMC1A. Thus, it is plausible that inhibiting fork progression could transiently alter Separase, MCM2 and cohesin distribution patterns, driving their localization where DNA synthesis stalls. This notion is supported by the observations that the level of MCM2 increased in response of stalled replication (57) and cohesin accumulates at replication sites and is crucial for the recovery of stalled forks (58). Taken together, these data indicate that MCM2, cohesin and Separase are critical for protection of the forks from collapse in presence of low-concentration aphidicolin treatment, which resulted in moderately reduced fork speed.
Separase, MCM2 and cohesin are highly enriched at regions 1–3 kb upstream transcription start sites. This suggests that initiation of DNA replication might be mechanistically linked to transcription through an overlapping set of DNA elements. This view is supported by experimental evidence showing a close relationship between transcription and replication initiation, since several replisome proteins have been found in the vicinity of transcriptional promoters in mammalian cells (59–63). It has been shown that the absence of replication factor C (RFC)-CTF18 clamp loader (RFCCTF18), nucleotide depletion, polymerase inhibition, and DNA damage reduce fork velocity (50,64,65). In contrast, we find that the replication dynamics is highly accelerated in human cells by applying Separase silencing and sensitive single-molecule assays. In fact, the average velocity of individual forks increased by more than 150% in Separase-depleted cells (0.90 kb/min against 0.57 kb/min), and at the same time inter-origin distances increased significantly (from 65.2 kb to 98.7 kb). In agreement, the length of unidirectional forks also increased in Separase-depleted cells, with the consequent reduction of their number. Furthermore, in Separase-silenced cells the inhibition of DNA synthesis induced by aphidicolin treatment reduced the number of cells in S-phase. At the same time, the increased proportion of cells in G1-phase may be due to the combination of a failed intra-S checkpoint activation and increased replication rate, leading to faster progression through the cell cycle. The enrichment of Separase, MCM2 and cohesin in promoter regions suggests that Separase could be required to stop replication fork in active genes and its removal would result in increased fork speed. On the whole, our results imply that Separase is a component of replication machinery and modulates replication fork speed.
Separase silencing caused a robust acetylation of cohesin's SMC3 subunit. Slow replication speed was previously observed in cells expressing a non-acetylatable SMC3 and in Roberts syndrome cells mutated in ESCO2 gene, responsible for SMC3 acetylation (50). Altogether, these data indicate that SMC3 acetylation is a critical determinant of fork processivity since its modification leads to cohesin modification allowing the switch from slow- to quick-moving replication fork.
It has been shown that as a consequence of perturbed DNA replication, the intra-S-phase checkpoint delays the progression of S-phase (66). However, we found that Separase silencing caused the abrogation of Chk1 function. Since Chk1 is involved in DNA damage induced by replication stress, the loss of this checkpoint is responsible for overcoming mitotic arrest so that cells entered mitosis but failed to segregate their chromosomes properly, leading to genomic instability manifested as increased aneuploidy, chromosome aberrations and micronucleus formation.
Altogether, our findings indicate a novel function for Separase in conjunction with DNA replication factors and provide a molecular mechanism for the means by which Separase maintains genomic stability (Figure 5). In our model, Separase controls replication fork speed by interacting with the MCM complex and cohesin. It has been shown that DNA damage induces the dissociation of cohesin mediated by Separase promoting local accessibility to repair factors (67). Since we found that chromatin-bound cohesin increased following siRNA treatment, Separase silencing would prevent the dissociation of cohesin and its persistence on chromosomes may be a signal for SMC3 acetylation so that replication forks switch to a quick-processive configuration failing to block the cell cycle. This failure ultimately induces genomic instability. This model is further supported by the recent observation that cohesin remains associated with chromatin in S-phase and this does not affect the passage of replication forks (68). The detailed mechanisms of cohesin acetylation and checkpoint overcome following Separase inhibition are enthralling questions for the future. However, we cannot exclude that the replication-transcription conflict may also be a source of genome instability (69–71). Indeed, these processes routinely interfere with each other and an intriguing possibility is that Separase depletion affects the highly orchestrated processes devoted to minimizing the negative impact of replication-transcription conflict on genome stability. Of note, the expression of Separase is down-regulated in several tumors as shown in Gene Expression across Normal and Tumor tissue databases (http://mgrc.kribb.re.kr/GENT) as well as the level of Separase protein (http://www.proteinatlas.org). This study suggests interesting implications for the interplay between Separase and DNA replication machinery in preserving genomic stability. In addition to providing the first evidence that Separase controls replication fork speed, our study suggests that the loss of this regulatory mechanism causes genomic instability, with consequences for cancer development.
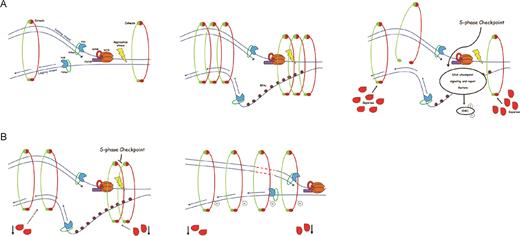
A model for genomic instability caused by replication fork progression mediated by Separase. (A) During S-phase, in presence of replication stress or DNA damage, Separase cleaves the cohesin ring promoting local accessibility to DNA repair enzymes. (B) However, the down-regulation of Separase prevents cohesion dissolution allowing cohesin to remain on chromatin. This facilitates the acetylation of SMC3 so that fork replication becomes more processive. In addition, due to Chk1 and pS345-Chk1 down-regulation, the S-phase checkpoint response is not able to slow or stop fork progression. As a consequence, cells fail to repair DNA, inducing genomic instability.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Dr Maurizio Fanciulli (Regina Elena National Cancer Institute, Rome, Italy) for the Chk1 vector.
FUNDING
Associazione Italiana Ricerca sul Cancro (AIRC, IG17374) and InterOmics Flagship project (to A.M.). Funding for open access charge: Associazione Italiana Ricerca sul Cancro.
Conflict of interest statement. None declared.
Footnotes
Present address: Antonella Russo, Department of Molecular Medicine, University of Padua, Padua, Italy.
REFERENCES
Author notes
These authors contributed equally to this work as second authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments