





ABSTRACT
C3 glomerulonephritis (C3GN) is caused by alternate complement pathway over-activation. It frequently progresses to end-stage renal disease, recurs in two-thirds of transplants and in half of these cases progresses to allograft loss. There is currently no proven treatment for C3GN.
We describe a family segregating pathogenic alleles of complement factor H and I (CFH and CFI). The only member carrying both mutations developed C3GN. Prolonged delayed graft function after deceased donor transplantation, heavy proteinuria and isolated C3 hypocomplementemia prompted an allograft biopsy confirming diagnosis of recurrent C3GN.
This is the first report of early recurrence of C3GN in an allograft in a patient with known mutations in complement regulatory genes and no preexisting para-proteinemia. Complement activation resulting from ischemia-reperfusion injury from prolonged cold ischemia time unabated in the setting of deficiency of two major complement regulators likely led to the early and severe recurrence. In atypical hemolytic uremic syndrome, the terminal complement cascade activation in the sentinel event initiating endothelial injury; blockade at the level of C5 convertase with eculizumab is uniformly highly effective in management. C3 glomerulopathies (C3GN and dense deposit disease) are a more complex and heterogeneous group. The relative degree of dysregulation at the levels of C3 and C5 convertases and therefore response to eculizumab varies among patients. In our patient, the clinical response to eculizumab was dramatic with recovery of allograft function and complete resolution of proteinuria. We review all cases of recurrent C3 glomerulopathy treated with eculizumab and discuss how complement biomarkers may aid in predicting response to therapy.
INTRODUCTION
C3 glomerulopathy (C3G) describes a group of rare renal disorders characterized by glomerular deposition of complement component C3 in the absence of significant amounts of immunoglobulins (Ig). The underlying abnormality is uncontrolled activation of alternate pathway (AP) of the complement system [1]. Two major subgroups that are recognized based on electron microscopic (EM) appearance are C3 glomerulonephritis (C3GN) and dense deposit disease (DDD). Despite the differing appearance of the deposits, recent mass spectrometry data show that in both diseases the dominant glomerular antigen in these deposits is C3 with varying contributions of other complement components [2]. Both diagnoses recur in most patients after transplantation, leading to graft loss in up to half of affected cases [3, 4]. Treatment options for these disorders are quite limited. Non-specific immunomodulatory therapies such as corticosteroids, cyclophosphamide, calcineurin inhibitors and rituximab have not proven effective. Eculizumab, a recombinant, fully humanized hybrid IgG2/IgG4 monoclonal antibody targeted against human complement protein C5 blocks generation of the lytic C5b-9 membrane attack complex (MAC). It has revolutionized the management of atypical hemolytic uremic syndrome (aHUS), another disorder of complement dysregulation, where it is effective in the treatment and prevention of disease in native kidneys and also after transplantation [5, 6]. However, it has not proven to be uniformly effective in C3GN.
Herein, we describe a family segregating pathogenic mutants of the alleles for complement factor H (CFH) and I (CFI) genes (Figure 1), pathogenesis of C3GN and recurrent disease after transplantation in the only member carrying both mutations, and the mechanism by which eculizumab was effective in this case.
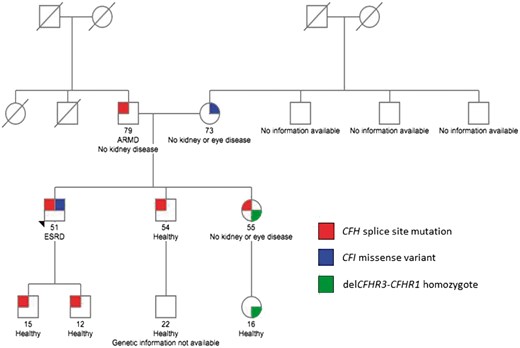
Family pedigree. Note that the proband is the only person to carry both the CFH and CFI genetic variants. Both siblings of the proband were excluded as potential donors because they also had inherited the paternally carried CFH splice site mutation. The deletion of CFHR3-CFHR1 is a common copy number variation in the Caucasian population. Homozygosity for this variation is associated with an increased risk for aHUS due to the development of fH autoantibodies. ARMD, age-related macular degeneration; ESRD, end-stage renal disease.
CASE PRESENTATION
A 51-year-old Caucasian man on hemodialysis for 4 years underwent deceased donor renal transplantation (DDRT). Native kidney biopsy at age 42 had revealed a membranoproliferative pattern of injury, with sparse and ill-defined, subendothelial and mesangial deposits reactive for C3, negative for IgG and IgA, and very weakly positive for IgM, characteristic of C3GN (Figure 2). His medications prior to transplantation included amlodipine, valsartan and sevelamer.
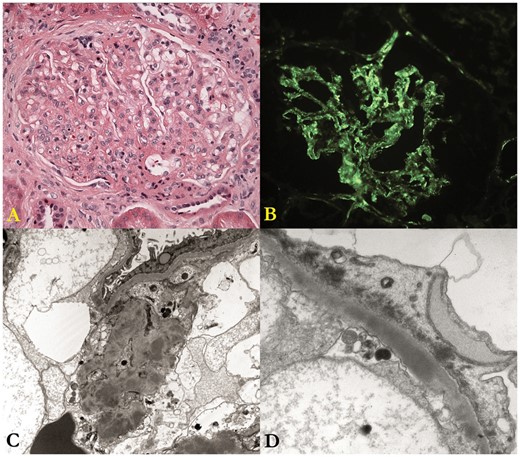
Native kidney biopsy. (A) The glomerulus shows moderate mesangial hypercellularity and mild focal thickening of the capillary walls; the structural changes are best characterized as a membranoproliferative pattern of injury. (B) There is diffuse, fine-granular deposition of C3 along the glomerular capillary walls and in the mesangium; the deposits are only very weakly positive for IgM and negative for IgG and IgA (not shown). (C) This low-magnification electron micrograph shows many discrete mesangial electron dense deposits. (D) This electron micrograph shows a large, confluent and ill-defined intramembranous dense deposit and marked effacement of the foot processes of the glomerular visceral epithelial cell.
Pretransplant complement pathway evaluation and family pedigree
Pretransplant testing was completed to screen for acquired and genetic drivers of disease. Autoantibodies like C3 nephritic factors (C3NeFs), factor H autoantibody (FHAAs) and monoclonal gammopathy of renal significance were excluded. Genetic evaluation revealed a pathogenic variant in the CFH gene, c.790+1G>A, which alters the canonical donor splice site of intron 6, the consequence of which is a novel factor H (fH) protein shorter than normal fH by one short consensus repeat (SCR), SCR4. The absence of SCR4 is predicted to reduce binding to C3b, reduce factor I (fI)-mediated C3b cofactor activity, and reduce the C3-convertase decay-accelerating activity of fH. While not documented in association with aHUS or C3GN, this same variant [labeled as CFH 1: 196648924 (IVS6 +1 G>A)] has been reported in a study of patients with age-related macular degeneration, another disorder of complement pathway over-activation [7]. Interestingly, segregation analysis showed that the CFH splice-site mutation was inherited from the patient’s father, whose medical history was notable for age-related macular degeneration in the left eye (with Drusen and an abnormal Amsler grid test). His renal function was completely normal. In addition, a missense variant was identified in exon 5 of CFI, c.719C>G, p.Ala240Gly. Pathogenic variants at and near this nucleotide have been reported in patients with aHUS and C3GN [8, 9]. This CFI missense variant was inherited from the patient’s mother, whose medical history was notable only for chronic lymphoid leukemia and hypertension well-controlled on one medication. The patient’s elder brother was evaluated as a potential kidney donor; although his renal function and ophthalmic examination were normal, he carried the CFH c.790+1G>A mutation and as a consequence, was deemed ineligible for donation. An elder sister also carried this mutation, in addition to homozygous deletion of the CFHR3-CFHR1 genes. This copy number variation is common in the European–American population, with homozygosity for del (CFHR3-CFHR1) occurring in ∼5%. Its prevalence is increased in the aHUS population and is associated with the development of fH autoantibodies (Figure 1).
Early recurrence of C3GN manifesting as delayed graft function
After 4 years of hemodialysis, the patient received a DDRT with an immunosuppression regimen consisting of anti-thymocyte globulin (6 mg/kg), steroids (withdrawal over 6 days), mycophenolate (2 gm/day) and tacrolimus (target trough 10–12 ng/mL). Due to acute kidney injury in the donor at time of demise (with a terminal serum creatinine of 5 mg/dL) and a prolonged cold ischemia time of over 18 h, delayed graft function was not unexpected. The patient was anuric prior to transplantation and although urine output improved to the non-oliguric range (∼800 mL/day), he developed heavy proteinuria (urine protein-to-creatinine ratio 8–15), had poor solute clearance and remained dialysis dependent 2 weeks posttransplantation. These findings, together with low serum C3 [55 mg/dL (normal: 90–180 mg/dL)] and a normal C4 [28 mg/dL (normal: 10–40 mg/dL)] raised concerns for recurrent C3GN, which was validated by an allograft biopsy on post-operative day (POD) 14. There was widespread acute tubular injury, and although glomeruli showed only mild mesangial expansion, coarse mesangial and capillary wall C3 deposition was seen; IgG and IgA were negative. EM revealed sub-epithelial, sub-endothelial and mesangial deposits typical of C3GN (Figure 3 A–D).
![Allograft biopsies performed on POD 14 (A–D) and POD 50 (E–H). The POD 14 biopsy is remarkable for glomeruli without proliferation (periodic acid–Schiff) (A), associated with 3+ deposition of C3, trace staining for IgM and no staining for IgG and IgA (B). EM shows intermediately electron dense granular deposits in the mesangium (C) and subendothelium (D). Diagnosis: recurrent C3GN with associated acute tubular necrosis (not shown). After 36 days later, the POD 50 shows similar glomerular changes by light microscopy (hematoxylin and eosin) (E), and C3 immunoflourescence (F). This time there was also low-level deposition for IgG2 and IgG4 (G) as well as Kappa, secondary to the eculizumab therapy. EM revealed a similar pattern of deposition [mesangial deposits enclosed by circle], (H) although the amount of extramesangial deposition was thought to be somewhat less than in the prior biopsy.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/ndt/33/12/10.1093_ndt_gfx369/1/m_gfx369f3.jpeg?Expires=1716323192&Signature=rkVv8es6vbnLhOsQ5ht8RzsU0TQzHVcBwcpNiPjfCDrm5BfUZBLgA7CKONIY0JWCJLUbmbr6ZHw~zSejQj03uhacewTqgnnNaZo13O31VslKLnWOXGOFYsTkxBVmHZPxqcIkifTgBECCKqOtvWaH56KX2ZVbjNje8RbMTlcYkHc9UZyHGVqss7bOzj6D9jnn6zEzfYVa3rPQdvT3kV8cgWZeU9Nuxi7X1H0eMWroshEoYgIHXDdbKKUOdxq2Pq9bKs38TwbeWE84H8VLrWefVVii-4iTySCnoCrB60ic9HFU7aKoKC4r9Iqdi3xkJ-3sADX32qmI3nddiMDgU0xT5A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Allograft biopsies performed on POD 14 (A–D) and POD 50 (E–H). The POD 14 biopsy is remarkable for glomeruli without proliferation (periodic acid–Schiff) (A), associated with 3+ deposition of C3, trace staining for IgM and no staining for IgG and IgA (B). EM shows intermediately electron dense granular deposits in the mesangium (C) and subendothelium (D). Diagnosis: recurrent C3GN with associated acute tubular necrosis (not shown). After 36 days later, the POD 50 shows similar glomerular changes by light microscopy (hematoxylin and eosin) (E), and C3 immunoflourescence (F). This time there was also low-level deposition for IgG2 and IgG4 (G) as well as Kappa, secondary to the eculizumab therapy. EM revealed a similar pattern of deposition [mesangial deposits enclosed by circle], (H) although the amount of extramesangial deposition was thought to be somewhat less than in the prior biopsy.
Effective treatment with eculizumab
Based on his genotype, we suspected terminal complement pathway over activity and initiated treatment with eculizumab. Given his immunocompromised state he was revaccinated against Streptococcus pneumoniae and Hemophilus influenzae B, and as recommended for eculizumab-mediated complement blockade, received the polyvalent meningococcal vaccine and amoxicillin 500, twice daily. Eculizumab treatment was started at 900 mg intravenously (IV) weekly for four doses, followed by biweekly maintenance dosing at 1200 mg IV beginning on week 5. Proteinuria improved dramatically after the first dose and no dialysis was required after the second dose. Following five doses of eculizumab, there was significant recovery in allograft function as evidenced by complete resolution of proteinuria and improvement of serum creatinine to 2.1 mg/dL (Figure 4).
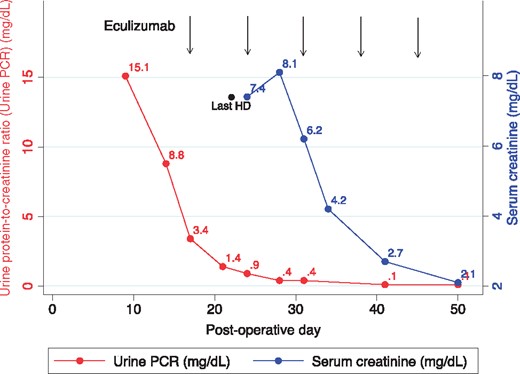
Response to eculizumab therapy. The response to eculizumab therapy (timing of administration shown by the vertical lines) was dramatic and followed quickly by a precipitous decrease in the urine protein-to-creatinine ratio (PCR) and within 3 weeks, a similarly dramatic improvement in serum creatinine. HD, hemodialysis.
Repeat allograft biopsy on POD 50 (5 days after fifth dose of eculizumab) showed considerably less tubulo-interstitial injury as compared with the prior biopsy. Glomerular findings by light microscopy were unchanged. Immunofluorescence (IF) was notable for persistent 2+ to 3+ staining for C3 and the new finding of 1+ mesangial IgG and kappa positivity. IgG subtype staining revealed 0-to-trace positivity for IgG2 and trace-to-1+ positivity for IgG4; IgG1, IgG3 and lambda were negative. These results are consistent with tissue-bound eculizumab (Figure 3 E–H). Given ongoing AP over-activation with persistently low C3 levels (range: 54–77 mg/dL) and evident effectiveness of treatment with eculizumab, he has continued on this medication at 1200 mg IV every 2 weeks. Three years after transplantation, his allograft is functioning well with a serum creatinine of 1.1 mg/dL and no abnormal proteinuria. Repeat allograft biopsies at 1 and 2 years posttransplant confirmed essentially the same findings as at POD 50. There have been no complications related to eculizumab therapy thus far.
DISCUSSION
The index patient discussed above is the first reported case of an early recurrence of C3GN in a patient with known mutations in complement regulatory genes, in absence of a paraproteinemia. In the largest reported series to date of 21 patients with C3GN who underwent renal transplantation from the Mayo Clinic, 14 (66.7%) developed recurrent C3GN. The median time to recurrence was 28 months (ranging from 9 days to over 11 years); the only patient who had delayed graft function and diagnosis at POD 9 had a monoclonal gammopathy, presumably signifying the presence of aberrant proteins that impacted complement regulation [3].
Role of ischemia-reperfusion injury and concomitant CFH and CFI mutations in early C3GN recurrence
Multiple factors colluded in this patient and led to an early and aggressive recurrence that manifested as delayed graft function. It is well recognized that time-dependent changes occur in deceased organ donors; these can compromise normal function of donated organs. Specific to the kidney are the increased synthesis of C3 [10] and the upregulation of damage-associated molecular patterns (DAMPs) that activate mannan-associated serine protease-2 (MASP-2), which cleaves excessive C3 [11, 12]. Further fueling C3 activation is the fact that the kidney in its new host is stimulated to upregulate C3 synthesis due to the stress of severe ischemia-reperfusion injury (IRI) compounded by being immunologically mismatched [13]. Notably, an acute and transient increase in C5b-9 has been documented subsequent to reperfusion of kidneys from deceased donors, especially those with donation after cardiac death, but not in transplant from living donors [14]. This contained activation of C5-9 is a reflection of how these patients are able to control C3 activation. However, in our patient, it is likely that this ‘storm’ of C3 activation in the new transplant was magnified exponentially by the recipient’s mutations in CFH and CFI. fH dissembles the C3-cleaving enzymes (C4b2a and more importantly C3bBb), as well as serving as a cofactor for the C3b inactivator, that is, fI, which itself is already compromised due to its mutated allele. These mutations synergized for allowing excessive C3b generation as well as allowing C3b to abnormally persist. Thus, once triggered by IRI, complement activation unabated in the setting of defective two most important regulators of complement activity led to recurrence of C3GN in the allograft.
Treatment with eculizumab
Recent investigations have shown that eculizumab binds to C5 and blocks its binding to a second surface-bound C3b [15]. This explains why eculizumab may be effective in treating aHUS, a genetic disorder that often results from excessive activation of C3, or failure to inactivate C3b [5, 6]. It also explains why eculizumab is efficacious in our patient, who is affected by both activating too much C3 (fH mutation) and the abnormal persistence of C3b (fI mutation). However, it should be noted that the pathophysiology of C3G is less well understood and substantially more complex than aHUS. In many patients with C3G, uncontrolled complement activity upstream of the site of action of eculizumab plays a significant role in disease progression; however, the relative degrees of upstream and downstream complement dysregulation (at the sites of the C3 convertase and the C5 convertase, respectively) vary in C3G patients and may drive some of the observed phenotypic differences [16, 17]. This spectrum of pathophysiology may be clinically relevant for two reasons.
These differences likely explain the variation in response to eculizumab therapy in C3G transplant recipients that is reported in the literature. For example, in C3G patients in whom the dominant process is activation of C5 convertase and the terminal complement cascade, as evidenced by elevated levels of soluble C5b-9, eculizumab may be of therapeutic benefit. Conversely, in C3G patients in whom the dominant process is upstream dysregulation at the level of C3 convertase, as evidenced by elevated levels of C3 split products, the effect of eculizumab therapy would be less dramatic and continued complement dysregulation with continuing renal injury would occur.
Measurement of levels of soluble C5b-9 and C3 breakdown products may be useful in predicting response and guiding therapy to eculizumab or any C3 convertase inhibitors that may become available in the future. Unfortunately, this testing was not performed for our patient prior to initiation of treatment with eculizumab.
All reported cases in which eculizumab has been used for recurrent C3G are summarized in Table 1. In the present case, significant recovery in allograft function was noted using the dosing regimen recommended in the treatment of aHUS. At the time of this report, almost 3 years after transplantation, the patient’s renal function is stable with serum creatinine of 1.1 mg/dL and urine protein-to-creatinine of 0.1. His C3 remains low at 57 mg/dL and biopsies since initiation of eculizumab continue to show persistent 2–3+ staining for C3, not an unexpected finding since treatment with eculizumab should not be expected to alter C3 deposition [23]. Additionally noted is the persistence of IgG2, IgG4 and kappa deposition in the mesangium after treatment with eculizumab. Since eculizumab is a recombinant IgG2/4κ monoclonal antibody and IgG staining was not present on the pre-treatment allograft biopsy, we believe this finding represents tissue-bound eculizumab. This pattern of IF staining has been reported in a prior case series [23]. The long-term consequences of eculizumab deposition in the glomerulus require further investigation.
C3G transplant recipients
| Reference | Type of recurrent C3G | Underlying defect | Response to eculizumab (renal function and proteinuria) | Response to eculizumab (histopathology) |
|---|---|---|---|---|
| Bomback et al. 2012 [18] | DDD | No genetic or acquired abnormalities |
| Improved |
| C3GN | C3Nef |
| Improved | |
| C3GN | C3Nef, CD46 mutation |
| Unchanged | |
| McCaughan et al. 2012 [19] | DDD | C3Nef |
| Follow-up biopsy not performed |
| Gurkan et al. 2013 [20] | C3GN (DDD in native kidney) | C3Nef, CFH mutations |
| Worsened, with increased fibrosis and continuously active C3GN |
| Sánchez-Moreno et al. 2014 [21] | DDD | C3Nef |
| Improved |
| Le Quintrec et al. 2015 [22] | DDD | No genetic or acquired abnormalities |
| Improved |
| Present case | C3GN | CFH and CFI mutations |
| Improved tubulo-interstitial injury likely representing recovering from ischemic injury; persistent 2–3+ C3 deposition |
| Reference | Type of recurrent C3G | Underlying defect | Response to eculizumab (renal function and proteinuria) | Response to eculizumab (histopathology) |
|---|---|---|---|---|
| Bomback et al. 2012 [18] | DDD | No genetic or acquired abnormalities |
| Improved |
| C3GN | C3Nef |
| Improved | |
| C3GN | C3Nef, CD46 mutation |
| Unchanged | |
| McCaughan et al. 2012 [19] | DDD | C3Nef |
| Follow-up biopsy not performed |
| Gurkan et al. 2013 [20] | C3GN (DDD in native kidney) | C3Nef, CFH mutations |
| Worsened, with increased fibrosis and continuously active C3GN |
| Sánchez-Moreno et al. 2014 [21] | DDD | C3Nef |
| Improved |
| Le Quintrec et al. 2015 [22] | DDD | No genetic or acquired abnormalities |
| Improved |
| Present case | C3GN | CFH and CFI mutations |
| Improved tubulo-interstitial injury likely representing recovering from ischemic injury; persistent 2–3+ C3 deposition |
C3G transplant recipients
| Reference | Type of recurrent C3G | Underlying defect | Response to eculizumab (renal function and proteinuria) | Response to eculizumab (histopathology) |
|---|---|---|---|---|
| Bomback et al. 2012 [18] | DDD | No genetic or acquired abnormalities |
| Improved |
| C3GN | C3Nef |
| Improved | |
| C3GN | C3Nef, CD46 mutation |
| Unchanged | |
| McCaughan et al. 2012 [19] | DDD | C3Nef |
| Follow-up biopsy not performed |
| Gurkan et al. 2013 [20] | C3GN (DDD in native kidney) | C3Nef, CFH mutations |
| Worsened, with increased fibrosis and continuously active C3GN |
| Sánchez-Moreno et al. 2014 [21] | DDD | C3Nef |
| Improved |
| Le Quintrec et al. 2015 [22] | DDD | No genetic or acquired abnormalities |
| Improved |
| Present case | C3GN | CFH and CFI mutations |
| Improved tubulo-interstitial injury likely representing recovering from ischemic injury; persistent 2–3+ C3 deposition |
| Reference | Type of recurrent C3G | Underlying defect | Response to eculizumab (renal function and proteinuria) | Response to eculizumab (histopathology) |
|---|---|---|---|---|
| Bomback et al. 2012 [18] | DDD | No genetic or acquired abnormalities |
| Improved |
| C3GN | C3Nef |
| Improved | |
| C3GN | C3Nef, CD46 mutation |
| Unchanged | |
| McCaughan et al. 2012 [19] | DDD | C3Nef |
| Follow-up biopsy not performed |
| Gurkan et al. 2013 [20] | C3GN (DDD in native kidney) | C3Nef, CFH mutations |
| Worsened, with increased fibrosis and continuously active C3GN |
| Sánchez-Moreno et al. 2014 [21] | DDD | C3Nef |
| Improved |
| Le Quintrec et al. 2015 [22] | DDD | No genetic or acquired abnormalities |
| Improved |
| Present case | C3GN | CFH and CFI mutations |
| Improved tubulo-interstitial injury likely representing recovering from ischemic injury; persistent 2–3+ C3 deposition |
CONCLUSIONS
The high rate of disease recurrence in allografts frequently leads to graft failure in patients with C3G, underscoring the importance of a comprehensive genetic and functional evaluation of the complement system in these patients. The complexity and heterogeneity of complement dysregulation and pattern of tissue injury in C3 glomerulopathies highlights the importance of detailed studies of the complement system and of identifying biomarkers that would determine the best therapy in an individual patient, especially as additional anti-complement therapies (for example, those targeting the complement pathway at level of C3 convertase), become available.
FUNDING
Funds from a philanthropic donation were used to conduct this research. R.J.S. reports grants from NIH R01 DK110023, during the conduct of this study.
CONFLICTS OF INTEREST STATEMENT
None declared.
REFERENCES
Author notes
N.G. and Y.Z. are co-first authors.
R.J.S. and M.P. are co-senior authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments