





Abstract
The APPRIS database (http://appris-tools.org) uses protein structural and functional features and information from cross-species conservation to annotate splice isoforms in protein-coding genes. APPRIS selects a single protein isoform, the ‘principal’ isoform, as the reference for each gene based on these annotations. A single main splice isoform reflects the biological reality for most protein coding genes and APPRIS principal isoforms are the best predictors of these main proteins isoforms. Here, we present the updates to the database, new developments that include the addition of three new species (chimpanzee, Drosophila melangaster and Caenorhabditis elegans), the expansion of APPRIS to cover the RefSeq gene set and the UniProtKB proteome for six species and refinements in the core methods that make up the annotation pipeline. In addition APPRIS now provides a measure of reliability for individual principal isoforms and updates with each release of the GENCODE/Ensembl and RefSeq reference sets. The individual GENCODE/Ensembl, RefSeq and UniProtKB reference gene sets for six organisms have been merged to produce common sets of splice variants.
INTRODUCTION
It has been estimated that 95% of multi-exon human genes produce alternatively spliced messenger RNA (1,2) transcripts. These alternative transcripts, if translated, would generate a range of alternative proteins that are often strikingly different from the constitutive gene product and that would add to the repertoire of cellular functions (3,4). However, the cellular role of alternative splicing is a controversial topic (5–7) and the functional importance of any potential alternative protein isoforms is an open question (7).
APPRIS (8,9) was developed within the GENCODE (10) consortium to cope with the challenge of annotating alternatively spliced protein-coding transcripts with functional information. The database employs a series of modules to map protein structure and functional features and cross-species conservation to all reference splice isoforms. Unlike the other maintained databases that annotate alternative splice isoforms with functional information (11,12), APPRIS concentrates only on the most reliably predicted features, including the presence of Pfam domains (13) and highly conserved functional residues (14).
Information from APPRIS is fed back to the GENCODE manual annotators to inform gene models. However, the main role of APPRIS is the annotation of a main (principal) isoform for individual coding genes (15). APPRIS selects principal isoforms based on the presence or absence of evolutionary evidence such as conserved functional and structural motifs. Principal isoforms are those with the most preserved structural and functional features and those with the greatest cross species conservation, while alternative isoforms often have non-conserved exons and structure or function features that are damaged or missing (15). APPRIS core modules almost always agree on the principal isoform.
Historically researchers and annotators have had to resort to choosing the longest annotated CDS as the reference variant for individual coding genes (16). We have shown that this simple solution is not ideal (17) and as databases expand and more alternative transcripts are annotated the fix will become less viable. Highest Connected Isoforms (18), from RNAseq data and from protein–protein interaction and structural information have been proposed as an answer, but we have shown that these isoforms agree with the main functional isoform less often than the longest isoforms (5). The APPRIS principal isoforms are the splice variants that best represent the gene (17).
Although APPRIS was developed for use with GENCODE/Ensembl annotations (10,19), there are other manually annotated reference sets, in particular RefSeq (RefSeqGene) (20) and UniProtKB (16). The RefSeq, GENCODE/Ensembl and UniProtKB annotations are not identical and many gene models or predicted proteins are present in one or more reference sets, but not in others. For that reason we have extended APPRIS to the RefSeqGene and UniProtKB annotations in the case of vertebrate genomes (human, mouse, zebra-fish, rat, pig and chimpanzee). In addition, we have made improvements to core methods in the APPRIS pipeline, implemented the UCSC Track Hub to enhance annotation access and created Docker images to help execute the annotation pipeline.
THE DATABASE
APPRIS annotates splice isoforms with protein structural and functional features, and data from cross-species alignments. The database uses these features to select a single reference isoform for each protein-coding gene, here termed the principal isoform. This principal isoform has the most conserved protein features and the most evidence of cross-species conservation. At the same time isoforms that have lost conserved protein features or do not have cross-species conservation are flagged as alternative.
Currently the APPRIS annotation pipeline comprises six modules (9). Matador3D detects similarity to structural homologs in the PDB (21); firestar (14) predicts functionally important amino acid residues; SPADE identifies Pfam functional domains via the PfamScan algorithm (13); CORSAIR carries out BLAST (22) searches against vertebrate protein sequences to determine the number of orthologs that align correctly and without gaps; THUMP makes unanimous predictions of trans-membrane helices from three predictors (23–25); and CRASH predicts the presence and location of signal peptides using the SignalP and TargetP programs (26,27). APPRIS maps protein features to all coding transcripts. The databases implied in each method (PDB, Pfam, non-redundant sequence database, etc.) are updated periodically to get most correct annotations.
Refinements to core methods
All modules in APPRIS are continually revised against the GENCODE annotation of the human reference gene set. As a result we have been able to improve the performance of each of the core modules in APPRIS. The gold standard set for principal isoforms are those genes with just one CCDS variant (consensus coding sequence, 28). Tests have shown that unique CCDS variants (transcripts annotated consistently by RefSeq and Ensembl/GENCODE manual annotators) and APPRIS principal isoforms are both highly reliable predictors of the dominant cellular isoform, so they should select the same reference isoform for the vast majority of genes.
Comparison between unique CCDS isoforms for each gene and those selected by the individual APPRIS modules shows that there is almost complete agreement between the two, both at the time of the initial database publication (8) and with the current version of APPRIS. The more recent APPRIS principal isoforms disagree with the unique CCDS isoforms less often and with the exception of CORSAIR (98.92%), all methods have more than 99% agreement with unique CCDS variants (Supplementary Figure S1).
Reliability scores
Many experiments require every studied gene to have a single representative, so APPRIS now automatically selects a principal isoform for every single coding gene. However, not all APPRIS principal isoforms are alike. Principal isoforms are tagged with a score from 1 to 5 depending on the reliability of the selection, with 1 being the most reliable. ‘PRINCIPAL:1’ isoforms are determined solely using information from the APPRIS core modules. For those genes where the modules cannot make a unique selection, APPRIS uses external data such as the CCDS annotation and the GENCODE Consortium Transcript Support Level (29). Where all else fails, the longest not previously rejected isoform is selected as the principal (‘PRINCIPAL:5’). Splice variants rejected as principal isoforms by the APPRIS core modules are labeled as ‘MINOR’, while those variants not rejected by the core modules, but rejected using external information are labeled as ‘ALTERNATIVE’ (for more details on the reliability scores see the Supplementary Data).
Additional features
Annotations are stored in a MySQL relational database and these can be downloaded via the APPRIS web site. The human and mouse annotations are available through GENCODE, and Ensembl exports APPRIS principal isoforms of human, mouse, zebra-fish, rat and pig within its website, BioMart data-mining tool and API. Furthermore, APPRIS annotations can be visualized in the UCSC Genome Browser (30) from its own Public Track Hub. In addition, users can extract APPRIS annotations for specific reference sets (Ensembl, RefSeqGene, UniProt) via the APPRIS WebServer and WebServices (9). All the APPRIS source code is available in a GitHub public-repository (https://github.com/appris/appris/) offering a distributed version control.
The APPRIS pipeline is executed on the Linux (Ubuntu) system but it can be run on Windows, Mac OS X or Unix-based systems using the Docker image (appris/core) provided by the software container platform, Docker (http://www.docker.com). The APPRIS-Docker image is stored in the public Docker Hub (https://hub.docker.com/).
New APPRIS annotations
The APPRIS database has increased in size to cover six different vertebrate genomes (human, mouse, rat, pig, zebra-fish, chimpanzee), and two invertebrate genomes (Drosophila and Caenorhabditis elegans). The human GENCODE gene set (release 24) recognizes 20 250 protein-coding genes and APPRIS determines a P1 isoform for 76.8% of these genes (see Figure 1A). The mouse GENCODE gene set (release M12) has 22 538 protein-coding genes and 82.6% are tagged with a P1 isoform (see Figure 1B). More than 90% of the genes in the Ensembl annotation of three vertebrate species (rat, pig, chimpanzee) have P1 isoforms. The number is higher for these species because the majority of genes have a unique CDS (see Supplementary Figure S2).
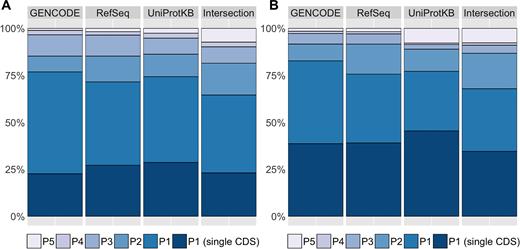
Bar-plots with the percentage of genes identified with the final annotations of APPRIS for the human (A) and mouse (B) species house in database. APPRIS identifies a principal isoform (Pn) for each gene that are tagged with numbers from 1 to 5, with 1 being the most reliable. Isoforms in genes with a unique protein representative (single CDS) are automatically categorized as P1. The APPRIS Database annotates the protein-coding genes in all public sets GENCODE, RefSeq and UniProtKB. In addition, we established a common gene set (Intersection) with the GENCODE, RefSeq, and UniProtKB reference sets.
RefSeqGene and UniProtKB annotations
APPRIS has now been extended to the other main public genome annotation, RefSeqGene and to the UniProtKB proteome. RefSeqGene human (release 107) currently houses 20 066 protein-coding genes, while the UniProtKB human proteome (release 2016_06) has 21 608 genes. APPRIS identifies a P1 principal isoform for 71.5% of genes in the RefSeqGene set, and 74.3% of genes in the UniProtKB proteome (see Figure 2A).
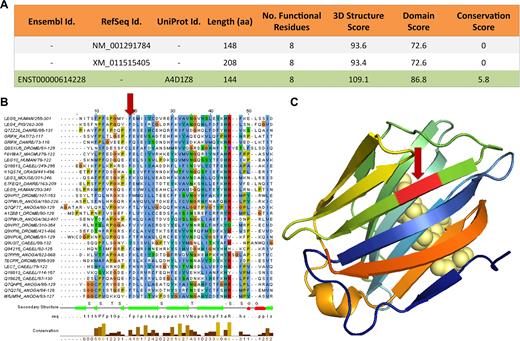
APPRIS annotations for gene GRIFIN. (A) APPRIS results for the three protein-coding variants from the gene reference sets, GENCODE (Ensembl), RefSeq and UniProtKB. APPRIS chooses isoform ENST00000614228+A4D1Z8 as the principal isoform (highlighted in green), which belongs to Ensembl and UniProtKB. A selection based on the 3D structure, the functional domains and the conservation in related species. (B) Alignment for a section of the Pfam galectin family of proteins. The red arrow shows where 8 extra residues in the RefSeq variants would disrupt a region of the galectin functional domain of GRIFIN. (C) The 3D structure of 4LBJ (human galectin-3 CRD) that has 29% identity with variants ENST00000614228+A4D1Z8. The galectins are a family of proteins defined by their binding specificity for β-galactoside sugars (displayed in light yellow spheres). The red arrow shows where the 8 extra residues would have to insert into the structure, breaking a β-sheet.
The pipeline of annotations in the RefSeqGene set is identical to that of GENCODE/Ensembl, but two of the modules used in the pipeline (Matador3D, and CORSAIR) have had to be modified for use with the UniProtKB and Intersection (see below) gene sets because the original versions of these modules made use of genomic coordinates.
Intersection gene sets
We have also created merged gene sets for vertebrate species by cross-referencing the GENCODE/Ensembl, RefSeqGene and UniProtKB reference sets. For the human genome we established a common gene set (Intersection) with the GENCODE (release 24), RefSeqGene (release 107) and UniProtKB (version 2016_06) reference sets. The initial cross-reference was generated with the data-mining tool, BioMart (31) and from there we re-annotated the cross-database relationships manually. For the remaining species we generated common gene sets with the BioMart tool, although these relationships are not yet manually annotated. The version and the number of genes for each reference set are shown in Supplementary Table S1.
There were a total of 22 207 protein-coding genes in the human intersection reference set composed of GENCODE (release 24), RefSeqGene (release 107) and UniProtKB (version 2016_06) genes. Just 5132 (23.1%) of these genes have a single CDS variant, while APPRIS determined P1 principal isoforms for 9204 (41.4%) of the genes (see Figure 1A).
The merged Intersection gene set allows us to identify principal isoforms missing in the individual gene sets. For example the principal isoform from the merged set for GRIFIN is annotated in GENCODE (ENST00000614228) and UniProtKB (A4D1Z8), but not in RefSeqGene (See Figure 2). This principal isoform is chosen because it maps better to known 3D structures, has an unbroken Pfam domain and has orthologous sequences in vertebrate species. In contrast, the domain in the RefSeqGene isoforms is broken and neither isoform has cross-species conservation. The 8-residue insertion in the two RefSeqGene variants breaks a Pfam functional domain (Figure 2B) and 3D structure (Figure 2C). The C-terminal extension in the GENCODE/Ensembl/UniProtKB principal isoform (but not in the RefSeqGene variants) is also established in mammals (see Supplementary Figure S3).
DISCUSSION
APPRIS annotate alternatively spliced protein isoforms with protein structural and functional information and cross-species conservation using a range of computational prediction methods. It also selects one of these isoforms to be the representative protein sequence for each coding gene.
We have shown that a single representative protein reflects the biological reality of the cell: most coding genes have a single dominant protein isoform (5,7,17) and this seems to be true regardless of cell type (17). This dominant protein isoform is almost always the APPRIS principal isoform: APPRIS principal isoforms overwhelmingly coincide with the manually annotated unique CCDS variants and with the main isoforms detected in large-scale proteomics experiments (17). In fact where dominant isoforms could be determined for all three methods, the agreement was 99.5% (17). Further corroboration of the importance of APPRIS principal isoforms comes from large-scale genetic variation studies, which show that exons from principal isoforms are under purifying selection. By way of contrast alternative exons are under neutral selection (5,32).
APPRIS principal isoforms have a wide range of uses. Designating a single alternative splice variant as principal is an important technical issue and is a critical first step for any genome-wide analysis. Large-scale analyses are highly dependent on the quality of input data; so principal isoforms should improve the reliability of these experiments. Determining whether an exon belongs to a principal or alternative variant is key in biomedical studies. APPRIS principal isoforms can also be useful when working with individual genes; since it is not always clear which splice isoform (or isoforms) is functionally important.
APPRIS principal isoforms and annotations are freely accessible to all via the APPRIS web page, via the APPRIS WebServices (9), and the Ensembl reference annotations for individual species.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [U41 HG007234, 2U41 HG007234]; Spanish Ministry of Economics and Competitiveness [BIO2015-67580-P]; Spanish National Institute of Bioinformatics (www.inab.org) [INB-ISCIII, PRB2 to J.M.R.]; ProteoRed [IPT13/0001-ISCIII-SGEFI/FEDER to J.V.]; Joint BSC-IRB-CRG Program in Computational Biology and Award Severo Ochoa [SEV 2015-0493 to A.V.]. Funding for open access charge: U.S. Department of Health and Human Services; National Institutes of Health; National Human Genome Research Institute [2U41 HG007234].
Conflict of interest statement. None declared.
Present addresses:
Jose Manuel Rodriguez, Cardiovascular Proteomics Laboratory, Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain.
Juan Rodriguez-Rivas, Barcelona Supercomputing Centre (BSC), Barcelona 08034, Spain.

{kind=link}
{kind=link}
Comments