





Introduction
Fibrosis, defined as an abnormal increase of tissue collagen fibres, is an important component of cardiac remodelling in patients with hypertension. Cardiac remodelling may in turn lead to increased myocardial stiffness, left ventricular (LV) dysfunctions, and ultimately to heart failure. A growing body of evidence indicates that the renin–angiotensin–aldosterone system (RAAS) plays an important role in the development of cardiac fibrosis [1–8]. Furthermore, experimental data suggest that angiotensin II (Ang II) and aldosterone can stimulate cardiac fibrosis independent of haemodynamic mechanisms through direct trophic effects on the myocardium, and probably also by inhibiting interstitial collagenases, thereby reducing collagen degradation [1,2,7,8]. However, less is known about the influence of the RAAS on cardiac fibrosis in humans. We report here the case of a patient with hypertension, and an activation of the RAAS, who demonstrated pronounced right ventricular (RV) fibrosis in the absence of pulmonary hypertension. Our observation suggests that an activation of the RAAS, per se, may promote the development of cardiac fibrosis independent of haemodynamic mechanisms in humans also.
Case
A 42-year-old Caucasian man was referred to the local hospital by his family practitioner because of newly diagnosed hypertension and a 1 week history of dyspnoea. The patient was previously healthy and had no family history of hypertension or cardiovascular disease. He was a heavy smoker, had a very modest alcohol intake, and denied drug abuse. On physical examination, his blood pressure (BP) was 180/120 mmHg and a pan-systolic cardiac murmur compatible with mitral regurgitation was noted. Total blood counts, serum creatinine and electrolytes, liver function tests, thyroid hormones, C-reactive protein, plasma glucose, urinary catecholamines and general urine examination were all normal (Table 1). However, plasma renin activity (PRA) and serum aldosterone levels were significantly elevated (Table 1). Chest X-ray revealed slight cardiac enlargement and pulmonary vascular congestion. Echocardiography showed LV hypertrophy and dilatation, moderate mitral regurgitation, and an LV ejection fraction of 0.40 (Table 2).
Laboratory data at presentation
| Variable | Patient data | Normal values |
|---|---|---|
| S-creatinine (mg/dl) | 1.0 | 0.7–1.3 |
| S-Na (mEq/l) | 135 | 136–144 |
| S-K (mEq/l) | 4.3 | 3.6–5.0 |
| tU-Na (mEq/24 h) | 260 | 50–150 |
| S-aldosterone (ng/dl) | 25 | 4–14 |
| PRA (ng/ml/h) | 4.5 | 0.68–1.36 |
| tU-VMA (mg/24 h) | 3.68 | <7 |
| Variable | Patient data | Normal values |
|---|---|---|
| S-creatinine (mg/dl) | 1.0 | 0.7–1.3 |
| S-Na (mEq/l) | 135 | 136–144 |
| S-K (mEq/l) | 4.3 | 3.6–5.0 |
| tU-Na (mEq/24 h) | 260 | 50–150 |
| S-aldosterone (ng/dl) | 25 | 4–14 |
| PRA (ng/ml/h) | 4.5 | 0.68–1.36 |
| tU-VMA (mg/24 h) | 3.68 | <7 |
S-Na, serum sodium concentration; S-K, serum potassium concentration; tU-Na, urinary sodium excretion; PRA, plasma renin activity; and tU-VMA, urinary vanillylmandelic acid excretion.
Note: to convert sodium and potassium in mEq/l to mmol/l, multiply by 1; creatinine in mg/dl to µmol/l, multiply by 88.4; aldosterone in ng/dl to nmol/l multiply by 0.02774; and VMA in mg/24 h to µmol/24 h multiply by 5.71.
Laboratory data at presentation
| Variable | Patient data | Normal values |
|---|---|---|
| S-creatinine (mg/dl) | 1.0 | 0.7–1.3 |
| S-Na (mEq/l) | 135 | 136–144 |
| S-K (mEq/l) | 4.3 | 3.6–5.0 |
| tU-Na (mEq/24 h) | 260 | 50–150 |
| S-aldosterone (ng/dl) | 25 | 4–14 |
| PRA (ng/ml/h) | 4.5 | 0.68–1.36 |
| tU-VMA (mg/24 h) | 3.68 | <7 |
| Variable | Patient data | Normal values |
|---|---|---|
| S-creatinine (mg/dl) | 1.0 | 0.7–1.3 |
| S-Na (mEq/l) | 135 | 136–144 |
| S-K (mEq/l) | 4.3 | 3.6–5.0 |
| tU-Na (mEq/24 h) | 260 | 50–150 |
| S-aldosterone (ng/dl) | 25 | 4–14 |
| PRA (ng/ml/h) | 4.5 | 0.68–1.36 |
| tU-VMA (mg/24 h) | 3.68 | <7 |
S-Na, serum sodium concentration; S-K, serum potassium concentration; tU-Na, urinary sodium excretion; PRA, plasma renin activity; and tU-VMA, urinary vanillylmandelic acid excretion.
Note: to convert sodium and potassium in mEq/l to mmol/l, multiply by 1; creatinine in mg/dl to µmol/l, multiply by 88.4; aldosterone in ng/dl to nmol/l multiply by 0.02774; and VMA in mg/24 h to µmol/24 h multiply by 5.71.
Blood pressure and echocardiography data before and after PTRA
| 10 months pre-PTRA | 2 months pre-PTRA | 4 months post-PTRA | 30 months post-PTRA | Normal values | |
|---|---|---|---|---|---|
| Blood pressure (mmHg) | 180/120 | 150/95 | 120/75 | 120/80 | <140/90 |
| Enalapril treatment | Started | Yes | Yes | No | – |
| EF (Teichholtz) | 0.40 | 0.45 | 0.50 | 0.63 | >0.55 |
| MR | 2/4 | 1/4 | None | None | None |
| LV EDD (mm) | 65 | 63 | 60 | 55 | 47–60 |
| IVS (mm) | 13 | 11 | 9 | 8 | 9–12 |
| LVPW (mm) | 13 | 11 | 9 | 8 | 8–12 |
| LA diameter (mm) | 45 | 43 | 40 | 35 | 32–48 |
| 10 months pre-PTRA | 2 months pre-PTRA | 4 months post-PTRA | 30 months post-PTRA | Normal values | |
|---|---|---|---|---|---|
| Blood pressure (mmHg) | 180/120 | 150/95 | 120/75 | 120/80 | <140/90 |
| Enalapril treatment | Started | Yes | Yes | No | – |
| EF (Teichholtz) | 0.40 | 0.45 | 0.50 | 0.63 | >0.55 |
| MR | 2/4 | 1/4 | None | None | None |
| LV EDD (mm) | 65 | 63 | 60 | 55 | 47–60 |
| IVS (mm) | 13 | 11 | 9 | 8 | 9–12 |
| LVPW (mm) | 13 | 11 | 9 | 8 | 8–12 |
| LA diameter (mm) | 45 | 43 | 40 | 35 | 32–48 |
EF, ejection fraction; MR, mitral regurgitation; LV EDD, left ventricular end-diastolic diameter; IVS, interventricular septum thickness; LVPW, left ventricular posterior wall thickness; LA, left atrium. The enalapril dose used was 10 mg twice daily.
Blood pressure and echocardiography data before and after PTRA
| 10 months pre-PTRA | 2 months pre-PTRA | 4 months post-PTRA | 30 months post-PTRA | Normal values | |
|---|---|---|---|---|---|
| Blood pressure (mmHg) | 180/120 | 150/95 | 120/75 | 120/80 | <140/90 |
| Enalapril treatment | Started | Yes | Yes | No | – |
| EF (Teichholtz) | 0.40 | 0.45 | 0.50 | 0.63 | >0.55 |
| MR | 2/4 | 1/4 | None | None | None |
| LV EDD (mm) | 65 | 63 | 60 | 55 | 47–60 |
| IVS (mm) | 13 | 11 | 9 | 8 | 9–12 |
| LVPW (mm) | 13 | 11 | 9 | 8 | 8–12 |
| LA diameter (mm) | 45 | 43 | 40 | 35 | 32–48 |
| 10 months pre-PTRA | 2 months pre-PTRA | 4 months post-PTRA | 30 months post-PTRA | Normal values | |
|---|---|---|---|---|---|
| Blood pressure (mmHg) | 180/120 | 150/95 | 120/75 | 120/80 | <140/90 |
| Enalapril treatment | Started | Yes | Yes | No | – |
| EF (Teichholtz) | 0.40 | 0.45 | 0.50 | 0.63 | >0.55 |
| MR | 2/4 | 1/4 | None | None | None |
| LV EDD (mm) | 65 | 63 | 60 | 55 | 47–60 |
| IVS (mm) | 13 | 11 | 9 | 8 | 9–12 |
| LVPW (mm) | 13 | 11 | 9 | 8 | 8–12 |
| LA diameter (mm) | 45 | 43 | 40 | 35 | 32–48 |
EF, ejection fraction; MR, mitral regurgitation; LV EDD, left ventricular end-diastolic diameter; IVS, interventricular septum thickness; LVPW, left ventricular posterior wall thickness; LA, left atrium. The enalapril dose used was 10 mg twice daily.
Cardiomyopathy was suspected and a wide range of serological tests were carried out without revealing viral infection. Coronary angiography showed no signs of coronary artery disease. A right heart catheterization revealed signs of moderate LV dysfunction with a cardiac index of 2.3 l/min/m2 measured by thermodilution (normal range 2.8–4.2 l/min/m2), but no pulmonary hypertension (mean pulmonary artery pressure at rest 23 mmHg). Biopsies from the interventricular septum of the RV showed increased thickness of the endocardium, proliferation of subendocardial smooth muscle cells, regions with marked interstitial fibrosis, and hypertrophy and proliferation of cardiomyocytes (Figure 1). There were no histological signs of inflammation. Treatment was initiated with the loop diuretic furosemide (40–80 mg daily) and the angiotensin-converting enzyme (ACE) inhibitor enalapril (10 mg twice daily).
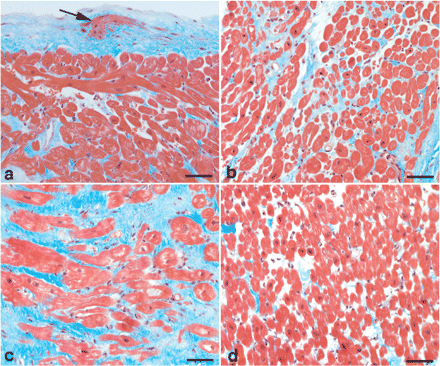
Light micrographs illustrate histopathological changes in endomyocardial biopsies taken from the septal wall of the right ventricle. (a) Increased thickness of endomyocardium and proliferation of subendocardial smooth muscle cells (arrow); (b) region with slight interstitial fibrosis; (c) region with marked interstitial fibrosis and partly hypertrophic cardiomyocytes and (d) region with normal appearance. Sections were stained by Masson trichrome. Bar = 50 µm.
Eight months after admission and the initiation of enalapril treatment, BP remained slightly elevated (150/95 mmHg) and echocardiography revealed only minor improvements in cardiac morphology and function (Table 2). To rule out renovascular hypertension, a renography was carried out that demonstrated a slower washout of the isotope on the left side suggestive of left renal artery stenosis. Renal angiography confirmed the diagnosis and revealed a tight atherosclerotic left renal artery stenosis, while the right renal artery appeared normal. After successful percutaneous transluminal renal angioplasty (PTRA), BP and cardiac abnormalities gradually normalized (Table 2). Six months after PTRA, treatment with enalapril and furosemide was discontinued and 2 months later PRA became normal, 1.16 ng/ml/h (normal range 0.68–1.36). Two years later, echocardiography revealed normalization of all previously pathological variables and BP was normal without pharmacological treatment (Table 2).
Discussion
Patients with hypertension develop abnormalities in cardiac structure and function that are often responsible for the morbidity and mortality associated with high BP. Morphologically, these changes involve both hypertrophy and proliferation of cardiomyocytes and interstitial fibrosis [5,6,9]. Compelling evidence indicates that cardiac fibrosis increases myocardial stiffness and promotes abnormalities in cardiac function that eventually lead to heart failure [3,4,8,10].
We here report a case of a patient with unilateral renal artery stenosis, and a pronounced activation of the RAAS as evidenced by elevated PRA and serum aldosterone levels, who presented with hypertension and LV failure. Notably, a biopsy from the interventricular septum of the RV of this patient demonstrated marked interstitial fibrosis in the absence of pulmonary hypertension. As opposed to the systemic circulation, the pulmonary vascular bed is a low-pressure circuit characterized by high compliance and a low resistance. Our findings suggest that RV fibrosis was not induced by haemodynamic factors, but rather by increased circulating levels of components of the RAAS, i.e. Ang II and aldosterone.
A growing body of evidence demonstrates an important role for the RAAS in the development of cardiac fibrosis, and also suggests that the pro-fibrotic effects are at least partially mediated through non-haemodynamic mechanisms. For instance, in a high-renin model of hypertension in rats caused by suprarenal aortic banding, Brilla et al. [1] found a significant increase of interstitial collagen in both the pressure-overloaded hypertrophied LV, and in the normotensive non-hypertrophied RV. However, these authors found no fibrosis in either ventricle when aortic banding was performed below the renal arteries (a low-renin model), despite a comparable increase in BP [1]. In addition, cardiac fibrosis can be induced experimentally by chronic administration of Ang II or aldosterone, and treatment with ACE inhibitors, Ang II type-1 (AT1) receptor antagonists, or aldosterone receptor antagonists, inhibits the development of myocardial fibrosis in these animal models [8].
Observations from clinical trials support the aforementioned experimental evidence [3–5]. In the Losartan Intervention For Endpoint (LIFE) reduction in hypertension trial, losartan was significantly more effective than conventional atenolol-based treatment in reducing myocardial collagen content assessed by echo-reflectivity and serum collagen markers [3,4]. Similarly, in patients with hypertensive heart disease, treatment with the ACE inhibitor lisinopril has been shown to decrease myocardial fibrosis, as determined histologically on endomyocardial biopsies, more effectively than hydrochlorothiazide despite comparable reductions in BP [5].
Interestingly, although both PRA and serum aldosterone levels were elevated in the present case, treatment with enalapril (20 mg/day) for 8 months caused only minor improvements in cardiac morphology or function despite reducing BP (Table 2). However, PTRA of the renal artery stenosis rapidly normalized BP and gradually reversed cardiac abnormalities observed on the echocardiogram (Table 2). This finding suggests that enalapril treatment did not completely block the formation of Ang II, probably due to the presence of parallel enzyme-systems like chymases and tissue plasminogen activator that can form Ang II directly from angiotensin I and angiotensinogen, respectively. Apparently, correction of the renal artery stenosis by PTRA produced a much more efficient interruption of RAAS than ACE inhibition in the present case. For the same reason, it is feasible to speculate that combined pharmacological blockade of the RAAS with an ACE inhibitor and an AT1 receptor antagonist would also have been more efficient than ACE inhibition alone in the present case. Finally, it should be underlined that although PTRA normalized echocardiographic abnormalities in the presented patient, we did not demonstrate a reversal of myocardial fibrosis, as no re-biopsy was performed.
In conclusion, our findings support the notion that an activation of the RAAS, per se, stimulates the development of cardiac fibrosis in humans. Consequently, treatment of hypertensive heart disease should not only focus on the reduction of BP, but also on the inhibition of the RAAS.
This study was supported by grants from the Swedish state under the LUA/ALF agreement.
Conflict of interest statement. None declared.
References
Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension.
Tokuda K, Kai H, Kuwahara F et al. Pressure-independent effects of angiotensin II on hypertensive myocardial fibrosis.
Devereux RB, Dahlof B, Gerdts E et al. Regression of hypertensive left ventricular hypertrophy by losartan compared with atenolol: the Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) trial.
Ciulla MM, Paliotti R, Esposito A et al. Different effects of antihypertensive therapies based on losartan or atenolol on ultrasound and biochemical markers of myocardial fibrosis: results of a randomized trial.
Brilla CG, Funck RC, Rupp H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease.
Brilla CG, Rupp H, Maisch B. Effects of ACE inhibition versus non-ACE inhibitor antihypertensive treatment on myocardial fibrosis in patients with arterial hypertension. Retrospective analysis of 120 patients with left ventricular endomyocardial biopsies.
Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone.
Querejeta R, Lopez B, Gonzalez A et al. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis.
Author notes
1Department of Nephrology, Institute of Internal Medicine, 2Department of Pathology, Institute of Laboratory Medicine and 3The Cardiovascular Institute, The Sahlgrenska Academy at Göteborg University, Göteborg, Sweden


{kind=link}
Comments