





Abstract
Background. Hydrochlorothiazide (HCT) is applied in the therapy of familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC) caused by claudin-16 (CLDN16) mutation. However, the short-term efficacy of HCT to reduce hypercalciuria in FHHNC has not yet been demonstrated in a clinical trial.
Methods. Four male and four female patients with FHHNC and CLDN16 mutation, under long-standing HCT therapy (0.4–1.2 mg/kg, median 0.9 mg/kg, dose according to calciuria), aged 0.7–22.4 years, were included in a clinical study to investigate the effect of HCT on calciuria. The study design consisted of three periods: continued therapy for 4 weeks, HCT withdrawal for 6 weeks and restart of therapy at the same dose for 4 weeks. Calciuria and magnesiuria were assessed weekly as Ca/creat and Mg/creat ratio, every 2 weeks in 24 h urine, and serum Mg, K and kaliuria (s-Mg, s-K and K/creat) at weeks 0, 6, 10 and 14. The data of each study period were averaged and analysed by Friedman and Wilcoxon test.
Results. Ca/creat was significantly reduced by HCT (median before/at/after withdrawal 0.76/1.24/0.77 mol/mol creat; n = 8, P<0.05). The reduction of Ca/24 h by HCT was not statistically significant (0.13/0.19/0.13 mmol/kg × 24 h; n = 5). Serum Mg (0.51/0.64/0.56 mmol/l; n = 8, P<0.05) and Serum K (3.65/4.35/3.65 mmol/l; n = 8, P<0.05) were significantly higher during withdrawal. However, Mg/creat (0.98/0.90/0.90 mol/mol creat; n = 8), Mg/24 h (0.14/0.12/0.18 mmol/kg × 24h; n = 5) and K/creat (6.3/8.4/6.2 mol/mol creat; n = 8) remained statistically unchanged during withdrawal.
Conclusions. We demonstrated that HCT is effective in reducing hypercalciuria due to CLDN16 mutation on a short-term basis. However, the efficacy of HCT to attenuate disease progression remains to be elucidated.
Introduction
Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis (FHHNC) is a rare autosomal recessive disease caused by mutations in the claudin-16 (CLDN16) gene [1,2]. CLDN16 is located in tight junctions of the thick ascending limb of Henle, permitting paracellular passage of Mg and Ca [1–3]. This paracellular pathway is the main location for Mg re-absorption in the kidney, and is driven by the positive luminal transepithelial voltage [4]. To date, 28 different mutations in the CLDN16 gene have been identified [1–3,5]. The loss of function mutations result in a selective alteration in paracellular Mg and Ca permeability [3].
The clinical presentation includes hypomagnesaemia, hypercalciuria and nephrocalcinosis. The disease progresses at a variable course to chronic and end-stage renal failure [5–8]. At diagnosis, the symptoms include urinary tract infections, polyuria–polydipsia, renal concrements, seizures and ocular and hearing impairment. Further biochemical abnormalities are hyperuricaemia, elevated levels for parathyroid hormone and hypocitraturia [5–10].
For treatment, hydrochlorothiazide (HCT) is applied in order to reduce hypercalciuria [5,6,11–13]. HCT is reported to effectively reduce hypercalciuria of various aetiologies [14,15]. However, the efficacy of HCT in FHHNC caused by CLDN16 mutation has not yet been demonstrated in a clinical trial. Since the nature of FHHNC is quite distinct from idiopathic hypercalciuria, data may not be transferred from one disease to another. Furthermore, it is not known to what degree HCT therapy can attenuate disease progression towards end-stage renal failure [5,6,8,10,11].
Concerning adverse effects, thiazide diuretics are well known to induce hypokalaemia. Additionally, although little is known about the effects of thiazide diuretics on renal magnesium excretion, chronic administration of thiazides may result in renal Mg wasting [4,14,16,17]. Hypomagnesaemia has been reported to be unresponsive to Mg administration in patients with CLDN16 mutation [5,10,11,13].
The aim of our clinical study was to investigate the short-term efficacy of HCT on hypercalciuria in patients with CLDN16 mutation, and to explore the significance of therapy-induced short-term adverse effects, such as hypomagnesaemia and hypokalaemia.
Methods
Study design
All clinical experimentation was conducted in accordance with the guidelines proposed in the Declaration of Helsinki. The local ethics committee approved this monocentre open trial. Written informed consent of the patients and/or their legal representatives was obtained. The central hypothesis was that HCT is effective in reducing hypercalciuria in patients with CLDN16 mutation. All patients had been under HCT treatment for at least 3 months before the study entry. Drug doses had been individually chosen and titrated to calciuria (HCT dose at study start and end, and concomitant medication are listed in Table 1). The study design consisted of three periods: continuation of HCT therapy at prior dose for 4 weeks (‘before withdrawal’), a wash-out phase of 2 weeks and further HCT withdrawal for 4 weeks (‘at withdrawal’), and restart of HCT therapy at the dose ultimately used for 4 weeks (‘after withdrawal’). During therapy periods, the titration of HCT dose to calciuria was performed if necessary (Table 1). During the study period, urinary calcium/creatinine ratio and magnesium/creatinine ratio (Ca/creat and Mg/creat) were assessed weekly. Calcium and magnesium excretion in 24 h (Ca/24 h and Mg/24 h) was measured every 2 weeks. At weeks 0, 6, 10 and 14, serum magnesium, serum potassium and potassium/creatinine ratio, (s-Mg, s-K and K/creat) were examined. Further biochemical data of the patients, as serum creatinine, glomerular filtration rate (estimated following the Schwartz formula) and serum calcium are listed in Table 1.
Clinical data and biochemical values of the patients before study start
| Patient no. | sex | Age (years) | s-creat (mg/dl) at study start/end | GFR (ml/min/1.73 m2) at study start/end | s-Ca (mmol/l) at study start/end | Results of CLDN16 gene analyse | HCT dose (mg/kg) at study start/end | Concomitant medication (dose per day) at study start/end |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 0.7 | 0.36/0.45 | 90/72 | 2.85/2.59 | Heterozygous | 0.7/1.4 | Mg 1.2 mmol/kg |
| Leu151Phe | Fluoride 0.25 mg | |||||||
| Ferrum (II) 50 mg | ||||||||
| 2 | F | 5.3 | 1.06/1.09 | 57/56 | 2.24/2.49 | Heterozygous | 0.7/1.5 | Mg 0.6 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| 3 | M | 8.8 | 1.07/1.19 | 64/57 | 2.53/2.41 | Heterozygous | 0.9/0.9 | Mg 0.5 mmol/kg |
| Leu151Phe | Triamteren 50mg | |||||||
| 4 | M | 9.8 | 0.92/0.85 | 77/83 | 2.30/2.45 | Heterozygous | 0.8/1.0 | Mg 0.4 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| Methylphenidan 10 mg | ||||||||
| 5 | F | 14.3 | 1.07/1.09 | 84/82 | 2.07/2.20 | Homozygous | 0.8/0.8 | Mg 0.3 mmol/kg |
| Leu151Phe | Triamteren 100 mg | |||||||
| 6 | M | 14.8 | 1.36/1.53 | 92/81 | 2.22/n.d. | Homozygous | 0.8/0.8 | Mg 0.4 mmol/kg |
| Leu151Phe | Atenolol 100 mg | |||||||
| Colecalciferol 2000 IU | ||||||||
| 7 | F | 19.7 | 1.04/1.27 | 87/71 | 2.20/2.37 | Compound | 1.2/1.2 | Mg 0.4 mmol/kg |
| Heterozygous | Triamteren 150 mg | |||||||
| Leu145Pro/Leu151Phe | Losartan 25 mg | |||||||
| 8 | F | 22.3 | 1.07/1.10 | 87/85 | 1.93/2.13 | Compound | 0.4/0.4 | Mg 0.5 mmol/kg |
| Heterozygous | Triamteren 50 mg | |||||||
| Leu145Pro/Leu151Phe | Enalapril 5 mg |
| Patient no. | sex | Age (years) | s-creat (mg/dl) at study start/end | GFR (ml/min/1.73 m2) at study start/end | s-Ca (mmol/l) at study start/end | Results of CLDN16 gene analyse | HCT dose (mg/kg) at study start/end | Concomitant medication (dose per day) at study start/end |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 0.7 | 0.36/0.45 | 90/72 | 2.85/2.59 | Heterozygous | 0.7/1.4 | Mg 1.2 mmol/kg |
| Leu151Phe | Fluoride 0.25 mg | |||||||
| Ferrum (II) 50 mg | ||||||||
| 2 | F | 5.3 | 1.06/1.09 | 57/56 | 2.24/2.49 | Heterozygous | 0.7/1.5 | Mg 0.6 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| 3 | M | 8.8 | 1.07/1.19 | 64/57 | 2.53/2.41 | Heterozygous | 0.9/0.9 | Mg 0.5 mmol/kg |
| Leu151Phe | Triamteren 50mg | |||||||
| 4 | M | 9.8 | 0.92/0.85 | 77/83 | 2.30/2.45 | Heterozygous | 0.8/1.0 | Mg 0.4 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| Methylphenidan 10 mg | ||||||||
| 5 | F | 14.3 | 1.07/1.09 | 84/82 | 2.07/2.20 | Homozygous | 0.8/0.8 | Mg 0.3 mmol/kg |
| Leu151Phe | Triamteren 100 mg | |||||||
| 6 | M | 14.8 | 1.36/1.53 | 92/81 | 2.22/n.d. | Homozygous | 0.8/0.8 | Mg 0.4 mmol/kg |
| Leu151Phe | Atenolol 100 mg | |||||||
| Colecalciferol 2000 IU | ||||||||
| 7 | F | 19.7 | 1.04/1.27 | 87/71 | 2.20/2.37 | Compound | 1.2/1.2 | Mg 0.4 mmol/kg |
| Heterozygous | Triamteren 150 mg | |||||||
| Leu145Pro/Leu151Phe | Losartan 25 mg | |||||||
| 8 | F | 22.3 | 1.07/1.10 | 87/85 | 1.93/2.13 | Compound | 0.4/0.4 | Mg 0.5 mmol/kg |
| Heterozygous | Triamteren 50 mg | |||||||
| Leu145Pro/Leu151Phe | Enalapril 5 mg |
CLDN16, claudin-16; GFR, glomerular filtration rate (calculated according to Schwartz formula); HCT, hydrochlorothiazide; Leu, leucin; Mg, magnesium; Phe, phenylalanin; s-Ca, serum-calcium; s-creat., serum-creatinine.
Clinical data and biochemical values of the patients before study start
| Patient no. | sex | Age (years) | s-creat (mg/dl) at study start/end | GFR (ml/min/1.73 m2) at study start/end | s-Ca (mmol/l) at study start/end | Results of CLDN16 gene analyse | HCT dose (mg/kg) at study start/end | Concomitant medication (dose per day) at study start/end |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 0.7 | 0.36/0.45 | 90/72 | 2.85/2.59 | Heterozygous | 0.7/1.4 | Mg 1.2 mmol/kg |
| Leu151Phe | Fluoride 0.25 mg | |||||||
| Ferrum (II) 50 mg | ||||||||
| 2 | F | 5.3 | 1.06/1.09 | 57/56 | 2.24/2.49 | Heterozygous | 0.7/1.5 | Mg 0.6 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| 3 | M | 8.8 | 1.07/1.19 | 64/57 | 2.53/2.41 | Heterozygous | 0.9/0.9 | Mg 0.5 mmol/kg |
| Leu151Phe | Triamteren 50mg | |||||||
| 4 | M | 9.8 | 0.92/0.85 | 77/83 | 2.30/2.45 | Heterozygous | 0.8/1.0 | Mg 0.4 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| Methylphenidan 10 mg | ||||||||
| 5 | F | 14.3 | 1.07/1.09 | 84/82 | 2.07/2.20 | Homozygous | 0.8/0.8 | Mg 0.3 mmol/kg |
| Leu151Phe | Triamteren 100 mg | |||||||
| 6 | M | 14.8 | 1.36/1.53 | 92/81 | 2.22/n.d. | Homozygous | 0.8/0.8 | Mg 0.4 mmol/kg |
| Leu151Phe | Atenolol 100 mg | |||||||
| Colecalciferol 2000 IU | ||||||||
| 7 | F | 19.7 | 1.04/1.27 | 87/71 | 2.20/2.37 | Compound | 1.2/1.2 | Mg 0.4 mmol/kg |
| Heterozygous | Triamteren 150 mg | |||||||
| Leu145Pro/Leu151Phe | Losartan 25 mg | |||||||
| 8 | F | 22.3 | 1.07/1.10 | 87/85 | 1.93/2.13 | Compound | 0.4/0.4 | Mg 0.5 mmol/kg |
| Heterozygous | Triamteren 50 mg | |||||||
| Leu145Pro/Leu151Phe | Enalapril 5 mg |
| Patient no. | sex | Age (years) | s-creat (mg/dl) at study start/end | GFR (ml/min/1.73 m2) at study start/end | s-Ca (mmol/l) at study start/end | Results of CLDN16 gene analyse | HCT dose (mg/kg) at study start/end | Concomitant medication (dose per day) at study start/end |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 0.7 | 0.36/0.45 | 90/72 | 2.85/2.59 | Heterozygous | 0.7/1.4 | Mg 1.2 mmol/kg |
| Leu151Phe | Fluoride 0.25 mg | |||||||
| Ferrum (II) 50 mg | ||||||||
| 2 | F | 5.3 | 1.06/1.09 | 57/56 | 2.24/2.49 | Heterozygous | 0.7/1.5 | Mg 0.6 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| 3 | M | 8.8 | 1.07/1.19 | 64/57 | 2.53/2.41 | Heterozygous | 0.9/0.9 | Mg 0.5 mmol/kg |
| Leu151Phe | Triamteren 50mg | |||||||
| 4 | M | 9.8 | 0.92/0.85 | 77/83 | 2.30/2.45 | Heterozygous | 0.8/1.0 | Mg 0.4 mmol/kg |
| Leu151Phe | Triamteren 50 mg | |||||||
| Methylphenidan 10 mg | ||||||||
| 5 | F | 14.3 | 1.07/1.09 | 84/82 | 2.07/2.20 | Homozygous | 0.8/0.8 | Mg 0.3 mmol/kg |
| Leu151Phe | Triamteren 100 mg | |||||||
| 6 | M | 14.8 | 1.36/1.53 | 92/81 | 2.22/n.d. | Homozygous | 0.8/0.8 | Mg 0.4 mmol/kg |
| Leu151Phe | Atenolol 100 mg | |||||||
| Colecalciferol 2000 IU | ||||||||
| 7 | F | 19.7 | 1.04/1.27 | 87/71 | 2.20/2.37 | Compound | 1.2/1.2 | Mg 0.4 mmol/kg |
| Heterozygous | Triamteren 150 mg | |||||||
| Leu145Pro/Leu151Phe | Losartan 25 mg | |||||||
| 8 | F | 22.3 | 1.07/1.10 | 87/85 | 1.93/2.13 | Compound | 0.4/0.4 | Mg 0.5 mmol/kg |
| Heterozygous | Triamteren 50 mg | |||||||
| Leu145Pro/Leu151Phe | Enalapril 5 mg |
CLDN16, claudin-16; GFR, glomerular filtration rate (calculated according to Schwartz formula); HCT, hydrochlorothiazide; Leu, leucin; Mg, magnesium; Phe, phenylalanin; s-Ca, serum-calcium; s-creat., serum-creatinine.
Patients
Four male and four female patients from five families, aged 0.7–22.4 years (median 12.0 years), with FHHNC caused by CLDN16 mutation, were enrolled. Inclusion criteria for the study were CLDN16 mutation in at least one allele, hypomagnesaemia, hypercalciuria, nephrocalcinosis, and optionally chronic renal failure. Clinical diagnosis of FHHNC and CLDN16 gene analysis were obtained between early infancy and age 18.5 years. Gene analysis was performed by Konrad and coworkers [5]. Out of eight patients, two were homozygous for Leu151Phe, two patients were compound heterozygous for Leu145Pro/Leu151Phe and four patients were heterozygous for Leu151Phe (Table 1). Figure 1 shows the pedigrees of the five families that the patients belong to.
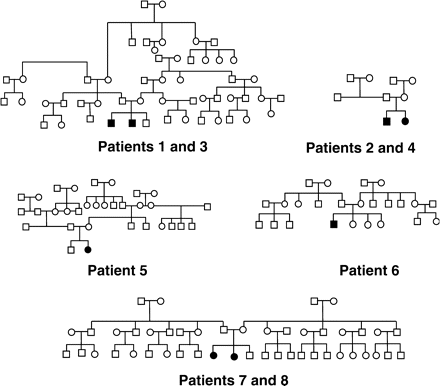
Pedigrees of patients 1–8.
All patients had hypomagnesaemia, hypercalciuria and bilateral nephrocalcinosis. At the start of the study, patients had a glomerular filtration rate of 57–92 ml/min/1.73 m2 (calculated according to Schwartz formula; median 85.5 ml/min/1.73 m2). The patients had been treated for at least 3 months (patients 3–8 for more than a year) with HCT alone (patients 1, 6), or in combination with triamteren because of hypokalaemia (patients 2–5, 7, 8). The HCT dose was individually chosen and titrated to calciuria (0.4–1.2 mg/kg body weight, median 0.9 mg/kg). In case of persistent hypercalciuria, the thiazide dose was increased to 125%. All patients received oral Mg according to their s-Mg levels and individual tolerance (0.3–1.2 mmol/kg body weight, constant dose for each patient throughout the study).
Reference values
Age-specific reference values were based on literature [18–24]:
upper normal values for Ca/creatinine ratio: 2.2 (patient 1), 0.8 (patient 2) and 0.7 mol/mol creatinine (patients 3–8);
upper normal values for Mg/creatinine ratio: 2.2 (patient 1), 1.0 (patient 2), 0.9 (patients 3 and 4) and 0.6 mol/mol creatinine (patients 5–8);
potassium/creatinine: female, 1.7–8.7; male, 1.3–8.4 mol/mol;
lower normal value for s-Mg: 0.70 mmol/l;
lower normal value for s-K: 3.70 mmol/l and
upper normal values for calciuria/24 h and magnesiuria/24 h: 0.1 mmol/kg body weight per 24 h.
Statistical analysis
In each of the three periods of the study (before, at and after withdrawal of HCT), the weekly assessed data of the patients were averaged, excluding the wash-out phase. Because of the small sample size, the statistical analysis consisted of non-parametric tests. The differences between the three phases were analysed by Friedman test. Post hoc analysis was performed using Wilcoxon test for paired data. Statistical significance was defined as two-tailed P-value of ≤0.05.
Data were analysed a second time separately for patients 1–4 (heterozygote for CLDN16 mutation) and patients 5–8 (homozygote or compound heterozygote).
Results
Before withdrawal, HCT was administered at a median dose of 0.9 mg/kg body weight (0.4–1.2 mg/kg) and after withdrawal at a median dose of 1.0 mg/kg (0.4–1.5 mg/kg). The differences in HCT dose result from a dose increase in patients 1, 2 and 4 due to persistent hypercalciuria.
Calciuria
Ca/creatinine was significantly reduced by HCT-therapy (median before/at/after withdrawal 0.76/1.24/0.77 mol/mol creatinine; n = 8, P<0.05, Figure 2). A separate analysis of heterozygous patients (patients 1–4) and homozygous/compound heterozygous patients (patients 5–8) showed similar results (heterozygous patients: median 0.91/1.48/0.90 mol/mol creatinine, n = 4, P<0,05; homozygous/compound heterozygous patients: median 0.70/1.13/0.71 mol/mol creatinine, n = 4, P<0,05). Patients 1–4 with the higher Ca/creatinine values were younger than patients 5–8 and therefore had higher normal values (age 0.7–9.8 vs 14.3–22.3 years). Before HCT withdrawal, five out of eight patients were normocalciuric or slightly hypercalciuric (up to 0.10 mol/mol creatinine above reference value). Three out of eight patients were hypercalciuric, with values 0.15–0.31 mol/mol above age-specific reference. At HCT withdrawal, seven patients were hypercalciuric, with values 0.22–1.01 mol/mol above age-specific reference. After restart of HCT therapy, six patients returned normocalciuric or slightly hypercalciuric. Two patients were hypercalciuric with values 0.17–0.27 mol/mol above the age-specific reference. The two patients with the highest calcium excretion (patients 1 and 2) were the youngest patients with a higher normal value compared with the older patients.
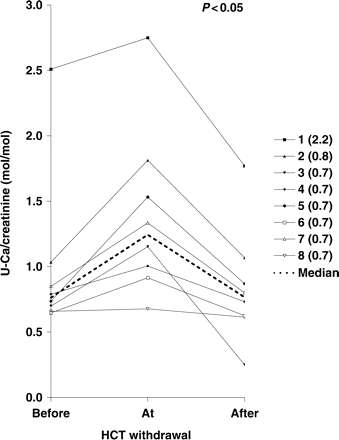
Urinary calcium/creatinine ratio (mol/mol creatinine) for patients 1–8, and median value before/at/after HCT withdrawal. The upper normal value is indicated after each patient number.
Twenty-four hour urine was only obtained from five patients because of age-related limitations. Reduction of Ca/24 h by HCT was not statistically significant, but showed the same tendency as Ca/creatinine (median 0.13/0.19/0.13 mmol/kg × 24 h; n = 5). A separate analysis for the homozygous/compound heterozygous patients showed nearly identical results (median 0.14/0.19/0.14 mmol/kg × 24 h; n = 4). Because of age, we only had 24 h urine from one heterozygous patient.
Serum magnesium and urinary magnesium excretion
S-Mg was significantly higher during HCT withdrawal (median before/at/after withdrawal 0.51/0.64/0.56 mmol/l; n = 8, P<0.05, Figure 3). Again, separate analysis showed similar results (heterozygous patients: median 0.56/0.66/0.59 mmol/l, n = 4, P<0,05; homozygous/compound heterozygous patients: median 0.45/0.61/0.58 mmol/l, n = 4, P<0.05). All patients had hypomagnesaemia during HCT therapy (values 0.03–0.31 mmol/l below reference). Hypomagnesaemia was less pronounced at HCT withdrawal (0.06–0.15 mmol/l below reference). However, only two patients achieved normomagnesaemia. In all patients except patient 6, values for s-Mg decreased after restart of HCT therapy. None of the patients showed any clinical symptoms of hypomagnesaemia such as muscle cramps, cardiac palpitations or seizures throughout the study.
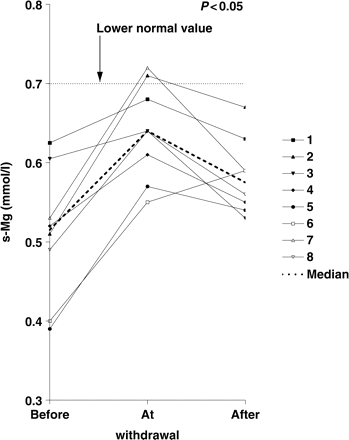
Serum magnesium (mmol/l) for patients 1–8, and median value before/at/after HCT withdrawal. The lower normal value is indicated by the dotted line in the figure.
Mg/creatinine remained statistically unchanged during withdrawal (median 0.98/0.90/0.90 mol/mol creatinine; n = 8). The same results could be shown in the separate analysis (heterozygous patients: median 1.46/1.40/1.27 mol/mol creatinine, n = 4; homozygous/compound heterozygous patients: median 0.85/0.85/0.88 mol/mol creatinine, n = 4). There was no increase of magnesiuria after restart of HCT therapy. Furthermore, almost all patients showed hypermagnesiuria throughout the study (except patient 1 before/at/after withdrawal, and patients 3 and 4 after withdrawal).
Mg/24 h also remained statistically unchanged during HCT withdrawal. However, there was a tendency towards increased magnesiuria under HCT therapy (median before/at/after withdrawal 0.14/0.12/0.18 mmol/kg × 24 h; n = 5). As we assessed 24 h urine only from one heterozygous patient, the results of the separate analysis did not relevantly differ.
Serum potassium and kaliuria
Serum potassium was significantly higher during HCT withdrawal (median before/at/after withdrawal 3.65/4.35/3.65 mmol/l; n = 8, P<0.05, Figure 4). These results were reproduced in the separate analysis (heterozygous patients: median 3.60/4.15/3.40 mmol/l, n = 4, P<0.05; homozygous/compound heterozygous patients: median 3.65/4.50/3.65 mmol/l, n = 4, P<0.05). Half of the patients were hypokalaemic under HCT therapy at study start (values 0.2–0.5 mmol/l below reference). All patients achieved normokalaemia at HCT withdrawal. Furthermore, all patients showed an increase of s-K values at withdrawal. After restart of HCT therapy, four patients went back into hypokalaemia (values 0.1–0.8 mmol/l below reference). None of the patients showed any symptoms of hypokalaemia such as fatigue, constipation or cardiac palpitation throughout the study.
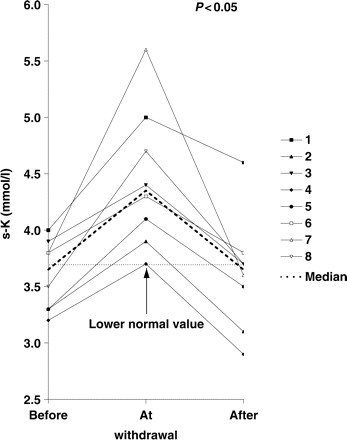
Serum potassium (mmol/l) for patients 1–8, and median value before/at/after HCT withdrawal. The lower normal value is indicated by the dotted line in the figure.
K/creat remained statistically unchanged during withdrawal (median 6.3/8.4/6.2 mol/mol creatinine; n = 8). The separate analysis showed the same results (heterozygous patients: median 7.0/9.4/6.7 mol/mol creatinine, n = 4; homozygous/compound heterozygous patients: median 5.7/8.4/6.2 mmol/l, n = 4).
Discussion
We investigated the acute effect of HCT on hypercalciuria on eight patients with FHHNC caused by CLDN16 mutation. The hypocalciuric effect of HCT is, as recent data suggest, caused by enhanced passive calcium transport in the proximal tubule [17]. Secondly, the question of whether clinically significant side effects of HCT therapy could be seen was evaluated.
Our patients were an inhomogeneous group regarding CLDN16 gene mutation. Patients 5–8 were homozygous or compound heterozygous for well-known mutations. In contrast, in patients 1–4 only one allele of the CLDN16 gene showed a mutation. All patients showed typical clinical manifestations of the disease. However, questions remain about genotype/phenotype correlation in CLDN16 mutation. A recent publication identifies heterozygous carriers of CLDN16 mutation at risk for hypercalciuria leading to renal stones or nephrocalcinosis [25]. In patients 1–4 (two pairs of siblings: patients 1/3 and patients 2/4), only one mutant allele could be detected. This raised the question of whether these four patients, despite their typical clinical disease, should be regarded as heterozygous carriers for CLDN16 mutation rather than FHHNC patients. As mutation analysis was performed by direct sequencing of the coding region and the exon–intron boundaries, it might well be that the second mutation was missed if it was located somewhere else in CLDN16. In addition, the phenotype (marked hypomagnesaemia and chronic renal failure in at least one sibling of each family) strongly suggests the diagnosis of FHHNC. Furthermore, haplotype analysis for CLDN16 region showed the same second haplotype in both sibling pairs, which is compatible with linkage analysis to the CLDN16 locus. This further supports the idea that both patients in the two families have the same disease.
The separate analysis of our heterozygous and homozygous/compound heterozygous patients showed the same results for all parameters as the analysis of the entire group. As a consequence, the results for all patients will be discussed together.
We could demonstrate a significant reduction of urinary calcium/creatinine ratio under HCT therapy. However, the effect was less pronounced in 24 h urine excretion. The lack of statistical significance might mainly be due to the fact that 24 h urine was only obtained from five patients because of age-related limitations.
The more prominent reduction of calciuria observed in Patients 1 and 3 could be due to an increased HCT dose in Patient 1. Furthermore, a habituation effect to thiazide therapy could play a role, resulting in an enhanced hypocalciuric effect after restarting therapy.
All patients were hypomagnesaemic under HCT therapy, and six patients at HCT withdrawal. However, hypomagnesaemia was less pronounced during HCT withdrawal. Neither under HCT therapy nor at withdrawal did our patients show any symptoms of hypomagnesaemia. Neither muscular cramps, seizures nor cardiac palpitations during the observation period were seen, which might indicate a good adaptation. We could not demonstrate any significant change in magnesiuria under HCT therapy in our patients. Like serum magnesium, serum potassium significantly dropped during HCT therapy, which did not result in clinical symptoms such as fatigue or palpitations. However, we did not see a significantly decreased tubular potassium re-absorption in our patients under HCT therapy.
Chronic use of thiazide diuretics in patients with idiopathic hypercalciuria is reported to induce hypokalaemia (through increased urinary potassium excretion), as well as renal magnesium wasting [4,14,16,17]. However, almost all our patients showed hypermagnesiuria even at HCT withdrawal as a feature of their CLDN16 mutation. In addition, the high dose of magnesium supplements might add to the degree of magnesium excretion. It is conceivable that patients with CLDN16 mutation only develop a transient increase of magnesiuria and kaliuria after start of HCT therapy that might be counter-regulated by the low serum electrolyte levels, and result in a new steady state. This seems to be a possible explanation for statistically unchanged magnesiuria and kaliuria with or without thiazide treatment. Furthermore, renal magnesium re-absorption underlies a series of regulating factors, for example hormonal control, Mg availability or biochemical changes like metabolic acidosis or hypokalaemia [4]. Additionally, it cannot be excluded that thiazide diuretics may have an effect upon calcium, magnesium or potassium excretion beyond the kidney. For instance, a decrease in fecal calcium excretion under HCT therapy has been reported in patients with idiopathic hypercalciuria [14]. Finally, the exchange of potassium and hydrogen ions in the tubuli might be altered, which could be examined by changes in the acid–base status. Taken together, several factors may account for our observation that despite the significant impact of HCT therapy on serum magnesium and potassium levels, this effect cannot be demonstrated in urinary magnesium and potassium excretion. Nevertheless, thiazide-induced potassium and magnesium depletion may enhance the risk of cardiac events (due to prolonged QT interval). Additionally, hypokalaemia is known to induce renal injury via an increase in renin secretion, stimulation of the sympathetic nervous system, and alteration of nitric oxide metabolism [26]. For these reasons, regular cardiac monitoring by electrocardiogram recording and regular monitoring of serum electrolyte levels is highly advisable [27,28].
Conclusion
We could demonstrate that HCT is effective in reducing hypercalciuria in FHHNC due to CLDN16 mutation. The therapy appears not to be associated with acute adverse effects of clinical significance. Our data indicate that HCT therapy can be used in patients with CLDN16 mutation to reduce hypercalciuria. However, long-term safety and the efficacy of HCT therapy to attenuate disease progression and prolong renal survival remains to be analysed.
The authors would like to thank Mrs Sibylle Pötz for her excellent assistance throughout the study period and clinical work.
Conflict of interest statement. None declared.
References
Simon DB, Lu Y, Choate KA et al. Paracellin-1, a renal tight junction protein required for paracellular [Mg2+] resorption.
Weber S, Hoffmann K, Jeck N et al. Familial hypomagnesaemia with hypercalciuria and nephrocalcinosis maps to chromosome 3q27 and is associated with mutations in the PCLN-1 gene.
Blanchard A, Jeunemaitre X, Coudol P et al. Paracellin-1 is critical for magnesium and calcium reabsorption in the human thick ascending limb of Henle.
Quamme GA. Renal magnesium handling: new insights in understanding old problems.
Weber S, Schneider L, Peters M et al. Novel Paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis.
Wolf MTF, Dötsch J, Konrad M, Böswald M, Rascher W. Follow-up of five patients with FHHNC due to mutations in the Paracellin-1 gene.
Benigno V, Canonica CS, Bettinelli A, von Vigier RO, Truttmann AC, Bianchetti MG. Hypomagnesemia hypercalciuria nephrocalcinosis: a report of nine cases and a review.
Kari JA, Farouq M, Alshaya HO. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis.
Tajima T, Nukae J, Fujieda K. Two heterozygous mutations of CLDN16 in a Japanese patient with FHHNC.
Praga M, Vara J, Gonzalez-Parra E et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis.
Kuwertz-Broking E, Frund S, Bulla M, Kleta R, August C, Kisters K. Familial hypomagnesemia-hypercalciuria in 2 siblings.
Monnens L, Starremans P, Bindels R. Great strides in the understanding of renal magnesium and calcium reabsorption.
Mourani C, Khallouf E, Akkari V, Akatcherian C, Cochat P. Early hypomagnesemia, hypercalciuria and nephrocalcinosis: two cases in a family.
Yendt ER, Gagne RJA, Cohanim M. The effects of thiazides in idiopathic hypercalciuria.
Sato K, Hasegawa Y, Nakae J et al. Hydrochlorothiazide effectively reduces urinary calcium excretion in two Japanese patients with gain-of-function mutations of the calcium-sensing receptor-gene.
Dai LJ, Friedman PA, Quamme GA. Cellular mechanisms of chlorothiazide and cellular potassium depletion on [Mg2+] uptake in mouse distal convoluted tubule cells.
Nijenhuis T, Vallon V, van der Kemp AWCM, Loffing J, Hoenderop JGJ, Bindels RJM. Enhanced passive Ca2+ reabsorption and reduced [Mg2+] channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia.
Matos V, van Melle G, Boulat O, Markert M, Bachmann C, Guignard J-P. Urinary phosphate/creatinine, calcium/creatinine, and magnesium/creatinine ratios in a healthy pediatric population.
Hicks JM, Bailey J, Bjorn S. Pediatric reference ranges for plasma magnesium.
Greeley C, Snell J, Colaco A. Pediatric reference ranges for electrolytes and creatinine.
Geven WB, Monnens LAH, Willems JL. Magnesium metabolism in childhood.
Hoppe B, Michalk D. Harnsteinerkrankungen im Kindesalter.
Fuentes-Arderiu X, Mas-Serra R, de-No-Lengaran C et al. Multicentre physiological reference values for some urinary component-to-creatinine (creatininium) concentration ratios.
Tasic V, Dervisov D, Koceva S, Weber S, Konrad M. Hypomagnesemia with hypercalciuria and nephrocalcinosis: case report and a family study.
Suga S-I, Phillips MI, Ray PE et al. Hypokalemia induces renal injury and alterations in vasoactive mediators that favour salt sensitivity.
Foglia PEG, Bettinelli A, Tosetto C et al. Cardiac work up in primary renal hypokalaemia-hypomagnesaemia (Gitelman syndrome).
Author notes
1Klinik für Kinder und Jugendliche der Friedrich-Alexander-Universität, Loschgestr. 15, D-91054 Erlangen, 2Universitäts-Kinderklinik, Inselspital, CH-3010 Bern and 3Institut für Medizininformatik, Biometrie und Epidemiologie der Friedrich-Alexander-Universität, Waldstr. 6, D-91054 Erlangen, Germany


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments