





Abstract
In 2020, the Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease (DAPA-CKD) trial first demonstrated that inhibition of the sodium–glucose transporter-2 (SGLT2) with dapagliflozin attenuates the progression of chronic kidney disease (CKD) with proteinuria in patients with or without diabetes at an unprecedented effect size. These results have far-reaching implications for a series of traditional concepts in Nephrology. It now became obvious that CKD with and without diabetes involves a predominant SGLT2-driven pathophysiology compared with the other pathogenic pathways currently under consideration. As SGLT2 inhibition is similarly efficacious in diabetic and non-diabetic CKD with proteinuria, treating CKD rather than ‘diabetic nephropathy’ becomes the central paradigm. Indeed, in older adults with type 2 diabetes, CKD is rather of multifactorial origin. As the DAPA-CKD trial included more patients with immunoglobulin A nephropathy (IgAN) than any of the previous IgAN trials, dual renin-angiotensin/SGLT2 inhibition may become the new standard. The same applies for patients with podocytopathy-related focal segmental glomerulosclerosis lesions. From now on, IgAN and podocytopathy trials without SGLT2 inhibition as background therapy and without glomerular filtration rate decline as primary outcome criterion will be of limited value. These and other potential implications will trigger broad discussions and secondary research activities with conclusions difficult to predict today. However, one is for sure: Nephrology after the DAPA-CKD trial will be not the same as it was before. Finally!
INTRODUCTION
Chronic kidney disease (CKD) is defined by persistent structural or functional abnormalities of the kidneys that frequently lead to a progressive loss of kidney function as defined by a decline in glomerular filtration rate (GFR) [1]. CKD affects hundreds of million people around the globe often without knowing, as the progression of CKD leaves patients largely asymptomatic up to when they reach the stage of kidney failure. In addition, advanced stages of CKD are a major risk factor for cardiovascular (CV) morbidity and mortality. Indeed, many CKD patients experience or die from CV events before reaching the stage of kidney failure. Therefore, the current management of CKD includes:
identifying and controlling the underlying cause of CKD. However, frequently the specific cause remains unknown or CKD is multifactorial. Thus, specific treatments are either not available or elicit only little effect on CKD progression;
controlling shared pathomechanisms of CKD progression such as activation of the renin–angiotensin–aldosterone system (RAAS), but not correcting hyperuricaemia [2]. Inflammation and fibrosis remain attractive. Ageing-related nephron loss and obesity are well recognized in this context but difficult to target;
avoiding exposure to nephrotoxins and preventing episodes of acute kidney injury (AKI); and
controlling hypertension, hyperlipidaemia and CKD-related anaemia to reduce the CV risk.
Nevertheless, CKD frequently progresses to kidney failure [3], which remains a death sentence in many parts of the world due to limited accessibility and affordability of kidney replacement therapies. Thus, finding better ways to prevent CKD progression and related CV events are a major global medical need.
In 2020, the Dapagliflozin and Prevention of Adverse Outcomes in Chronic Kidney Disease (DAPA-CKD) trial demonstrated protective effects on the progression of CKD at an unprecedented effect size of around 40% [4]. The effect size for preventing CV death was 19%. While subgroup analysis of this trial and the results of other CKD trials with sodium–glucose transporter-2 (SGLT2) inhibitors (SGLT2is) are pending, the data available so far already imply that the existing concepts in CKD, hierarchy of pathomechanisms and priorities for possible treatment targets deserve reconsideration (Table 1).
PATHOPHYSIOLOGY OF CKD
Pathophysiology of CKD before DAPA-CKD
Kidney lifespan and nephron number
In healthy people, ageing-related nephron loss prompts a moderate kidney sclerosis and atrophy with a moderate decline in GFR that assures a kidney lifespan exceeding total lifespan (Figure 1) [5]. In contrast, poor nephron endowment at birth and/or injury-related nephron loss shorten kidney lifespan, especially in patients suffering from persistent kidney injuries or excessive functional overload, e.g. in pregnancy, obesity or sodium- and protein-rich diets [1]. Each lost nephron increases the workload on the remnant nephrons accelerating also their demise, up to when nephron number is too low to avoid uraemia. Unfortunately, a marker of nephron number suitable for clinical practice is not yet available [6].
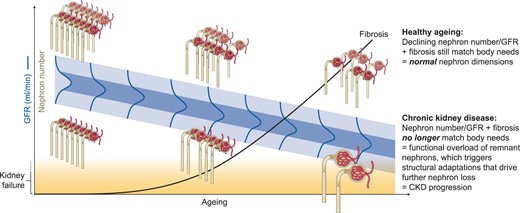
Decline of GFR with age. Already at young age, the level of GFR follows a Gaussian distribution (green) within the population depending on the number of nephrons (endowment, nephrons in grey) at the end of kidney development. Ageing implies a linear nephron loss and fibrosis (black line) for everyone, but those with poor nephron endowment may reach the point where nephron number no longer matches the metabolic needs of the body (kidney failure indicated in yellow), also depending on body size and weight. Healthy kidney ageing is also obvious from the lack of adaptive changes in kidney dimensions indicating that a declining number of nephrons is still in balance with the metabolic needs of the ageing body. A mismatch in nephron number and metabolic needs induces adaptive changes indicated by an increase in nephron dimensions. In advanced CKD, only few ‘megalonephrons’ remain. The current KDIGO classification ignores healthy ageing and defines many elderlies with reduced GFR as suffering from CKD, which many feel to be inappropriate. Proteinuria may be one non-invasive way to distinguish CKD from healthy kidney ageing in patients with a reduced GFR.
Progressive increase of remnant nephron dimensions
Healthy kidney ageing implies nephron loss at normal dimensions of the remnant nephrons as an indicator of absent functional overload [5] (Figure 1). Thus, low GFR in the elderly occurs in the absence of proteinuria most likely does not represent ‘disease’, whether or not this is in conflict with the current CKD classification [7]. In contrast, any adaptive increase in nephron dimensions represents a structural adaptation to functional nephron overload, i.e. glomerular hyperfiltration and tubular hyperreabsorption [7]. Up to a certain threshold, this compensation does not drive further nephron loss and CKD progression, e.g. in most living kidney donors [8]. What determines this threshold? The tipping point is when the degree of glomerular hyperfiltration-related podocyte shear stress ultimately leads to podocyte detachment, proteinuria and first focal segmental and later diffuse global glomerulosclerosis [9, 10]. The same principle likely also applies to the proximal tubule, where metabolic overload may promote irreversible detachment and loss of proximal tubular cells via the urine, ultimately promoting tubule atrophy. Thus, any mismatch between nephron number and body size, body weight or other conditions of increased metabolic demands that trigger an excessive adaptation of nephron dimensions promote the progression of CKD and hence shorten kidney lifespan [1].
Biomarkers of CKD pathophysiology: estimated GFR (eGFR) and proteinuria
The Kidney Disease: Improving Global Outcomes (KDIGO) defines the stages of CKD via estimated GFR (eGFR) and barrier function (proteinuria) [11], which has proven useful despite significant drawbacks. For example, apart from misclassifying healthy kidney ageing as CKD, eGFR may not adequately cover distal nephron excretory mechanisms, because eGFR formulas were designed to predict measured GFR instead of creatinine clearance [12]. This, by concept, makes it more difficult for drugs that act mainly on interstitial fibrosis around the distal tubules to prove efficacy in terms of CKD progression compared with drugs that act mainly on glomerular pathomechanisms. In contrast, proteinuria and especially albuminuria, with the exception of proximal tubule dysfunction, indicates leakage through to the glomerular filtration barrier [13]. However, whether glomerular proteinuria indicates the activity of uncontrolled primary glomerulopathies or podocytopathies, adaptive hyperfiltration or glomerular scarring is often difficult to determine in clinical practice [13]. Indeed, some use proteinuria as a marker of disease activity, e.g. in lupus nephritis or podocytopathy with minimal change lesions, some as marker of scarring, e.g. in podocytopathy with lesions of focal segmental glomerulosclerosis (FSGS) and some as marker of glomerular hyperfiltration, e.g. in CKD with diabetes, although in a single patient the respective contribution of each mechanism remains speculative [13].
Inflammation and fibrosis
Kidney pathology and transcriptome analysis consistently reveal inflammation and fibrosis to predict a progression of CKD, fueling the assumption that association would imply causation. Indeed, inflammation and fibrosis as possible targets for CKD progression have stimulated generations of researchers to explore the cellular and molecular details of kidney inflammation and fibrogenesis. However, clinical trials that would demonstrate a clinically significant contribution to CKD progression are scarce. Whether this relates to the lower sensitivity of eGFR and proteinuria to mirror the functional relevance of targeting inflammation and fibrosis or to a lack of causation for CKD progression remains unknown.
Pathophysiology of CKD after DAPA-CKD
SGLT2is have been developed as antidiabetic drugs and their potent effects on CV morbidity, heart failure and CKD progression in patients with type 2 diabetes (T2D) came as unexpected [14–16]. While the diuretic effect may partially explain the beneficial effects on heart failure, the strong effect size on kidney outcomes requires a different explanation. For example, diabetes is unique in involving SGLT2 for a deactivation of the tubuloglomerular feedback and thereby installing a persistent glomerular hyperfiltration and hypertension, known to accelerate proteinuria, glomerulosclerosis and the progression of CKD [17, 18]. However, among the 1398 patients with non-diabetic CKD in the DAPA-CKD trial, the primary outcome showed a hazard ratio (HR) of 0.5 (95% confidence interval 0.35–0.72) for participants without diabetes [4]. These results undisputedly identify the SGLT2i dapagliflozin as a renoprotective agent at an unprecedented potency on top of RAAS inhibition [4]. Effect size was consistently large for other relevant endpoints including decline in GFR [HR = 0.53 (0.42–0.67)], kidney failure as defined by a GFR <15 mL/min/1.73 m2 [HR = 0.64 (0.5–0.82)] and death from CV causes [HR = 0.81 (0.58–1.12)] [4]. Likely, other SGLT2i will demonstrate similar results as the consistent results of these drugs in diabetes trials suggest a class-effect.
The discussions about what mechanism-of-action could account for this enormous effect size in the absence of hyperglycaemia as the apparent driver of inactivation of the tubuloglomerular feedback have just started. One possibility is that the sensitivity for a deactivation of the tubuloglomerular feedback is different in CKD kidneys compared with healthy kidneys [19]. Indeed, CKD is associated with impaired autoregulation of glomerular perfusion and a higher impact of systemic blood pressure (BP) on intraglomerular pressure and proteinuria [20]. Secondly, in the absence of diabetes, 300 g of glucose, normally filtered by a full number of nephrons, in CKD is filtered by a much lower number of nephrons. This implies that the amount of glucose exposure to each remnant nephron increases significantly and can drive deactivation of tubuloglomerular feedback just as in the diabetic kidney [21]. Thirdly, SGLT2 is a central transporter involved in the functional overload of remnant nephrons, which have to reabsorb excessive amounts of sodium, protein and other solutes of the glomerular filtrate, which contribute to the metabolic stress on tubular epithelial cells of CKD kidneys especially with concomitant proteinuria [21]. SGLT2 inhibition may considerably relieve this metabolic stress from proximal tubular cells, which may explain better kidney function performance and outcomes.
The strong effect size of dapagliflozin on non-diabetic CKD strongly suggests that haemodynamic and metabolic effects play a major upstream role in the pathophysiology of CKD progression, which puts into question the significance of the previously favoured pathomechanisms of ‘inflammation’ and ‘fibrosis’ (Figure 2). Numerous experimental studies document that inhibiting leucocyte recruitment or activation or blocking fibroblast proliferation or activation reduces interstitial fibrosis, although few experimental models actually evaluated the respective impact on GFR. Those that did usually found an effect size in rodent models [22–24] far below what has been found with modulators of kidney haemodynamics and glomerular hyperfiltration, e.g. RAASis [25]. One explanation may be that the concept of ‘inflammation’ as defined by the presence of immune cells is no longer valid. It has become increasingly clear that chronic tissue remodelling involves immune cells with predominately anti-inflammatory and pro-repair functions [26]. Here and there, drugs are presented as ‘anti-fibrotic’ drugs although they elicit also other or even pleiotropic effects [27]. The latter also applies to the mineralocorticoid receptor antagonist finerenone [28], hence any effect reached in the Finerenone in Reducing Kidney Failure and Disease Progression in Diabetic Kidney Disease (FIDELIO-CKD) trial cannot be specifically attributed to blocking renal fibrogenesis [29]. Indeed, FIDELIO-CKD reported an early dip of GFR as a sign of haemodynamic effect similar to the SGLT2is [30]. Similarly, targeting kidney inflammation gave only moderate effect sizes in rodent models on excretory kidney function [31], translating into small effects in humans [32].
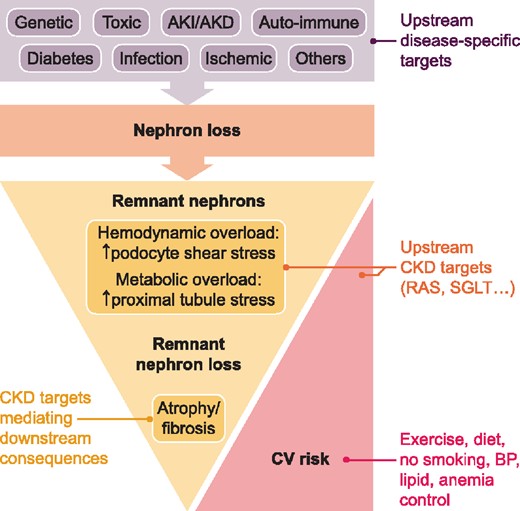
Updated concept of CKD progression. Nephron loss is the defining pathophysiology underlying the progression of CKD. Many different insults and diseases can trigger nephron loss and identifying these upstream triggers offers the opportunity for treatments targeting these upstream causative mechanisms to limit any further disease-specific nephron loss. However, all CKD patients benefit from targeting the unspecific mechanisms of further nephron loss, which all relate to the adaptive changes initiated by a mismatch of metabolic needs and remnant nephron capacity. These adaptive changes affect the glomerulus (single nephron hyperfiltration, increase in dimensions, podocyte shear stress and podocyte detachment) and the proximal tubule (single nephron hyperreabsorption, oxidative stress and loss of tubular epithelial cells, e.g. as frequently seen in the urine of patients with nephrotic syndrome). Functional overload leads to demise of more and more nephrons that further increase more and more the functional overload on the fewer and fewer remaining nephrons. Reducing the functional overload on the remnant nephrons is an upstream unspecific CKD treatment target, e.g. by drugs that reduce hyperfiltration and hyperreabsorption. Targeting the consequences of nephron loss, i.e. tissue remodelling involving inflammation and fibrosis, are downstream targets that can affect CKD progression at a limited effect size. Reducing the CKD-related CV risk is another major treatment target. Obviously, drugs with dual effects on CKD progression and as well as CV risk, such as inhibitors of the renin–angiotensin system (RAS) and of SGLTs, are of particular importance in the management of CKD. AKD, acute kidney disease.
DIABETIC NEPHROPATHY VERSUS CKD WITH DIABETES
‘Diabetic nephropathy’ before DAPA-CKD
The global rise of obesity and diabetes increased the awareness of ‘diabetic nephropathy’, a well-known microvascular complication upon 10–15 years of type 1 diabetes (T1D). The concept of ‘diabetic nephropathy’ was repurposed in patients with T2D despite obvious differences in the time between onset of diabetes and kidney disease, and biopsy studies documenting numerous alternative or combined kidney pathologies underlying the so-called ‘diabetic nephropathy’ [33]. The term ‘diabetic kidney disease’ was not of help, because it still implied diabetes as the central causative pathomechanism. Experimental studies focused on hyperglycaemia as the presumed direct driver of mesangial expansion, podocyte dysfunction, inflammation and glomerulosclerosis, because the haemodynamic alterations of kidney perfusion were difficult to study ex vivo [34]. In addition, the vast majority of preclinical studies were performed in lean rodent models of T1D instead of obese models of hyperinsulinaemic T2D [35]. Companies kept searching for cures for a doubtful global prevalence of ‘diabetic nephropathy’, and clinical trials on ‘diabetic nephropathy’ regularly included patients with CKD of unknown cause probably sharing little pathophysiology with the preclinical models [33, 36]. Consequently, most trials failed, with the exception of RAASis showing a moderate effect size [37]. Finally, SGLT2is consistently demonstrated a robust nephroprotective effect, first in trials with primary CV outcomes [16, 38, 39]. Subsequently, the Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) trial confirmed this concept for a primary kidney endpoint [15]. SGLT2is appeared to elicit specific effects on the pathogenesis of ‘diabetic nephropathy’. However, the weak effects on body weight, glucose control and BP could not explain the strong effect size on kidney endpoints. As a first hypothesis, restoration of the tubuloglomerular feedback was named as the central mechanism-of-action [18], followed by numerous others that also better explain the associated benefits on heart failure and CV events [40].
‘Diabetic nephropathy’ after DAPA-CKD
The results of the DAPA-CKD trial overt that the mechanisms-of-action of SGLT2 inhibition are not specific to ‘diabetic nephropathy’ and that SGLT2is are potent in patients with CKD even despite diabetes [4]. Thus, patients included into trials of ‘diabetic nephropathy’ are CKD patients with diabetes and the main treatment target is CKD and not ‘diabetic nephropathy’ [33]. Indeed, for patients with CKD and diabetes, the term ‘diabetic nephropathy’ should no longer be used unless the predominant role of diabetes for the CKD is proven by kidney biopsy. As a consequence, the 2020 KDIGO guideline on diabetes treatment in CKD declares the term ‘diabetic nephropathy’ as outdated and avoids it in the context of CKD in older adults with T2D. In many patients, diabetes is mostly a risk factor for an accelerated progression of CKD because diabetes augments hyperfiltration-related podocyte loss and glomerulosclerosis but T2D is frequently not the main cause of CKD [33]. As such, diabetes should be treated as one of many risk factors for CKD progression, e.g. with antidiabetic drugs but specific targeting of ‘diabetic nephropathy’ seems inappropriate. CKD itself has its own shared molecular targets and SGLT2 is now a treatment target for CKD and no longer only for ‘diabetic nephropathy’ (Figure 2). The efforts of stakeholders in the field such as academic and industry researchers and regulatory authorities should drop the concept of ‘diabetic nephropathy’ in favour of addressing CKD to improve the outcome of patients with CKD and diabetes. It will be important to also implement these new insights in medical teaching at the medical schools, medical congresses and the medical literature to overcome the conceptual hurdle of ‘diabetic nephropathy’ for the global challenge of CKD in obese patients with diabetes.
IgA NEPHROPATHY
IgA nephropathy before DAPA-CKD
Immunoglobulin A nephropathy (IgAN) is a common form of immune complex glomerulonephritis involving loss of tolerance, adaptive immunity and sometimes, systemic manifestations [41]. IgAN is a well-recognized form of progressive CKD with distinct clinical and histopathological risk factors for disease progression. An aberrant immune response is the central upstream pathomechanism and indeed, rapid-progressive forms of IgAN respond well to immunosuppressive therapy. In analogy, numerous trials tested the concept that steroids and other immunosuppressive drugs could elicit relevant protective effects on the progression of IgAN, albeit with disappointing results at the cost of significant side effects [42–44]. Indeed, the STOP IgA trial revealed that the rigorous conservative run-in treatment consisting of usual measures of CKD control almost entirely controlled the clinical problem, with few patients showing further GFR decline [42]. Thus, in chronic forms of IgAN, the idea of targeting CKD has gained supporters, although many keep adhering to the idea of targeting the upstream immunopathogenesis of IgAN and numerous trials with immunosuppressive drugs are currently being conducted [45, 46].
IgAN after DAPA-CKD
The DAPA-CKD trial included 271 patients with IgAN and could hence be considered the largest-ever IgAN trial so far [4]. Therefore, the DAPA-CKD trial has the potential to provide robust evidence on the role of SGLT2-mediated progression of CKD as a therapeutic target in IgAN. While a full subgroup analysis of primary and secondary outcomes in patients with IgAN has not been published, it was reported at Kidney Week 2020 that these results will be in line with the other forms of nondiabetic CKD. This would not only confirm the conclusion previously taken from the STOP IgAN trial that patients with IgAN benefit more from treating CKD than from targeting the immune system [42], but even reveal additive effects of dual RAAS/SGLT2 inhibition. In slowly progressing IgAN, the contribution of the immune system is too small for the benefits to outweigh the risks of immunosuppressive drugs [43]. As another consequence, the strong putative effect size of SGLT2is on the progression of IgAN puts the significance of all ongoing trials in doubt that did not include SGLT2 inhibition as a standard-of-care. It is likely that similar to the trials performed in the pre-RAASi era, it will remain uncertain whether drug effects can replicate on top of SGLT2 inhibition. From now on, IgAN trials will have to implement dual RAAS/SGLT2 inhibition as a background therapy and use GFR decline (instead of proteinuria) as the primary outcome.
PODOCYTOPATHIES WITH FSGS LESIONS
Podocytopathies with FSGS lesions before DAPA-CKD
FSGS has traditionally been considered and treated as a disease entity. However, the combination of proteinuria and FSGS lesions at kidney biopsy defines simply the presence of a podocyte injury that can result from numerous different causes, each of which define a specific diagnosis and eventually require a specific treatment [13]. Standard treatment for FSGS used to include glucocorticoids and other immunosuppressive drugs. In particular, calcineurin inhibitors are used as first-line immunosuppressive agents; however, only a variable subset of patients undergoes complete proteinuria remission [47]. Individuals not undergoing complete remission frequently develop progressive CKD. RAASis have become pivotal to control proteinuria, underlining the importance of haemodynamic effects in CKD progression in these patients. Consistently, sparsentan, a small molecule compound combining endothelin type A receptor blockade with angiotensin II type 1 receptor antagonism, showed more potent reduction in proteinuria after 8 weeks of treatment, versus irbesartan [48]. However, proteinuria and not GFR decline defines the primary outcome of most FSGS trials. Thus, there is a paucity of evidence on the progression of CKD, the ultimate clinically relevant outcome.
Podocytopathies with FSGS lesions after DAPA-CKD
The DAPA-CKD trial included 116 patients with a podocytopathy with FSGS lesions on a previous kidney biopsy. Although a subgroup analysis of primary and secondary outcomes in patients with podocytopathies with FSGS lesions is not yet available, it is tempting to speculate that all patients with FSGS lesions will benefit in terms of CKD progression. It will be important to work out whether those that respond to SGLT2 inhibition are those that did not benefit from immunosuppressive treatments before that are anyway the majority. Having available a drug that could delay progression of CKD in patients with podocytopathy with FSGS lesions and that could add on to the effects of RAASis would address a major unmet medical need for this group of patients. Future trials on podocytopathies with FSGS lesion should stratify patients by the underlying diagnosis and prioritize GFR decline as a primary endpoint over proteinuria.
OTHER FORMS OF CKD NOT INCLUDED IN THE DAPA-CKD TRIAL
CKD without proteinuria
The DAPA-CKD trial did not include patients without proteinuria, probably because proteinuria identifies patients at risk for progressive loss of kidney function. Whether patients with a low GFR, e.g. with ageing-related nephron loss or ischaemic nephropathy, can also benefit from SGLT2 inhibition is an important question, but remains currently unknown. In contrast, the ongoing EMPA-Kidney trial also included such patients and will ultimately address this important question.
T1D
Also excluded were patients with T1D but it is likely that patients with T1D benefit to the same extent as patients with T2D, albeit with a different safety profile.
Polycystic kidney disease
The mechanisms of disease progression in patients with polycystic kidney disease relate to genetic, mechanic and factors of fluid composition, which differs from other forms of CKD [49]. RAASis rather help to control hypertension. In addition, as a preferred marker of disease progression, kidney dimensions have a prominent role compared with GFR or proteinuria. Animal studies do not suggest a beneficial effect on cyst growth. Studies testing SGLT2 inhibition exclusively in this group of patients have to be awaited.
Lupus nephritis and ANCA-associated vasculitis
These two autoimmune diseases impact on the kidney with a persistent role of the immune system in immunopathology and disease flares. Management regularly includes immunosuppressive drugs and drug dosing varies frequently in the early phase, which makes these patients difficult to handle in a CKD trial. Studies exclusively focusing on these groups of patients, preferably conducted in the rather stable maintenance phase, have to be awaited.
SUMMARY
The finding that SGLT2 inhibition can attenuate the progression of diabetic as well as non-diabetic CKD with proteinuria at such a strong effect size is a milestone discovery in medicine. It gives hope to those that fear the morbidity and mortality related to CKD, those that fear the impact of kidney replacement therapy on their lives and especially those with CKD that do not have access to or cannot afford kidney replacement therapy. This discovery will create a new impulse for the field of Nephrology that has fallen behind others in terms of research activity and progress, and that has lost attractiveness for young doctors. The data prompt a revision of dogmas, management algorithms and research strategies, some of which have actually become hurdles for progress in Nephrology. As such, the results of the DAPA-CKD trial envision a new era of enthusiasm, dynamic changes and unexpected research opportunities for the next generation of nephrologists as well as clinical and basic science researchers. The ambitions do not stop with SGLT2 inhibition. The GFR of dapagliflozin-treated participants of the DAPA-CKD trial still declined with time, hence there is room for other innovative treatments beyond the dual blockade of the RAAS/SGLT2. For example, triple therapy by adding finerenone or drugs that enhance the production of novel podocytes from local podocyte progenitors may further attenuate glomerulosclerosis, nephron loss and GFR decline [50]. A new era in CKD research has just begun. Proud to be a part of it.
FUNDING
H.-J.A. was supported by the Deutsche Forschungsgemeinschaft (AN372/24-1).
CONFLICT OF INTEREST STATEMENT
H.-J.A. received consultancy fees from Boehringer, AstraZeneca and Bayer.
How SGLT2 inhibition affects fundamental concepts of CKD
| Disease entity | Traditional concepts | Revised concepts |
|---|---|---|
| CKD without diabetes | Reduce CV risk: RAASi, control BP, lipids and anaemia Slow down CKD progression: RAASi, bicarbonate Avoid nephrotoxins Numerous pathomechanisms of CKD progression under study | Reduce CV risk: RAASi/SGLT2i, control BP, lipids and anaemia Slow down CKD progression: RAASi/SGLT2i, bicarbonate Avoid nephrotoxins Effect size identifies SGLT2-driven CKD progression as a central upstream pathomechanism in CKD without diabetes |
| CKD with diabetes | Global pandemic of ‘diabetic nephropathy’, with diabetes as the major cause of CKD—SGLT2i for ‘diabetic nephro-pathy’ Numerous pathomechanisms of progression of diabetic nephropathy under study | T1D: diabetic nephropathy T2D: diabetic nephropathy only in a subset of patients, while many with other or multiple causes of CKD. Diabetes as a risk factor of CKD progression RAASi/SGLT2i to reduce CV risk and progression of CKD Effect size identifies SGLT2-driven progression as a central upstream pathomechanism in CKD with diabetes |
| IgAN | CKD therapy with RAASi for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Immunosuppressive drugs for patients at risk for progression of CKD, which existed in sufficient numbers IgAN trials with RAASi as only run-in and background therapy Numerous pathomechanisms of progression of diabetic nephropathy under study Proteinuria as trial endpoint | CKD therapy with RAASi/SGLT2i for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Probably a lot fewer patients at risk for CKD progression will remain that could benefit from immunosuppression Limited value of IgAN trials without SGLT2i as run-in/background therapy Subgroup analysis pending but effect size may identify SGLT2-driven progression of IgAN as a central pathomechanism of CKD progression GFR decline as trial endpoint |
| Podocytopathy with FSGS lesions | CKD the consequence of immune-mediated podocyte injury Immunosuppression for all patients Adjunct CKD therapy with RAASi Proteinuria as the study endpoint | Subgroup analysis pending but effect size may identify SGLT2-driven progression as a central pathomechanism of CKD progression in FSGS RAASi/SGLT2i as first line therapy Immunosuppression for selected patients with suspected immune pathogenesis GFR decline as trial endpoint |
| Disease entity | Traditional concepts | Revised concepts |
|---|---|---|
| CKD without diabetes | Reduce CV risk: RAASi, control BP, lipids and anaemia Slow down CKD progression: RAASi, bicarbonate Avoid nephrotoxins Numerous pathomechanisms of CKD progression under study | Reduce CV risk: RAASi/SGLT2i, control BP, lipids and anaemia Slow down CKD progression: RAASi/SGLT2i, bicarbonate Avoid nephrotoxins Effect size identifies SGLT2-driven CKD progression as a central upstream pathomechanism in CKD without diabetes |
| CKD with diabetes | Global pandemic of ‘diabetic nephropathy’, with diabetes as the major cause of CKD—SGLT2i for ‘diabetic nephro-pathy’ Numerous pathomechanisms of progression of diabetic nephropathy under study | T1D: diabetic nephropathy T2D: diabetic nephropathy only in a subset of patients, while many with other or multiple causes of CKD. Diabetes as a risk factor of CKD progression RAASi/SGLT2i to reduce CV risk and progression of CKD Effect size identifies SGLT2-driven progression as a central upstream pathomechanism in CKD with diabetes |
| IgAN | CKD therapy with RAASi for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Immunosuppressive drugs for patients at risk for progression of CKD, which existed in sufficient numbers IgAN trials with RAASi as only run-in and background therapy Numerous pathomechanisms of progression of diabetic nephropathy under study Proteinuria as trial endpoint | CKD therapy with RAASi/SGLT2i for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Probably a lot fewer patients at risk for CKD progression will remain that could benefit from immunosuppression Limited value of IgAN trials without SGLT2i as run-in/background therapy Subgroup analysis pending but effect size may identify SGLT2-driven progression of IgAN as a central pathomechanism of CKD progression GFR decline as trial endpoint |
| Podocytopathy with FSGS lesions | CKD the consequence of immune-mediated podocyte injury Immunosuppression for all patients Adjunct CKD therapy with RAASi Proteinuria as the study endpoint | Subgroup analysis pending but effect size may identify SGLT2-driven progression as a central pathomechanism of CKD progression in FSGS RAASi/SGLT2i as first line therapy Immunosuppression for selected patients with suspected immune pathogenesis GFR decline as trial endpoint |
How SGLT2 inhibition affects fundamental concepts of CKD
| Disease entity | Traditional concepts | Revised concepts |
|---|---|---|
| CKD without diabetes | Reduce CV risk: RAASi, control BP, lipids and anaemia Slow down CKD progression: RAASi, bicarbonate Avoid nephrotoxins Numerous pathomechanisms of CKD progression under study | Reduce CV risk: RAASi/SGLT2i, control BP, lipids and anaemia Slow down CKD progression: RAASi/SGLT2i, bicarbonate Avoid nephrotoxins Effect size identifies SGLT2-driven CKD progression as a central upstream pathomechanism in CKD without diabetes |
| CKD with diabetes | Global pandemic of ‘diabetic nephropathy’, with diabetes as the major cause of CKD—SGLT2i for ‘diabetic nephro-pathy’ Numerous pathomechanisms of progression of diabetic nephropathy under study | T1D: diabetic nephropathy T2D: diabetic nephropathy only in a subset of patients, while many with other or multiple causes of CKD. Diabetes as a risk factor of CKD progression RAASi/SGLT2i to reduce CV risk and progression of CKD Effect size identifies SGLT2-driven progression as a central upstream pathomechanism in CKD with diabetes |
| IgAN | CKD therapy with RAASi for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Immunosuppressive drugs for patients at risk for progression of CKD, which existed in sufficient numbers IgAN trials with RAASi as only run-in and background therapy Numerous pathomechanisms of progression of diabetic nephropathy under study Proteinuria as trial endpoint | CKD therapy with RAASi/SGLT2i for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Probably a lot fewer patients at risk for CKD progression will remain that could benefit from immunosuppression Limited value of IgAN trials without SGLT2i as run-in/background therapy Subgroup analysis pending but effect size may identify SGLT2-driven progression of IgAN as a central pathomechanism of CKD progression GFR decline as trial endpoint |
| Podocytopathy with FSGS lesions | CKD the consequence of immune-mediated podocyte injury Immunosuppression for all patients Adjunct CKD therapy with RAASi Proteinuria as the study endpoint | Subgroup analysis pending but effect size may identify SGLT2-driven progression as a central pathomechanism of CKD progression in FSGS RAASi/SGLT2i as first line therapy Immunosuppression for selected patients with suspected immune pathogenesis GFR decline as trial endpoint |
| Disease entity | Traditional concepts | Revised concepts |
|---|---|---|
| CKD without diabetes | Reduce CV risk: RAASi, control BP, lipids and anaemia Slow down CKD progression: RAASi, bicarbonate Avoid nephrotoxins Numerous pathomechanisms of CKD progression under study | Reduce CV risk: RAASi/SGLT2i, control BP, lipids and anaemia Slow down CKD progression: RAASi/SGLT2i, bicarbonate Avoid nephrotoxins Effect size identifies SGLT2-driven CKD progression as a central upstream pathomechanism in CKD without diabetes |
| CKD with diabetes | Global pandemic of ‘diabetic nephropathy’, with diabetes as the major cause of CKD—SGLT2i for ‘diabetic nephro-pathy’ Numerous pathomechanisms of progression of diabetic nephropathy under study | T1D: diabetic nephropathy T2D: diabetic nephropathy only in a subset of patients, while many with other or multiple causes of CKD. Diabetes as a risk factor of CKD progression RAASi/SGLT2i to reduce CV risk and progression of CKD Effect size identifies SGLT2-driven progression as a central upstream pathomechanism in CKD with diabetes |
| IgAN | CKD therapy with RAASi for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Immunosuppressive drugs for patients at risk for progression of CKD, which existed in sufficient numbers IgAN trials with RAASi as only run-in and background therapy Numerous pathomechanisms of progression of diabetic nephropathy under study Proteinuria as trial endpoint | CKD therapy with RAASi/SGLT2i for all patients Immunosuppression for rapidly progressive forms/IgA vasculitis Probably a lot fewer patients at risk for CKD progression will remain that could benefit from immunosuppression Limited value of IgAN trials without SGLT2i as run-in/background therapy Subgroup analysis pending but effect size may identify SGLT2-driven progression of IgAN as a central pathomechanism of CKD progression GFR decline as trial endpoint |
| Podocytopathy with FSGS lesions | CKD the consequence of immune-mediated podocyte injury Immunosuppression for all patients Adjunct CKD therapy with RAASi Proteinuria as the study endpoint | Subgroup analysis pending but effect size may identify SGLT2-driven progression as a central pathomechanism of CKD progression in FSGS RAASi/SGLT2i as first line therapy Immunosuppression for selected patients with suspected immune pathogenesis GFR decline as trial endpoint |


{kind=link}
{kind=link}
Comments