





Abstract
Ribosomes of different species share an evolutionarily conserved core, exhibiting flexible shells formed partially by the addition of species-specific ribosomal RNAs (rRNAs) with largely unexplored functions. In this study, we showed that by swapping the Saccharomyces cerevisiae 25S rRNA genes with non-S. cerevisiae homologs, species-specific rRNA variations caused moderate to severe pre-rRNA processing defects. Specifically, rRNA substitution by the Candida albicans caused severe growth defects and deficient pre-rRNA processing. We observed that such defects could be attributed primarily to variations in expansion segment 7L (ES7L) and could be restored by an assembly factor Noc2p mutant (Noc2p-K384R). We showed that swapping ES7L attenuated the incorporation of Noc2p and other proteins (Erb1p, Rrp1p, Rpl6p and Rpl7p) into pre-ribosomes, and this effect could be compensated for by Noc2p-K384R. Furthermore, replacement of Noc2p with ortholog from C. albicans could also enhance the incorporation of Noc2p and the above proteins into pre-ribosomes and consequently restore normal growth. Taken together, our findings help to elucidate the roles played by the species-specific rRNA variations in ribosomal biogenesis and further provide evidence that coevolution of rRNA expansion segments and cognate assembly factors specialized the ribosome biogenesis pathway, providing further insights into the function and evolution of ribosome.
INTRODUCTION
Ribosomes are responsible for protein production in all living cells. Despite being universally conserved, ribosomes exhibit substantial differences across species, partly due to variations in ribosomal RNAs (rRNAs), which serve as the central interface for hundreds of proteins, such as ribosome assembly factors (AFs) and ribosomal proteins (RPs), and these RNAs have evolved to exhibit high variability at the level of length and sequence, both within and among various species (1–4). Compared to prokaryotic rRNAs, eukaryotic rRNAs have evolved to insert enigmatic species-specific sequences designated expansion segments (ESs), which are the hot spots of variation among different eukaryotic rRNA species (1,2). For instance, the size of the ribosome varies from 3.3 MDa in Saccharomyces cerevisiae to 4.3 MDa in Homo sapiens, primarily owing to the augmentation of four ESs interspersed throughout the rRNA (2). In addition, ES/ES-like structures were also identified in the rRNAs of non-eukaryotic organisms, although they were not as prevalent in these species as in eukaryotes (5–8). Even in the same organism, heterogeneous rRNAs with varied sequences have also been observed, such as in Plasmodium, zebrafish, mice and Homo sapiens (9–11). However, the function of pervasive rRNA variants in specializing the universal ribosome and the underlying mechanism have not been fully elucidated (1–3,12–14). Functional dissection of the species-specific rRNA variations is crucial for understanding the general and unique aspects of rRNA-associated control.
Several studies have shown that ESs, where the rRNA variations are concentrated, could serve as scaffolds for the regulatory factors in modulating ribosome biogenesis and function (12–19). Ramesh et al. showed that the deletion of most yeast ESs led to abnormalities in various steps of pre-rRNA processing, identifying the roles played by certain ESs in regulating the specific step of ribosome biogenesis (12). For example, deletion of ES7L led to accumulation of the 27SA3 pre-rRNA in early processing steps, while partial deletions of ES27L resulted in an increase in 27SB pre-rRNA or a slight inefficiency in the 7S pre-rRNA processing step (12,20). Recent high-resolution studies of pre-ribosome structures demonstrated that ES7L could provide binding sites for several assembly factors such as Nsa1p, Mak16p, Rpf1p and Rrp1p, in early ribosome assembly, and ES27L interacted with the Erb1p-Ytm1p complex or Arx1p in later ribosome assembly (15–18). In addition, several studies have reported that ES27L is involved in the recruitment of other regulatory factors such as methionine amino peptidases (MetAP) or the N-terminal acetyl transferase (Nat) complex, and participates in modulating translation fidelity and cotranslational modification of nascent peptides (13,14,19). It is of special interest to investigate the largely unknown auxiliary proteins recruited by ESs to determine how these unique structural features regulate ribosome synthesis and function. Moreover, do species-specific rRNA variations, primarily in the ESs, diversify ribosome biogenesis and function across different species? If so, what are the underlying mechanisms governing this process, and how do cells evolve to accommodate such variations to ensure proper ribosome synthesis and function?
In this study, we replaced 25S rRNA with counterparts from three other yeast species and analyzed the functionalities of these chimeric rRNAs in S. cerevisiae, which showed that species-specific variations among 25S rRNA variants could interfere with rRNA biogenesis. To obtain further insight, we specifically focused on a strain with 25S rRNAs from the fungal pathogen C. albicans. This strain displayed severe growth defects and specific 27S pre-rRNA processing deficiencies, which were primarily attributed to the variations in the 25S rRNA expansion segment 7L (ES7L). Furthermore, through suppressor screening and biochemical analysis, we demonstrated that ES7L replacement attenuated the incorporation of the ribosome assembly factor Noc2p into pre-ribosomes to perturb 27SA3 pre-rRNA processing. We found that a particular mutation of Noc2p could restore normal pre-rRNA processing and suppress growth defects. In addition, we demonstrated that substituting the Noc2p of S. cerevisiae with the C. albicans ortholog also rescued the defects. Taken together, our findings indicated that rRNA expansion segments and cognate assembly factors acted as coevolutionary modules to regulate ribosome biogenesis, providing further insights into the function and evolutionary principle of rRNAs.
MATERIALS AND METHODS
Yeast material and growth conditions
Strains used in this paper are listed in Supplementary Table S1. All yeast strains were cultured at 30°C. Yeast strains were grown in following medium. YPD medium contains 2% glucose, 2% tryptone and 1% yeast extract. YPGal medium is the same as YPD, containing 2% galactose rather than glucose. Synthetic glucose (SGlu) medium (0.67% yeast nitrogen base, 2% glucose) was supplemented with required amino acids. Synthetic galactose (SGal) medium contains the same components as SGlu, except that 2% glucose is replaced with 2% galactose. Solid medium is produced by adding 1.5% agar.
Construction of plasmids and yeast strains
Plasmids and oligos used in this paper are listed in Supplementary Tables S2 and S3, respectively. The DNA sequence of foreign 25S rRNA originated from Candida glabrata, Kluyveromyces lactis and Candida albicans were synthesized from overlapping oligos and used to replace the 25S rDNA in pRDN-wt to generate the chimeric rDNA constructs by Golden-Gate Assembly as described previously (21). The rDNA mutant plasmids with partial regions of 25S rRNA replaced by the corresponding parts of C. albicans were assembled by the same way. These DNAs were sequenced to be 100% identical to the design. The pGal-rDNA assay strain was a derivative of previously reported yeast strain L1521 that contains a complete deletion of the entire chromosomal rDNA cluster and is kept alive by the multicopy plasmid carrying a wild-type rDNA repeat (pRDN-wt-U, URA3) (22), and generated by replacing the plasmid pRDN-wt-U with the plasmid carrying 35S rDNA fused to the GAL7 promoter (pGal7-rDNA, LYS2) as described previously (23). The rDNA mutant strains were produced from the pGal-rDNA assay strain by transformation of the plasmids containing rDNA mutants. To remove the pGal7-rDNA plasmid, these rDNA mutant strains were continuously streaked on SGlu plates to select the colonies that could not grow on the medium without lysine, and the resultant Lys− strains were verified by the specific primers towards wild-type rDNA or DNA sequence of foreign 25S rRNAs. Coding sequences (CDS) of wild-type Noc2,Noc2 suppressor mutant or the C. albicans’ homolog were obtained individually by PCR amplification using genome of S. cerevisiae, suppressor strain or C. albicans as the template, and fused to the constitutive GAP promoter with or without human influenza hemagglutinin (HA) tag coding sequence at the C terminus of the expression cassette in pRS415. After transformation of these Noc2-containing plasmids into target strains, the entire CDS of Noc2 from the chromosome was replaced by the nourseothricin (NAT) resistance gene to construct Noc2 assay strains. The conditional null mutants of nop15, rpl4, rcl1 or rat1 were produced from the BY4741 background yeast by standard procedures as described previously (24).
Yeast spot assay
About 5 ml cultures of single colonies were incubated in appropriate media overnight at 30°C. The cultures were adjusted to the same OD600. Ten fold serial dilutions of the yeast strains were spotted on appropriate solid media and incubated at 30°C for several days before photography.
Sequence alignment and RNA secondary structure predication
Aligned 25S rRNA sequences of different yeast species were obtained from MEGA Xsoftware (25). We inspected the aligments by eye and performed some slight modifications. The 25S rRNA sequence of S. cerevisiae was mapped onto the secondary structure using RiboVision (26). The secondary structure of indicated helixs domain were predicated by RNAfold.
RNA extraction and assay
The yeast strains were collected by centrifugation at 3000 rpm for 10 min at 4°C from 50 ml culture when the turbidity reached OD600 = 1.0. Cells were washed twice with ice-cold ddH2O, adjusted to OD600 = 15.0, flash frozen in liquid nitrogen and stored at −80°C. Cell pellets were gently suspended in 600 μl TRIzol, followed by glass beads disruption with Mini-Beadbeater-96 for 5 min. Then liquid mixture was collected and added in 300 μl chloroform, followed by shaking for 3 min. After centrifugation at 12 400 rpm at 4°C for 10 min, upper layer liquid phase was collected and added in 300 μl isopropanol for precipitating RNA. Finally, RNA pellets were washed once with 700 μl 75% (v/v, DEPC H2O) cold ethanol and dissolved in 100 μl DEPC H2O. The RNAs were electrophoresed on 1.2% agarose gels and stained with ethidium bromide.
Analysis of pre-rRNA intermediates
Pre-rRNA intermediates were analyzed by a previously described 5′3′ RACE technique (27,28) with some modification. Briefly, as described in Figure 2C, 500 ng of total RNAs were firstly phosphorylated using a T4 PNK enzyme (New England Biolabs, M0201S), and the resulting RNAs were cyclized using a T4 RNA ligase (New England Biolabs, M0204S). Then, the cyclized rRNAs were converted to linear cDNAs using an HiScript® II reverse transcriptase (Vazyme, R233–01) with mixed primers of ZYO251, ZYO252–1 and ZYO131. The cDNA products were then applied as the templates for Q-PCR analysis which was performed in a step one plus (applied biosystems) using ChamQ SYBR qPCR Master Mix (Vazyme, Q311–02). The specific primers designed for certain pre-rRNA were listed in Supplementary Table S3. RNA levels were normalized to unrelated RNA (e.g. scr1). In general the assays included three biological and three technical replicates.
Affinity purification of pre-ribosomes
Affinity purification of pre-ribosomes was performed with C-terminally Flag-tagged Rpf2p as bait protein. Freshly streaked single colonies of the respective yeast strains were incubated in 5 ml YPD at 30°C overnight and subcultured in 100 ml YPD until an OD600 of 0.8. The yeast cells were harvested by centrifugation at 3500 g for 10min at 4°C, washed with ice-cold ddH2O twice, flash frozen in liquid nitrogen and stored at −80°C. Cell pellets were gently resuspended in 500 μl freshly prepared ice-cold lysis buffer [20 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 150 mM potassium acetate, 0.2% (v/v) Triton X-100 and 1 × cOmplete™ protease inhibitor cocktail], and cells were ruptured by shaking in screw-top tubes in the presence of glass beads. Lysates were cleared by centrifugation at 15 700 g for 10 min at 4°C. Supernatants were incubated with the 50% slurry of anti-FLAG agarose beads (25 μl beads/100 ml of logarithmic growth phase yeast cultures) on a rotating wheel at 4°C for 150 min. Beads were collected by centrifugation at 500 g for 2 min at 4°C and washed with ice-cold lysis buffer for six times by centrifugation at 2000 g for 1 min at 4°C. The beads were resuspended in 50 μl of protein loading buffer and boiled at 100°C for 15 min and the supernatant was collected by centrifugation at 8000 g for 3 min and analyzed by western blotting with antibodies as indicated in the respective figures.
Western blotting
Protein samples were separated on SDS-PAGE gels and transferred to PVDF membranes by wet electroblotting following the manufacturer’s protocol. Western blotting signals were developed using immobilon Western ECL Substrate (Millipore) and visualized by chemiluminescence system (Tanon, T5200Multi). Same amounts of whole cell extracts from the yeast strains were loaded for analyzing the expression levels of Flag-tagged Rpf2p and HA-tagged Noc2 proteins and equal loading was controlled by determination of the protein level of GAPDH. The pre-ribosome purification samples containing equal amounts of Flag-tagged Rpf2p were loaded for investigating the assembled ability of Noc2 proteins into pre-ribosomes. Western blot analysis was performed using the following antibodies: anti-Flag rabbit polyclonal antibody (Sigma Cat No. F7425, 1:5000, RRID: AB_439687), anti-HA mouse monoclonal antibody (Sigma Cat No. H3663, 1:10 000, RRID: AB_262051), anti-GAPDH rabbit polyclonal antibody (GeneTex Cat No. GTX100118, 1:5000, RRID: AB_1080976).
Genomic DNA preparation, sequencing and analysis
Suppressor strain was cultured in YPD medium at 30°C overnight. 5 ml yeast culture (∼5 × 108 cells) was harvested by centrifugation. The genomic DNA was prepared using the method as described previously (21). After washing in ddH2O, cells were resuspended in 300 μl breaking buffer [2% Triton X-100, 1% SDS, 100mM NaCl, 10 mM Tris-HCl (pH 8.0) and 1 mM EDTA (pH 8.0)]. After addition of 300 μl acid-washed glass beads (BioSpec, Cat. No.11079105, 0.5 mm dia.) and 300 μl phenol/chloroform/isoamylol (25:24:1), cells were ruptured by vigorous vortexing for 5 min. Then 200 μl ddH2O was added into the tube, followed with slight shaking and centrifuged at 12 000 rpm for 10 min. The aqueous upper phase was transferred to a 1.5 ml Eppendorf tube, followed with adding 1 ml of absolute ethanol and mixing the contents by inversion. After centrifugation at 12 000 rpm for 10 min, the pellet was dried in vacuum pump and dissolved in 400 μl TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). To digest the RNA, 3 μl of a 10 mg/ml RNaseA solution was added into the tubes, incubated at 37°C for 120 min. To precipitate the genomic DNA, 1 ml of absolute ethanol and 10 μl of 5 M ammonium acetate were added. The pellet was collected by centrifugation at 12 000 rpm for 10 min, dried in vacuum pump and resuspended in 100 μl of TE buffer. Finally, integrity of the genomic DNA was demonstrated by migration on a 1% agarose gel and the concentration was evaluated by NanoDrop (Thermo).
Genomic DNA libraries for high-throughput sequencing was performed on a HiSeq 2000 instrument (Illumina). Library preparation, sequencing and reads’ quality check were carried out by the next-generation sequencing (NGS) Platform of the Novogene. Clean reads were mapped to the S. cerevisiae reference genome (strain S288C) obtained from Ensembl by BWA program (29). The output SAM files were sorted and converted to BAM files with SAMtools (30). Single nucleotide variants (SNVs), as well as small insertions and deletions (Indels), were identified with GATK (31). The output file was a Variant Call Format (VCF) that contains position information for each SNVs. Then the results were filtered and viewed with SAMtools.
Yeast two-hybrid assay
The sequences encoding Noc2p and A3 factors were amplified from genome by PCR and cloned into yeast two-hybrid vectors, pDB-Leu and pPC86 plasmids respectively (32). The pDB-Leu plasmid contained Noc2p fused to the C terminus of Gal4 binding domain and the pPC86 plasmid harbored A3 factor fused to the activation domain. The yeast two-hybrid (Y2H) assay was performed by co-transforming these two plasmids into yeast strain and then spotting the obtained yeast strain with serial dilutions on the synthetic medium lacking uracil at 30°C, using empty vectors as the negative control and previously identified Y2H interactions as the positive control.
Coimmunoprecipitation assay
CDS of Noc2-His6 and Erb1–3 × HA were obtained individually by PCR amplification using genome of S. cerevisiae as the template and the sequences of His6 or 3 × HA tag were added by reversed primers. Then Noc2-His6 or Erb1–3 × HA were first cloned into pET-28a via NcoI and NotI sites, respectively. In order to construct the vector used for co-expression of Noc2-His6 and Erb1–3 × HA, the SpeI and SbfI sites were added to the downstream side of SphI site in pET-28a-Erb1–3 × HA. Subsequently, the expression cassette of Noc2-His6 including T7 promoter and T7 terminator was obtained by PCR amplification using pET-28a-Noc2-His6as the template and then cloned into pET-28a-Erb1–3 × HA via SpeI and SbfI sites. The obtained plasmids were individually transformed into Rosetta cells to yield the IPTG-inducible strains. The resulting transformed strains were selected on LB plates containing kanamycin and chloramphenicol. For coimmunoprecipitation, 10 ml cultures of single colonies were incubated in LB medium overnight at 37°C in the presence of 50 μg/ml kanamycin and 12.5 μg/ml chloramphenicol. The cultures were inoculated in 1:100 500 ml Terrific Broth (TB) medium (1.2% Tryptone, 2.4% Yeast Extract, 0.4% Glycero, 1.7 mM KH2PO4 and 7.2 mM K2HPO4) with the same antibiotics and grown at 37°C. When OD600 of cell culture reached ∼0.8, 0.2 mM isopropyl-β-d-thiogalactoside (IPTG) was added and cells were cultured at 18°C for another 20 h to induce protein production. The resulting culture was rapidly cooled down on ice for 10 min and then cells were collected by centrifugation at 5000 g for 10 min at 4°C. The harvested cells were washed by ice-cold lysis buffuer (20 mM Tris–HCl, pH 7.5, 150 mM KCl, 1 mM MgCl2 and 1 mM PMSF) twice and resuspended in 50 ml lysis buffer and disrupted by high pressure cooling crushing apparatus until the solution became clear. The cell lysate was cleared by centrifugation at 12 000 g for 20 min at 4°C. Then supernatant was collected and incubated with Ni-NTA agarose beads (Genscript) for 1 h at 4°C. The beads were washed three times with 10 ml ice-cold washing buffer (20 mM Tris–HCl, pH 7.5, 150 mM KCl, 1 mM MgCl2, 1 mM PMSF and 50 mM imidazole) to remove unbound protein. The protein was eluted by step-wise addition and removal of 1 ml ice-cold elution buffers (20 mM Tris–HCl, pH 7.5, 150 mM KCl, 1 mM MgCl2, 1 mM PMSF, 100, 200, 300, 500 or 800 mM imidazole) at 4°C. Protein samples were separated on SDS-PAGE gels and transferred to PVDF membranes for western blot analysis with a 1:10 000 dilution of anti-His6 (Proteintech) or anti-HA (Sigma) mouse monoclonal antibody.
Statistical analysis
Statistical analysis was performed by using Student’s t test (*P < 0.05, **P < 0.01, *** P < 0.001). All data represent the mean of n = 3 biologically independent samples and error bars show standard deviation. Tests and specific P values used are indicated in the figure legends.
RESULTS
25S rRNAs from different yeast species are largely replaceable
To evaluate the function of species-specific rRNA variants, 25S rRNA of S. cerevisiae (designated S.c) was substituted by that from three representative non-S. cerevisiae yeasts, namely, Candida glabrata (C.g), Kluyveromyces lactis (K.l) and Candida albicans (C.a) (Figure 1A) (33). To analyze these rRNA variants in vivo, we took advantage of a premier S. cerevisiae strain (pGal7-rDNA assay strain) in which all chromosomal ribosomal RNA genes were deleted and compensated for by a rescue plasmid containing an rDNA unit driven by a galactose-inducible GAL7 promoter (Figure 1B, left) (34–36). Accordingly, this strain was only able to grow on medium containing galactose as a carbon source. A second donor vector was constructed in which transcription of the rRNA variant or S.c rRNA was driven by the native RDN promoter (Figure 1B, middle). Next, the second donor was transformed into the pGal7-rDNA assay strain to investigate the functionalities of the rRNA mutants on glucose-based medium (Figure 1B, right).
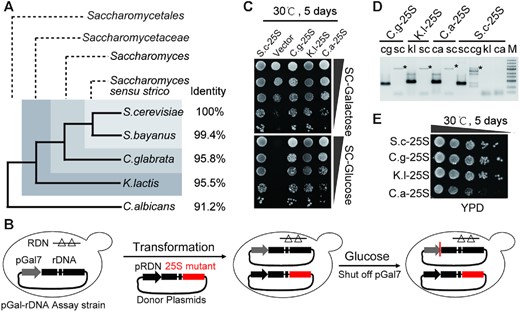
25S rRNAs exhibit considerable replaceability among different yeast species. (A) Phylogenetic relationship of several representative species in Saccharomycetales. ‘Identity’ denoted the nucleotide identities between S. cerevisiae and non-S. cerevisiae 25S rRNA sequences. (B) Schematic workflow to assay 25S rDNA mutants. Left- pGal-rDNA assay strain, in which all of the chromosomal rDNAs were deleted and complemented by the rescue plasmid containing a rDNA unit driven by a galactose-inducible GAL7 promoter (pGal7); Middle- donor plasmids, in which either the wild-type or the chimeric rDNA with the 25S rDNA replaced by the counterpart of non-S. cerevisiae yeasts were under control of the native RND promoter (pRDN). Right- pGal-rDNA assay strains transformed with donor plasmids were assayed on galactose- or glucose-containing medium (as shown in C). (C) Spot assays of the chimeric rDNA mutants on the synthetic complete medium containing either galactose (SC-Galactose) or glucose (SC-Glucose). (D) Electrophoresis patterns of the PCR products amplified by specific primers. ‘sc’ denoted the PCR products amplified by the primers specifically targeting the 25S rDNA of S. cerevisiae; ‘cg’, ‘kl’ or ‘ca’ denoted the products amplified by the primers specifically targeting the 25S rDNA of C. glabrata, K. lactis and C. albicans, respectively. ‘*’ indicates nonspecific amplicons. (E) Spot assays of the chimeric rDNA mutants on YPD medium.
Based on the above approach, the effects of species-specific rRNA variations on cell viability were examined. As expected, strains carrying S.c.25S but not the empty vector could survive on glucose medium (Figure 1C). Notably, all of the strains carrying chimeric rDNA mutants were also able to survive, indicating that these foreign 25S rRNAs could execute similar functions to the native rRNAs (Figure 1C). To further confirm this possibility, we obtained the strains C.g-25S, K.l-25S, C.a-25S and S.c-25S, which contained only the 25S rRNA genes from C.g, K.l, C.a and S.c, respectively, to avoid the possible interference of pGal7-rDNA. These strains were verified by PCR analysis, as evident by the presence of amplicons using primers specifically targeting foreign 25S rDNAs (cg, kl or ca) and the absence of bands with primers specifically targeting native 25S rDNAs (sc) (Figure 1D). Next, a spot assay was performed to compare the growth rates of these strains. The results showed that C.g.25S and K.l.25S possessed similar growth rates compared with that of the wildtype. In contrast, C.a.25S exhibited severe growth defects, resulting in small colonies on the YPD plate (Figure 1E). These results indicated that the 25S rRNAs from other species retained common functionalities essential for life. Indeed, although the primary sequences of these rRNAs exhibited considerable variations (with identities ranging from 91.2% to 95.8%), their secondary structures were largely retained by compensatory mutations between nucleotide pairs (including both Watson–Crick and noncanonical base pairs) (Supplementary Figure S1), possibly indicating that these 25S rRNA variants could support the growth of S. cerevisiae. This finding was also in keeping with a previous hypothesis that stressed the importance of conserving the secondary structure of rRNA for function (20,37,38).
25S rRNA substitutions affect pre-rRNA processing
To investigate whether replacements of 25S rDNA affect the maturation of 25S rRNA, total cellular RNA was separated on agarose gel and visualized by ethidium bromide staining (Figure 2A). Notably, C.a-25S exhibited a considerably reduced amount of 25S rRNA but a moderate decrease in18S rRNA, whereas relatively slight changes were observed in C.g-25S and K.l-25S, consistent with the growth phenotype. This result also suggested that 35S pre-rRNA processing might be affected in this mutant through a series of steps with several key pre-rRNA intermediates, as outlined in Figure 2B (see details in Supplementary Figure S2A).
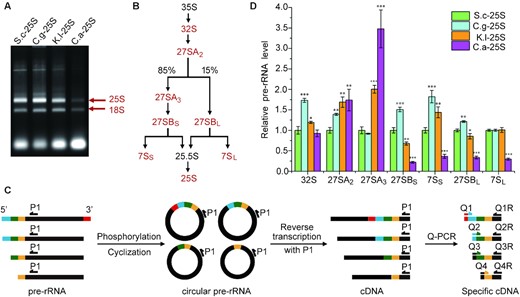
25S rRNA substitutions perturb pre-rRNA processing. (A) Electrophoresis patterns of total RNA. Mature 25S and 18S rRNAs were labeled by arrows, respectively. (B) Sketch of the 25S rRNA biogenesis pathway. The pre-rRNA intermediates analyzed in this study were marked in red. (C) Schematic workflow to quantify the pre-rRNA intermediates. First, total RNA was phosphorylated and circularized to produce circular pre-rRNA, in which 5′ end and 3′ end of the pre-rRNAs were ligated together. Then cDNAs were generated by reverse transcription with the primer P1 and further subjected to Q-PCR analysis with one specific primer (Q1,2…) designed to span across the 5′ and 3′ junction of certain circularized pre-rRNA and the other primer (Q1R,2R…) designed with compatible parameters for the Q-PCR experiment (see details in ‘Materials and Methods’ section). (D) Relative levels of key pre-rRNA intermediates in the 25S rRNA biogenesis pathway between strains with native and chimeric rDNA. Levels of the pre-rRNA intermediates in S.c-25S were set as 1.0. Error bars represent the standard deviation of three replicate reactions (t- test, *P < 0.05, **P< 0.01, ***P< 0.001).
To identify the potential defective steps during rRNA processing, several key pre-rRNA intermediates were converted to cDNA based on a previously established 5′ and 3′ RACE techniques and quantified by quantitative PCR (qPCR, Figure 2C) (27,28). As controls, four previously characterized genes (nop15, rpl4, rcl1 and rat1) that function in different steps of pre-rRNA processing were tested using conditional null mutants where the corresponding genes were expressed under the control of the galactose-inducible promoter (39). As expected, the growth of these strains was inhibited on the glucose-containing medium compared with that on the galactose-containing medium (Supplementary Figure S2B). qPCR results showed that Nop15p depletion clearly caused accumulation of 27SA3 accompanied by reduced levels of 27SBS and 7SS pre-rRNA intermediates (Supplementary Figure S2C), which was consistent with a previous finding that Nop15p was required for processing the 27SA3 pre-rRNA to 27SBS (40). Similar results were observed for the other mutants (Supplementary Figure S2D, S2E and S2F) (41–44).
Subsequently, this approach was used to assess the relative levels of pre-rRNA intermediates between S.c-25S and three other strains (C.g-25S, K.l-25S and C.a-25S) (Figure 2D). In C.g-25S, nearly all of the pre-rRNA intermediates, including 32S, 27SA2, 27SBS, 7SS and 27SBL pre-rRNAs, increased, indicating generally reduced efficiency of pre-rRNA processing. In K.l-25S, significantly increased amounts of 27SA2 and 27SA3 along with decreased amounts of downstream 27SB pre-rRNA were observed, suggesting that there were specific defects in the processing steps of 27SA2 and 27SA3. Notably, although pre-rRNA processing defects were observed in C.g-25S and K.l-25S, the amounts of mature rRNAs showed mild to moderate changes in these strains (Figure 2A), which was in keeping with previous findings that the pre-rRNA processing pathway was tolerant of perturbance to a certain extent (12,14,45). In contrast, C.a-25S exhibited dramatic accumulation of 27SA3 pre-rRNA accompanied by significant decreases in downstream 27SB and 7S pre-rRNAs, indicating that a strong and specific deficiency occurred in processing 27SA3 in C.a-25S. Taken together, these findings showed that interspecies substitutions of 25S rRNA could affect pre-rRNA processing.
ES7L replacement primarily accounts for the defects observed in C.a-25S
Next, we focused on studying C.a-25S, which exhibited strongly impaired growth and dramatic 27SA3 pre-rRNA processing defects. To specify the RNA domain responsible for the defects observed in C.a-25S, we systematically swapped the 25S RNA section by section from S.c to C.a. Notably, to achieve the best retention of the native structures in these 25S rRNA hybrids, intact RNA helix structures based on the well-established secondary structure of S.c 25S rRNA were kept when designing the replaced segments (Figure 3A). Two mapping groups were generated. In one group, the segment from helices (H) 29 to 78 (H29–78) and the segment H2–28 plus H78–101(H2–28 & 78–101) were substituted; in the other group, H2–28, H29–55 and H55–101 were replaced (Figure 3B). As the results showed, all strains with H2–28 replacement displayed strongly inhibited growth, suggesting that H2–28 was responsible for the growth defect. To further identify the core causal region within H2–28, another round of substitutions, including H2–24 and H25–28, was performed. The strain with H2–24 replaced (C.a H2–24) did not exhibit clear growth defects, while C.a H25–28 suffered from severe growth loss, indicating that variations in H25–28 contributed primarily to the impaired growth observed in C.a-25S. Notably, all of the variations in H25–28 between C.a and S.c 25S rRNA were located in the expansion segment 7L, which was an extension of H25 in the 25S rRNA and one of the most variable regions of eukaryotic rRNA (Supplementary Figure S1). Subsequently, we investigated the effect of ES7L replacement on rRNA synthesis. As shown in Figure 3C, the mature 25S rRNA decreased notably in C.a-ES7L. Moreover, strong accumulation of 27SA3 pre-rRNA accompanied by significant decreases in downstream 27SB and 7S pre-rRNAs was observed in C.a-ES7L, indicating that a severe defect occurred in 27SA3 pre-rRNA processing (Figure 3D). This result was in keeping with previous findings that ES7L functioned in 27SA3 pre-rRNA processing (12). Taken together, the growth and pre-rRNA processing defects observed in C.a-25S were nearly phenocopied by C.a-ES7L, suggesting that species-specific variations in ES7L were responsible for the defects observed in C.a-25S.
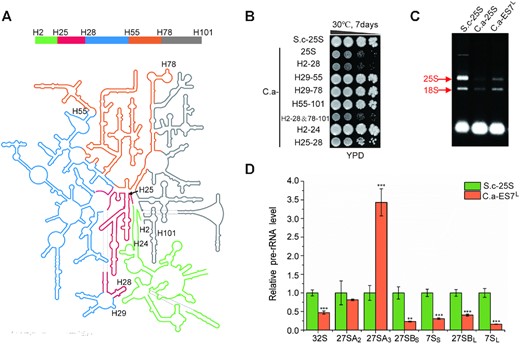
ES7L primarily accounts for defects observed in C.a-25S. (A) Secondary structure of the yeast 25S and 5.8S rRNAs. Domains swapped in this study were marked by different colors: H2–24- green; H25–28- red; H29–55- blue; H56–78- orange; H79–101- gray. (B) Spot assays of the 25S rRNA mutants with different domains replaced by the corresponding parts of C. albicans. S.c-25S was indicated as a control. (C) Electrophoresis of total RNA extracted from S.c-25S, C.a-25S and C.a-ES7L. Mature 25S and 18S rRNAs were labeled by arrows, respectively. (D) Relative levels of key pre-rRNA intermediates in the 25S rRNA biogenesis pathway between S.c-25S and C.a-ES7L. Levels of pre-rRNA intermediates in S.c-ES7L were set as 1.0. Error bars represent the standard deviation of three replicates (t-test, *P < 0.05, **P< 0.01, ***P< 0.001).
Noc2 suppressor mutation bypasses the defects in C.a-ES7L and C.a-25S
To elucidate the mechanism underlying the defects derived from ES7L or 25S rRNA substitution, we took advantage of the severe growth defect of C.a-25S to isolate suppressor mutations that enable C.a-25S to grow almost normally. To isolate the suppressor strain, the C.a-25S strain was incubated in YPD liquid medium to stationary phase, diluted (1:1000) and transformed into fresh YPD medium for another round of culturing. After each culturing cycle, an aliquot of cells was spread on YPD solid medium to identify fast-growing colonies (Figure 4A). After five rounds, one fast-growing strain was acquired (Figure 4B). To rule out the possibility of revertant mutants on the 25S rRNA gene, 25S rDNA in this strain was sequenced, and no intragenic mutation was identified (data not shown).
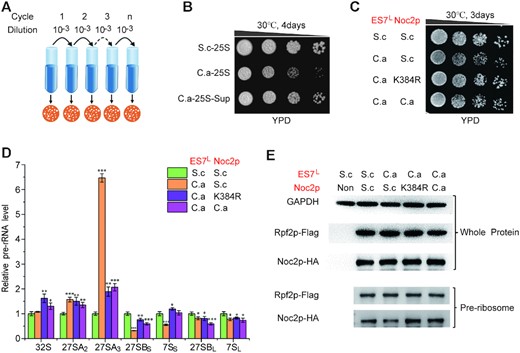
Defects in C.a-ES7L could be suppressed by noc2 mutation. (A) Schematic workflow to isolate suppressor strains of C.a-25S. (B) Growth analysis of the suppressor strains of C.a-25S. C.a-25S-Sup denoted the suppressor strains of C.a-25S. (C) Both noc2 mutant and C.a NOC2 rescue the growth defect in C.a-ES7L. ‘S.c’ denoted the corresponding component was derived from S. cerevisiae; ‘C.a’ denoted the corresponding component was derived from C. albicans; ‘K384R’denoted the Noc2 K384R mutant. (D) Relative levels of key pre-rRNA intermediates in the 25S rRNA biogenesis pathway between S.c-25S and the strains with or without ES7L and Noc2p engineered. ‘S.c’ denoted the corresponding component was derived from S. cerevisiae; ‘C.a’ denoted the corresponding component was derived from C. albicans; ‘K384R’ denoted the Noc2 K384R mutant; Levels of the pre-rRNA intermediates in S.c-25S were set as 1.0. Error bars represent the standard deviation of three replicate reactions (t-test, *P < 0.05, **P< 0.01, ***P< 0.001). (E) Western blot analysis of HA-tagged Noc2p in the whole cell extracts or in the pre-ribosomes purified by Flag-tagged Rpf2p. ‘Non’ denoted extracts from the strains with un-tagged Rpf2p and Noc2p, which was used to verify the specificity of antibodies. ‘GAPDH’ was used as a loading control.
To identify the mutations responsible for the restoration of the growth defect, whole genome sequencing (WGS) analysis of both the parental C.a-25S and the suppressor strain (C.a-25S Sup) was performed. A single missense mutation (K384R) in the open reading frame of the noc2 gene was found. Noc2p was previously reported to be an essential ribosome assembly factor that is involved in the maturation of pre-60S ribosomes and crucial for the 27S pre-rRNA processing (46,47). To investigate whether noc2 gene mutation actually accounted for the restoration of the growth defect, chromosomal noc2 was deleted and complemented by either the noc2 suppressor allele (noc2 K384R) or wild-type noc2 driven by the constitutive GAP promoter. As shown in Supplementary Figure S3A, the severe growth defect of C.a-25S was markedly restored, although not to that of wild-type, by Noc2p K384R. Furthermore, Noc2p K384R clearly improved the growth rate of C.a-ES7L to a level comparable to that of the wild-type strain (Figure 4C). In addition, in keeping with the improved growth, the levels of mature rRNAs were strongly increased in both C.a-ES7L and C.a-25S encompassing Noc2p K384R (Supplementary Figure S3B and S3C). Furthermore, Noc2p K384R strongly alleviated the accumulation of 27SA3 pre-rRNA and increased the amount of downstream pre-rRNA intermediates, such as 27SBS and 7SS (Figure 4D and Supplementary Figure S3D). These findings demonstrated that Noc2p K384R complemented the 27SA3 pre-rRNA processing defect derived from the interspecies exchange of ES7L, establishing a genetic relationship between ES7L and Noc2p in 27SA3 pre-rRNA processing.
ES7L coordinates incorporation of Noc2p into pre-ribosomes
Previous studies demonstrated that ES7L provided binding sites for specific assembly factors in ribosome biogenesis (15,48–51). Therefore, we tested whether ES7L could facilitate the recruitment of Noc2p into the assembly intermediates of ribosomes (pre-ribosomes), which were assayed by comparing the amounts of Noc2p in pre-ribosomes between S.c-25S and C.a-ES7L. Pre-ribosomes were purified by affinity-purification using Rpf2p as the bait protein. Rpf2p assembled early into 90S pre-ribosomes containing 35S pre-rRNA and remained associated with the pre-ribosomes until dissociation from the later pre-ribosome particles containing 27SB pre-rRNA (52). As shown in Figure 4E, the amount of Noc2p in pre-ribosomes was reduced in C.a-ES7L compared with S.c-25S, although Nop2p was equally expressed in both strains. These results indicated that replacing ES7L with the counterpart of C. albicans attenuated the incorporation of Noc2p into pre-ribosomes. In contrast, the Noc2p mutant (Noc2p K384R) could efficiently assemble into pre-ribosomes in C.a ES7L (Figure 4E). Moreover, similar results were obtained in strains with whole 25S rRNA replaced (Supplementary Figure S3E). These findings suggested that ES7L mediated the recruitment of Noc2p into pre-ribosomes, which was weakened by the ES7L substitution.
Noc2 ortholog from C. albicans rescues defects of ES7L substitution in S. cerevisiae
Since species-specific variations in ES7L perturbed the recruitment of Noc2p into pre-ribosomes, the question arose of whether Noc2p coevolved with ES7L to adapt to the variations in ES7L to ensure proper function. To address this question, S.c Noc2p in strains containing C.a-ES7L or C.a-25S rRNA was replaced by C.a Noc2p (57% amino acid identity compared to that of S.c Noc2p, see Supplementary Figure S4). Notably, we observed that the growth defect caused by C.a-ES7L or C.a-25S was largely recovered, similar to the suppressor strain with the Noc2p K384R mutation (Figure 4C and Supplementary Figure S3A). In addition, the amount of mature 25S rRNA in these strains was also upregulated to levels comparable to those of the wild-type strain (Supplementary Figure S3B and S3C). Moreover, when we quantified the amount of relevant pre-rRNAs, we observed that the presence of C.a Noc2p led to a substantial reduction in 27SA3 accumulation and an increase in 27SB and 7S pre-rRNAs (Figure 4D and Supplementary Figure S3D), indicating that the processing defects of 27SA3 pre-rRNA in these strains were greatly alleviated. Finally, we analyzed the assembly of C.a Noc2p and S.c Noc2p into the C.a-ES7L-containing pre-ribosomes. As shown in Figure 4E and Supplementary Figure S3E, the amount of Noc2p in pre-ribosomes was restored to that of the wild type in strains carrying either the C.a Noc2p or Noc2p K384R mutation, presumably resulting in the formation of pre-ribosomes with improved function. In addition, we observed that the levels of C.a Noc2p and S.c Noc2p K384R in S.c ES7L rRNA-containing pre-ribosomes were also increased (Supplementary Figure S5), which indicated that the increased affinity of C.a Noc2p and S.c Noc2p K384R toward pre-ribosomes was independent of ES7L.
ES7L and Noc2p coordinate together to facilitate the assembly of the A3 factor Erb1p into the early pre-ribosomes
In recent decades, approximately 200 assembly factors have been identified, and 12 of them have been assigned to the 27SA3 pre-rRNA processing step (designated A3 factors) (51). Of these A3 factors, Erb1p serves as a multivalent interaction hub that wraps around the pre-ribosome particles and interacts with different rRNA domains and other protein factors (15). Notably, Erb1p was previously shown to bind with ES7L in RNA-protein pull-down experiments using ES7L as a bait (49), which suggested a role played by ES7L in recruiting Erb1p into pre-ribosomes. To address this hypothesis, we analyzed the level of Erb1p assembled into pre-ribosomes. As shown in Figure 5A, Erb1p was greatly diminished in C.a-ES7L-containing pre-ribosomes, demonstrating that proper ES7L was required for the recruitment of Erb1p into pre-ribosomes. In addition, replacing ES7L also decreased the association of other protein factors (Rrp1p, Rpl6p and Rpl7p) with pre-ribosomes (Figure 5A), which bound directly or around ES7L in pre-ribosomes after Erb1p and were required for proper A3 processing (15,53). In contrast, the levels of Erb1p and other downstream proteins were increased in strains carrying either the C.a Noc2p or Noc2p K384R mutation, indicating the role played by Noc2p in coordinating the assembly of Erb1p and these factors into pre-ribosomes. In fact, Noc2p was previously found to interact with Erb1p based on a high-throughput yeast two-hybrid (Y2H) screening assay (54). Next, to explore the potential interaction between Noc2p and Erb1p (as well as 11 other A3 factors), a yeast two-hybrid (Y2H) assay was performed. Pairwise interactions were assayed based on whether they could drive marker gene expression and promote Y2H test strains growth on selective plates (Figure 5B and Supplementary Figure S6) (32). Of the 12 assayed pairwise interactions, the previously reported Erb1p-Noc2p interaction was recapitulated as the only positive interaction pair (Figure 5B). Furthermore, we successfully reconstituted the interaction between recombinant Noc2p and Erb1p produced in E. coli and showed that recombinant Erb1p was coimmunoprecipitated with Noc2p (Figure 5C), indicating that Noc2p and Erb1p interact directly with each other. In addition, the downstream protein Rpl6p showed a considerably decreased association with pre-ribosomes upon ES7L replacement and was reverted by Noc2p mutations (Figure 5A), indicating that ES7L swapping impaired Rpl6p assembly into pre-ribosomes, which could be functionally rescued by Noc2p mutations. To further evaluate potential physical interactions among Noc2p, Erb1p and Rpl6p, the Y2H assay was performed. We failed to detect binary interactions between Rpl6p and Noc2p or between Rpl6p and Erb1p in the experimental set-up used in this study (Supplementary Figure S7), suggesting that the effect of Noc2p on RpL6p assembly might be indirect. Taken together, these results suggest that ES7L and Noc2p coordinate to facilitate the recruitment of Erb1p and downstream factors into early pre-ribosomes, which is a critical pre-requisite for the subsequent processing of 27SA3 pre-rRNA.
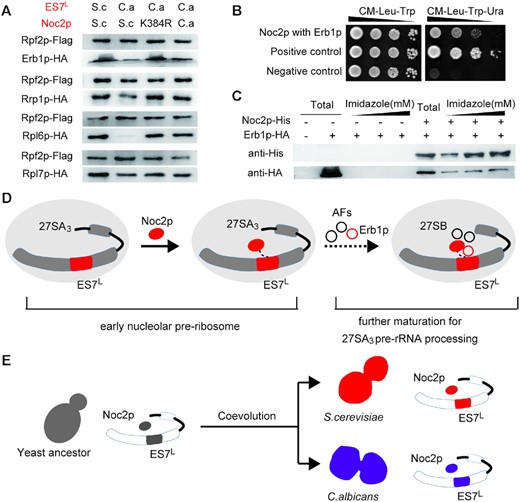
Proposed coevolution model of ES7L and Noc2p. (A) Western blot analysis of select AFs and r-proteins in the pre-ribosomes purified by Flag-tagged Rpf2p. (B) The yeast two-hybrid assay for interaction of Noc2p with Erb1p. (C) Validation of the interaction between Noc2p and Erb1p by coimmunoprecipitation. Western blot analysis of total lysate of cells (total) and immunoprecipitates eluted by different concentrations of imidazole (300, 500 and 800 mM) with anti-His6 and anti-HA antibody. (D) ES7L regulates the incorporation of Noc2p into early nucleolar pre-ribosomes, which later incorporated other AFs such as some well-known A3 factors (Erb1p, Rrp1 et al.) to promote further maturation of the pre-ribosomes to process 27SA3 pre-rRNA. (E) ES7L and Noc2p acted as a pair of coevolutionary module to specialize the ribosomal biogenesis pathway between S. cerevisiae and C. albicans.
DISCUSSION
Ribosomes in different species share an evolutionarily conserved RNA core, exhibiting flexible shells formed partially by the addition of species-specific rRNA moieties, which are thought to bestow unique characteristics to the ribosome (2). However, the physiological impacts of such species-specific rRNA variations and how cells evolved to accommodate such variations to ensure proper ribosome biogenesis and function have not been fully elucidated. In this study, we showed that despite high replaceability, eukaryotic 25S rRNAs specialized in the regulation of the pre-rRNA processing pathway across different yeast species. Specifically, the eukaryote-specific rRNA moiety ES7L and eukaryote-specific assembly factor Noc2p coevolved to ensure proper processing of the 27SA3 pre-rRNA in S. cerevisiae and the human pathogen C. albicans.
The biogenesis of a sufficiently stable and properly configured early 66S pre-ribosome involved numerous remodeling events driven in part by many assembly factors (AFs) and was a pre-requisite for processing 27SA3 to 27SB pre-rRNAs (51). Recent high-resolution pre-ribosome structures demonstrated that ES7L provided binding sites for some well-known A3 factors, such as Rpf1p, Erb1p and Rpl4p, which served structural roles to stabilize the pre-ribosomes to undergo further processing of the 27SA3 to 27SB pre-rRNAs (15,48–51). Of these A3 factors, Erb1p was successfully recruited by ES7L in RNA-protein pull-down assays, indicating a potential role of ES7L in recruiting Erb1p into pre-ribosomes. Indeed, our results demonstrated that ES7L regulated the association of Erb1p with pre-ribosomes, as ES7L replacement markedly decreased the level of Erb1p in pre-ribosomes. In this study, we found that ES7L regulated the association of another AF, Noc2p, in pre-ribosomes. Notably, concurrent K384R mutation of Noc2p or Noc2p replacement by the C. albicans ortholog reinforced the association of Noc2p with pre-ribosomes and restored the assembly of Erb1p into C.a-ES7L-containing pre-60S particles, probably via direct interaction, which successfully compensated for the unfavorable effects due to ES7L substitution. Notably, although K384 is conserved between S. cerevisiae and C. albicans, the residues adjacent to K384 show substantial variations in Ca. Noc2p (such as Q383C, L379Q, P378A and A388V), indicating that both mutations in K384 and adjacent residues could lead to topological changes in Noc2p and thus rescue the deficiency caused by ES7L replacement. Previous studies showed that Noc2p interacted with other large structural proteins, Noc1p and Rrp5p, to form a protein complex, which had already assembled in the ‘earliest’ nucleolar pre-ribosome particles before some of these A3 factors and provided a structural framework that might facilitate large-scale pre-ribosome folding before 27SA3 pre-rRNA processing (47,53,55). In this study, our results led us to hypothesize that ES7L and Noc2p could serve as functionally coordinated modules in the early 66S pre-ribosome, which facilitate the association of the A3 factor Erb1p through protein-RNA and protein–protein interaction networks to drive the compositional and conformational changes of the pre-ribosome to undergo further 27SA3 pre-rRNA processing (Figure 5D). However, based on the reported pre-60S structures, it is known that Erb1p is spanning through a large distance across the pre-ribosomes and the relative topological position between ES7L and Noc2p has not been elucidated. Accordingly, it remains possible that Noc2p and ES7L might coordinate together by nonphysical interaction (indirect functional complementation/adaptation) or via direct interaction and additional studies will be required. While the molecular details between ES7L and Noc2p remain unclear, multiple lines of contacts have been reported. Using ES7L as bait in RNA-protein pull-down experiments, a previous study identified the Noc2p partner protein Noc1p/Mak21p, rather than Noc2p, as an ES7L-binding protein (49). In addition, another known Noc1p-Noc2p partner protein, Rrp5, bound to a region of pre-rRNAs located close to ES7L and regulated 27SA3 pre-rRNA processing (47,55). Based on these results, although we cannot exclude a weak or transient direct interaction of Noc2p with ES7L, it appears feasible to incorporate these Noc2p partner proteins Noc1p and Rrp5p into the above ‘ES7L-Erb1p-Noc2p module’, which would serve as the bridging module between Noc2p and ES7L. Future work, especially a structural analysis of the Noc2p-containing pre-60S, might be performed to comprehensively explore these interaction networks and provide detailed mechanistic insight into how these protein and RNA components cooperate to drive the maturation of pre-60S.
The rRNA processing pathway was generally similar between S. cerevisiae and C. albicans (56). However, differences were observed previously with regard to this processing pathway (21,57). Based on the rRNA swap assay, our study demonstrated that ES7L substitution primarily accounted for the defects in C.a-25S, whereas the effects of variations in other ESs were mild, underscoring the role of ES7L in the specialization of the rRNA biogenesis pathway between C. albicans and S. cerevisiae. This finding makes sense, since ES7L is one of the largest and most variable ESs of the eukaryotic rRNA. However, considering that ESs not restricted to ES7L showed considerable variations among different species, a more comparative functional analysis of ESs from different species may give rise to a better understanding of the significance of these enigmatic species-specific rRNA moieties in the specialization of ribosomes. Notably, the results of our study demonstrated that the pre-rRNA processing defect due to ES7L substitution by C. albicans could be bypassed by concurrent replacement of the C. albicans Noc2 ortholog in S. cerevisiae, indicating that the eukaryote-specific rRNA moiety ES7L and the eukaryote-specific AF Noc2p might act as coevolutionary partners to specialize the ribosome biogenesis pathway between S. cerevisiae and C. albicans (Figure 5E). Furthermore, it has been previously pointed out that most of the AFs (>200) identified in S. cerevisiae were found throughout the eukaryotic tree of life (58), and emerging evidence has begun to uncover the role played by ESs in recruiting the eukaryote-specific assembly factors, such as Erb1p, Nsa1p, Rpf1p, Rrp1p, Rrp7p, Arx1p and Rlp7p (15–18,59,60). Hence, it is tempting to speculate that ESs may have coevolved with the AFs to drive the specialization of the ribosome biogenesis pathway across different eukaryotic species. More studies engineering species-specific rRNA elements in tandem with their cognate partners may help to elucidate the general and unique aspects of the ribosome and its regulation across different species in the future.
In conclusion, our findings demonstrated that the pervasive rRNA variations conferred specific characteristics to universal ribosomes across species, and such rRNA variations were accommodated by coevolved accessory AFs to ensure proper ribosome biogenesis and function, representing a potential evolutionary principle of naturally occurring ribosomal diversity. With these results, we foresee a new avenue to optimize engineered ribosomes (e.g. Ribo-T, which could incorporate multiple noncanonical amino acids into synthesized polypeptides but possess certain limitations, primarily due to imperfect biogenesis of the engineered ribosomes) by engineering the accessory AFs in tandem with rRNA (61–63).
DATA AVAILABILITY
MEGA X software implements many analytical methods and tools for molecular evolutionary analyses and can be downloaded from https://www.megasoftware.net free of charge. The 25S rRNA secondary structure of S. cerevisiae can be obtained from RiboVision (http://apollo.chemistry.gatech.edu/RiboVision/index.html). The RNAfold web server can predict secondary structures of single stranded RNA or DNA sequences (http://rna.tbi.univie.ac.at/). Ensemblis available at http://www.ensembl.org. SAMtoolsis available at http://samtools.sourceforge.net. GATKis available at https://software.broadinstitute.org/gatk/.
High-throughput sequencing files of genomic DNA have been deposited into the NCBI with the accession number PRJNA638206.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank members in Guanghou’s and Junbiao’s laboratory for constructive suggestions and thoughtful critiques of the work. We thank professor Wen Wang and professor Ruiwu Wang for materials sharing in the accomplishment of the experiments. We thank the editor Eric Westhof and anonymous reviewers for their careful work and thoughtful suggestions that have helped improve this paper substantially.
FUNDING
National Key Research and Development Program of China [2017YFA0505103]; National Natural Science Foundation of China [31971340, 31800080, 81901078]; Shenzhen Key Laboratory of Synthetic Genomics [ZDSYS201802061806209]; Shenzhen Science and Technology Program [KQTD20180413181837372]; Guangdong Provincial Key Laboratory of Synthetic Genomics [2019B030301006]. Funding for open access charge: National Key Research and Development Program of China [2017YFA0505103].
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments