





Abstract
Diagnostic workflows for glioblastoma (GBM) patients increasingly include DNA sequencing–based analysis of a single tumor site following biopsy or resection. We hypothesized that sequencing of multiple sectors within a given tumor would provide a more comprehensive representation of the molecular landscape and potentially inform therapeutic strategies.
Ten newly diagnosed, isocitrate dehydrogenase 1 (IDH1) wildtype GBM tumor samples were obtained from 2 (n = 9) or 4 (n = 1) spatially distinct tumor regions. Tumor and matched blood DNA samples underwent whole-exome sequencing.
Across all 10 tumors, 51% of mutations were clonal and 3% were subclonal and shared in different sectors, whereas 46% of mutations were subclonal and private. Two of the 10 tumors exhibited a regional hypermutator state despite being treatment naïve, and remarkably, the high mutational load was predominantly limited to one sector in each tumor. Among the canonical cancer-associated genes, only telomerase reverse transcriptase (TERT) promoter mutations were observed in the founding clone in all tumors. Reconstruction of the clonal architecture in different sectors revealed regionally divergent evolution, and integration of data from 2 sectors increased the resolution of inferred clonal architecture in a given tumor. Predicted therapeutic mutations differed in presence and frequency between tumor regions. Similarly, different sectors exhibited significant divergence in the predicted neoantigen landscape.
The substantial spatial heterogeneity observed in different GBM tumor sectors, especially in spatially restricted hypermutator cases, raises important caveats to our current dependence on single-sector molecular information to guide either targeted or immune-based treatments.
This study provides both a biological and a potentially clinical rationale for pursuing multisector molecular profiling in GBM patients. We demonstrate, using image-guidance directed sampling of 2–4 sectors of gadolinium-enhancing areas of IDH1 wildtype GBM tumors, that whole-exome sequencing of individual sectors reveals a spatially divergent mutational landscape. In 2 extreme cases of regional heterogeneity, we describe, for the first time, treatment-naïve tumors with region-specific hypermutator phenotypes. In remarkable contrast to the spatial diversity of the overall mutational landscape, we demonstrate that TERT promoter mutations are unique in being recurrent in all analyzed tumors and clonal in all tumor sectors. Finally, we examine the potential therapeutic consequences of multisector sequencing data and find that multisite analyses may be necessary to accurately characterize individual GBM tumors and identify meaningful therapeutic options.
Glioblastoma (GBM) is the most common malignant primary brain tumor in adults. The current standard-of-care treatment for GBM is maximal, safe, surgical resection followed by concomitant chemotherapy and radiation therapy.1 Despite recent advances, the overall prognosis of the majority of GBM patients remains poor, with a median survival of 15 months2 and a 5-year survival of 10%.1 However, clinical outcomes vary considerably among patients. Previous studies have demonstrated marked differences among tumors at the genomic3,4 and transcriptomic5,6 levels, which may underlie differences in both the natural history of a patient’s tumor as well as responses to treatment.2 For instance, patients with isocitrate dehydrogenase 1 (IDH1) mutant GBMs have a median survival at least 2 times longer than that of patients with IDH1 wildtype tumors,7 whereas isolated telomerase reverse transcriptase (TERT) promoter mutations are associated with significantly lower overall survival.8 Promoter methylation at the O6-methylguanine-DNA-methyltransferase (MGMT) locus predicts relative sensitivity to the drug temozolomide.9
More broadly, the genomic characterization of all cancers has led to the identification of tumor-specific alterations that may inform therapeutic options. Due to the urgent need for additional therapies in GBM, precision medicine has been introduced into the clinical space for this disease as well. The goal is to match specific tumor mutations to potentially therapeutic drugs and, more recently, to determine the neoantigen landscape as an aggregate biomarker either for immunotherapy or for vaccine approaches.10,11 The current clinical workflow in most academic hospitals includes the identification of potentially actionable mutations or neoantigens from a single tumor sector. For GBM located at sites that preclude open craniotomy for resection, stereotactic needle biopsy from a single tumor region is performed and analyzed for genomic and other molecular data. However, in addition to differences among individual patients’ tumors, many solid tumors harbor substantial intratumoral genetic heterogeneity,12,13 raising the important question of whether molecular characterization of a single sector in GBM sufficiently represents the genomic landscape of a tumor in a biologically and clinically meaningful way.
In this study, we performed whole-exome sequencing of multiple sectors of individual GBM tumors to more broadly characterize their mutational profiles, with implications for our understanding of tumor biology as it relates to clonal architecture and pretreatment evolutionary dynamics as well as the therapeutically actionable genomic landscape.
Methods
Tumor Samples
Adults with newly diagnosed, treatment-naive GBM undergoing craniotomy for tumor resection were included. During tumor resection, tumor samples (n = 9) were collected from regionally distinct areas that demonstrated gadolinium enhancement on MRI, and images of the biopsy locations were captured using the neuronavigation platform (Stealth, Medtronic). In one case, 4 adjacent sectors (each approximately 1 cm3) of an en bloc resected GBM tumor (B103) were sampled. This study was approved by the institutional review board at Washington University School of Medicine.
DNA Sequencing
Minimal tumor cellularity was 30% in all samples as determined by a pathologist. Matched tumor and blood DNA samples for each patient were subjected to whole-exome sequencing to identify somatic mutations, including single nucleotide variants (SNVs), insertions or deletions (indels), and copy number alterations (CNAs). Mean target coverage of 80–100× of coding bases in the exome was achieved for all samples except tumor B103, for which 54–90× mean coverage was achieved. The TERT promoter was not captured well, and therefore, a set of custom capture probes from IDT Technologies were used to target this region for additional sequencing, resulting in 4680× coverage of this locus (Supplementary Table S1).
Somatic Variant Detection
Sequences were aligned to reference build GRCh37-light using the bwa mem algorithm.14 Somatic SNVs and indels were detected using an ensemble of 6 different variant callers, the calls from which were unioned and then subjected to additional filtering and manual review. Copy number alterations were detected using Varscan 2.3.6.15 See Supplemental Methods for additional details.
Subclonal Inference and Clonal Evolution
Multidimensional subclonal inference was performed using copy-number neutral SNVs and SciClone16 version 1.1 (params: minimumDepth = 100, maximumClusters = 10). Variant clusters identified by SciClone were imported into ClonEvol (https://github.com/hdng/clonevol) to infer each tumor’s phylogeny.
Potentially Therapeutic Mutation and Neoantigen Prediction
DGIdb,17 the Drug-Gene Interaction database, was used to identify potentially druggable targets based on the lists of mutations and altered genes implicated. Major histocompatibility complex (MHC) class I neoantigen predictions were made using personalized variant antigens by cancer sequencing,18 which leverages 5 algorithms from the Immune Epitope Database and Analysis resource (iedb.org): netMHC, netmhcpan, pickpocket, smm, and smmpmbec. Predictions were retained if the best score had a half-maximal inhibitory concentration <500 nM and better binding of the mutant peptide than the wild type (fold change >1).
Compliance with Ethical Standards
Ethical Approval
All procedures performed were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Results
Sample Characteristics
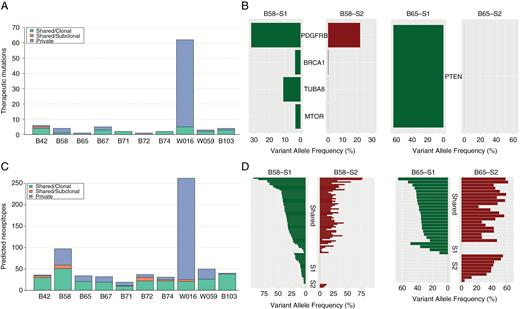
The cohort consisted of 10 patients with newly diagnosed, treatment-naïve IDH1 wildtype glioblastoma. Patient characteristics are shown in Table 1. The overall number of somatic mutations for each patient (aggregated from all samples) ranged from 112 to 1239, with a mean of 347 (Fig. 1A). The number of nonsynonymous mutations ranged from 47 to 556, with a mean of 132. Samples B58 and W016 harbored overall higher mutational loads than the remaining 8 tumors and were classified as hypermutators (see Mutational Heterogeneity between tumor regions). Excluding these samples reduces the mean number of total mutations to 191 and that of nonsynonymous mutations per tumor to 73. We observed recurrent somatic mutations in canonical GBM-associated genes, including the TERT promoter (10/10 cases, 100%), epidermal growth factor receptor (EGFR) (4/10, 40%), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) (3/10, 33%), and tumor suppressor protein 53 (TP53) (3/10, 33%) (Fig. 1B).3 Common CNAs included chromosome 7 amplifications (7/10 cases, 70%) and chromosome 10 deletions (10/10, 100%) (Supplementary Figure S1A). Paired samples from different sectors had mostly concordant CNAs (Supplementary Figure S1B, Supplementary Table S2), consistent with previous work suggesting that copy number events occur early in tumorigenesis.19
Clinical characteristics of the study cohort
| Patient | Age at Diagnosis, y | Sex | MGMT | IDH1 | Regions Sampled, n | Max. Tumor Diameter, cm | Tumor Volume, cm3 | Cellularity (S1/S2/S3/S4) | Necrosis (S1/S2/S3/S4) |
|---|---|---|---|---|---|---|---|---|---|
| B42 | 70.1 | F | 1 | WT | 2 | 5.3 | 13.5 | 75/75 | 10/10 |
| B58 | 66.4 | M | 0 | WT | 2 | 5.5 | 29.3 | 85/70 | 20/5 |
| B65 | 54 | F | 0 | WT | 2 | 5.6 | 26.5 | 60/80 | 5/0 |
| B67 | 60.4 | F | 0 | WT | 2 | 7.2 | 48.0 | 40/60 | 50/50 |
| B71 | 60.4 | M | 0 | WT | 2 | 4 | 13.2 | 65/70 | 50/70 |
| B72 | 76.6 | M | 1 | WT | 2 | 5.4 | 21.1 | 80/65 | 5/15 |
| B74 | 62.6 | F | 1 | WT | 2 | 5.2 | 23.5 | 70/70 | 10/70 |
| W016 | 56 | F | WT | 2 | 5.2 | 14.1 | 60/40 | 10/0 | |
| W059 | 71 | F | 1 | WT | 2 | 5 | 14.9 | 90/85 | 5/20 |
| B103 | 74.3 | M | 1 | WT | 4 | 5.5 | 27.5 | 70/45/70/70 | 50/5/10/5 |
| Patient | Age at Diagnosis, y | Sex | MGMT | IDH1 | Regions Sampled, n | Max. Tumor Diameter, cm | Tumor Volume, cm3 | Cellularity (S1/S2/S3/S4) | Necrosis (S1/S2/S3/S4) |
|---|---|---|---|---|---|---|---|---|---|
| B42 | 70.1 | F | 1 | WT | 2 | 5.3 | 13.5 | 75/75 | 10/10 |
| B58 | 66.4 | M | 0 | WT | 2 | 5.5 | 29.3 | 85/70 | 20/5 |
| B65 | 54 | F | 0 | WT | 2 | 5.6 | 26.5 | 60/80 | 5/0 |
| B67 | 60.4 | F | 0 | WT | 2 | 7.2 | 48.0 | 40/60 | 50/50 |
| B71 | 60.4 | M | 0 | WT | 2 | 4 | 13.2 | 65/70 | 50/70 |
| B72 | 76.6 | M | 1 | WT | 2 | 5.4 | 21.1 | 80/65 | 5/15 |
| B74 | 62.6 | F | 1 | WT | 2 | 5.2 | 23.5 | 70/70 | 10/70 |
| W016 | 56 | F | WT | 2 | 5.2 | 14.1 | 60/40 | 10/0 | |
| W059 | 71 | F | 1 | WT | 2 | 5 | 14.9 | 90/85 | 5/20 |
| B103 | 74.3 | M | 1 | WT | 4 | 5.5 | 27.5 | 70/45/70/70 | 50/5/10/5 |
Note. MGMT: 0 = methylated; 1 = unmethylated. IDH1 by R132H immunohistochemistry and exome sequencing.
Clinical characteristics of the study cohort
| Patient | Age at Diagnosis, y | Sex | MGMT | IDH1 | Regions Sampled, n | Max. Tumor Diameter, cm | Tumor Volume, cm3 | Cellularity (S1/S2/S3/S4) | Necrosis (S1/S2/S3/S4) |
|---|---|---|---|---|---|---|---|---|---|
| B42 | 70.1 | F | 1 | WT | 2 | 5.3 | 13.5 | 75/75 | 10/10 |
| B58 | 66.4 | M | 0 | WT | 2 | 5.5 | 29.3 | 85/70 | 20/5 |
| B65 | 54 | F | 0 | WT | 2 | 5.6 | 26.5 | 60/80 | 5/0 |
| B67 | 60.4 | F | 0 | WT | 2 | 7.2 | 48.0 | 40/60 | 50/50 |
| B71 | 60.4 | M | 0 | WT | 2 | 4 | 13.2 | 65/70 | 50/70 |
| B72 | 76.6 | M | 1 | WT | 2 | 5.4 | 21.1 | 80/65 | 5/15 |
| B74 | 62.6 | F | 1 | WT | 2 | 5.2 | 23.5 | 70/70 | 10/70 |
| W016 | 56 | F | WT | 2 | 5.2 | 14.1 | 60/40 | 10/0 | |
| W059 | 71 | F | 1 | WT | 2 | 5 | 14.9 | 90/85 | 5/20 |
| B103 | 74.3 | M | 1 | WT | 4 | 5.5 | 27.5 | 70/45/70/70 | 50/5/10/5 |
| Patient | Age at Diagnosis, y | Sex | MGMT | IDH1 | Regions Sampled, n | Max. Tumor Diameter, cm | Tumor Volume, cm3 | Cellularity (S1/S2/S3/S4) | Necrosis (S1/S2/S3/S4) |
|---|---|---|---|---|---|---|---|---|---|
| B42 | 70.1 | F | 1 | WT | 2 | 5.3 | 13.5 | 75/75 | 10/10 |
| B58 | 66.4 | M | 0 | WT | 2 | 5.5 | 29.3 | 85/70 | 20/5 |
| B65 | 54 | F | 0 | WT | 2 | 5.6 | 26.5 | 60/80 | 5/0 |
| B67 | 60.4 | F | 0 | WT | 2 | 7.2 | 48.0 | 40/60 | 50/50 |
| B71 | 60.4 | M | 0 | WT | 2 | 4 | 13.2 | 65/70 | 50/70 |
| B72 | 76.6 | M | 1 | WT | 2 | 5.4 | 21.1 | 80/65 | 5/15 |
| B74 | 62.6 | F | 1 | WT | 2 | 5.2 | 23.5 | 70/70 | 10/70 |
| W016 | 56 | F | WT | 2 | 5.2 | 14.1 | 60/40 | 10/0 | |
| W059 | 71 | F | 1 | WT | 2 | 5 | 14.9 | 90/85 | 5/20 |
| B103 | 74.3 | M | 1 | WT | 4 | 5.5 | 27.5 | 70/45/70/70 | 50/5/10/5 |
Note. MGMT: 0 = methylated; 1 = unmethylated. IDH1 by R132H immunohistochemistry and exome sequencing.
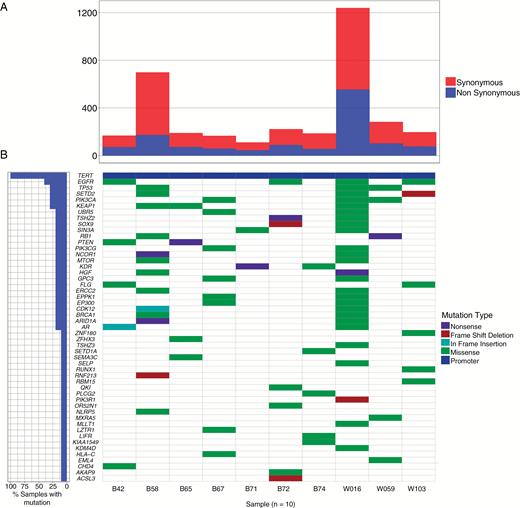
Somatic mutation landscape of treatment-naïve primary glioblastoma. (A) Overall mutation loads for 10 patients in the study cohort. (B) Waterfall plot highlighting somatic mutations in a subset of genes that are recurrently mutated in human cancers. The mutation frequencies for each gene are depicted in the chart on the left.
Mutational Heterogeneity Between Tumor Regions
We then compared somatic SNVs and indels in different tumor sectors for each patient to understand the extent of regional intratumoral heterogeneity (Fig. 2A). Mutations were classified as “private” if observed in only one sector of a tumor, and “shared” if present in 2 or more spatially distinct regions. Strikingly, 46% of mutations were private, while 54% were shared across sectors, indicating significant spatial heterogeneity (Fig. 2B–D, Supplementary Table S3).
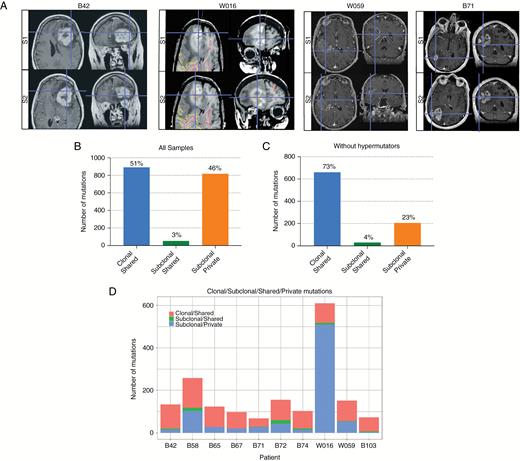
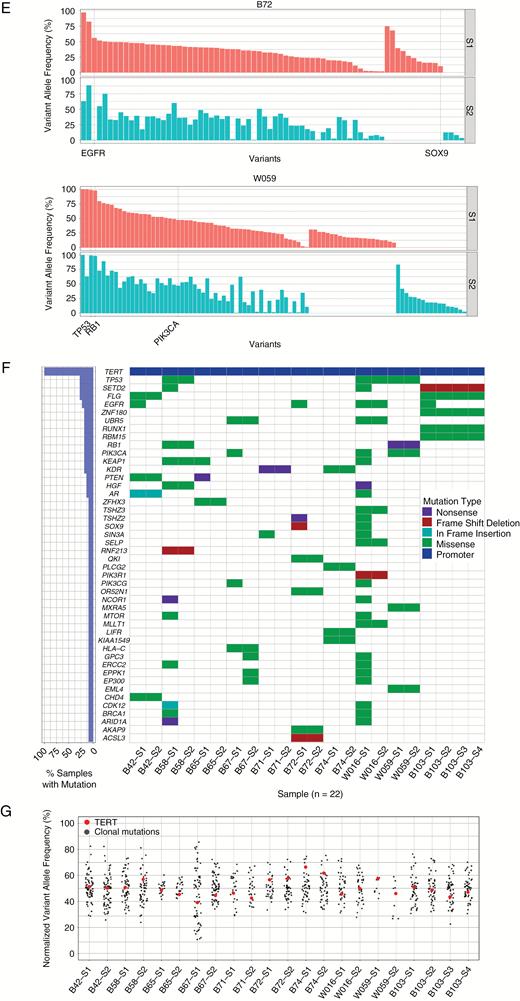
Mutational heterogeneity between tumor sectors. (A) Representative T1-weighted MRIs for patients B42, W016, W059, and B71 illustrating the coordinates of 2 distinct tumor biopsy regions (S1 and S2) in the axial and coronal planes. (B, C) Charts depicting the proportions of clonal-shared, subclonal-shared, and subclonal-private mutations for the entire study cohort (B) and 8 non-hypermutant cases (C). (D) Chart showing the overall numbers of clonal-shared, subclonal-shared, and subclonal-private mutations for each patient. (E) Representative VAF versus somatic mutation plots illustrating mutational heterogeneity between 2 distinct tumor sectors (S1 and S2) for patients B72 (left) and W059 (right). (F) Waterfall plot highlighting regional heterogeneity of somatic mutations in a subset of recurrently mutated cancer-associated genes. Mutation frequencies for each gene for all tumor regions sequenced are depicted on the left. (G) Plot of the VAFs of clonal mutations in each tumor sector normalized to the median VAF of all clonal mutations in that specific sector (black dots). The VAFs of TERT promoter mutations (G228A and G250A) are indicated in red. For patient W059, the TERT promoter region was amplified, thus VAFs after copy number correction are shown.
Using variant allele frequencies (VAFs) corrected for tumor purity, events were further classified as either clonal (present in all cancer cells) or subclonal (present in only a subset of cells). Fifty-one percent of mutations were clonal and shared between regions, 3% were subclonal and shared, and 46% were subclonal and private to one tumor sector (Fig. 2B). As both sectors appeared to share a common origin in all samples, events cannot be both clonal and private. The 2 hypermutator tumors influence these data due to a significantly higher load of subclonal private mutations. Excluding the hypermutator tumors resulted in 73% of mutations being classified as clonal and shared, 4% as subclonal and shared, and 23% as subclonal and private (Fig. 2C). In addition, we observed that even mutations present in both sectors may differ markedly in frequency, as observed in uncorrected VAF plots (Fig. 2E) for 2 representative cases, B72 and W059.
To better understand the impact of these differences between sectors, we focused on a subset of genes known to be recurrently mutated in cancer20–22 (Supplementary Table S4). In 8 of 10 cases, one or more mutations in cancer-related genes were private and not readily detectable in the other tumor sector(s) (Fig. 2F). These private mutations include 3 of the 4 EGFR mutations (M567I in B103, E320Q in B42, and T363I in B72) as well as both EP300 mutations (A1853T in B67, G322R in W016). This indicates that these mutations were acquired later in a tumor’s evolution. In contrast, all 3 samples with TP53 mutations harbored the mutation in all sectors (R248Q in B58, V173L in W016, and G245S in W059), suggesting that they are either initiating mutations (for B58 and W059, where they are high VAF and clonal) or acquired early after transformation (for W016 where the mutation has lower VAF and is subclonal). The only recurrent mutations observed in all sectors of all tumors were TERT promoter mutations (both G228A and G250A, in mutually exclusive fashion). In all cases, these TERT mutations were clonal, suggesting that they are likewise initiating events in GBM (Fig. 2G).
Treatment-Naïve Glioblastoma with Hypermutator Phenotype
Two patient tumors, W016 and B58, harbored extremely high mutational loads—1239 and 699 mutations, respectively. We classified them as hypermutated based on a statistical outlier test for overall mutation load across all samples and on the fact that they harbored mutations greater than 3 standard deviations above the trimmed mean (>409 SNVs) of the 8 other tumors, a definition consistent with the threshold of 400 SNVs used recently.23 In both cases, the hypermutator phenotype was restricted to a single sector, with the non-hypermutated sectors bearing mutation loads not significantly different from the mean number of mutations among the remaining 8 cases. In both cases, there were no germline hotspot missense mutations in DNA-damage associated genes that could potentially explain the increased mutation load (Supplementary Table S5). In Patient W016, over 90% of mutations were specific to one of the 2 sectors (S1) (Fig. 3A, B) and these consisted almost entirely of C to T transitions (Fig, 3E). The mutation spectrum of this sector did not match the oxo-8G profile (CCG > CAG)24 for all 1204 mutations (Supplementary Table S6), thus ruling out this artifact as a reason for increased mutation burden. We performed an exploratory analysis to determine if mutations in known DNA-damage repair genes might represent a major pathogenic event in W016 S1. W016 S1 harbored mutations in critical DNA repair associated genes ALKBH3, ATR, POLB, MLH3, and SPO11 (Supplementary Table S7), but due to the multiple plausible drivers at sufficient VAF, the exact initiating event in the hypermutated sector could not be determined. In tumor B58, sector S1 contained approximately 70% of the aggregate mutations from the 2 sequenced sectors (Fig. 3C, D). Unlike other tumors, a significant proportion of the mutations in both sectors of tumor B58 exhibited signature 3 in the Catalogue of Somatic Mutations in Cancer (COSMIC), a signature associated with germline breast cancer 1 and 2 (BRCA1 and BRCA2) mutations.25 Importantly, both sectors harbored a common somatic BRCA1 missense mutation, with a higher VAF in S1 relative to S2, raising the possibility that BRCA1 mutation drove hypermutation in S1. These 2 cases highlight both the occurrence and the potential for regional heterogeneity of the hypermutator phenotype in treatment-naïve GBM.
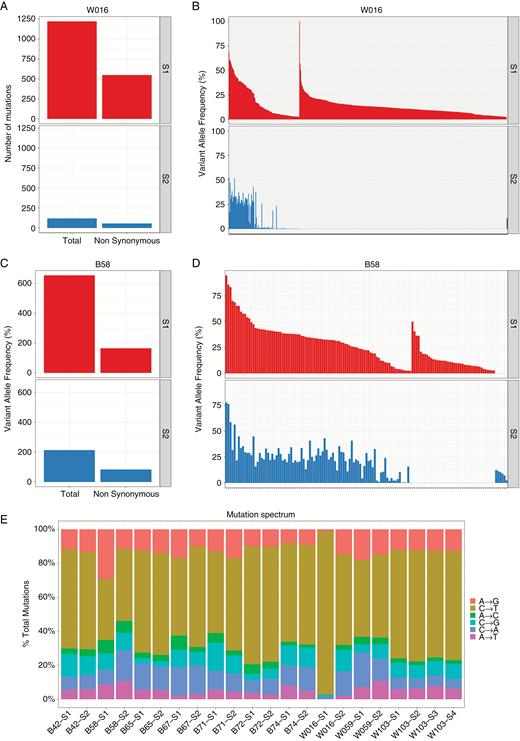
Treatment-naïve hypermutated GBMs. (A, C) Charts depicting the total numbers of overall and non-synonymous mutations in the hypermutant tumors, W106 (A) and B58 (C), in each sector sequenced S1(top) and S2 (bottom). (B, D) VAF versus somatic mutation plots for 2 distinct tumor sectors, S1 (top) and S2 (bottom), for W016 (A) and B58 (B). (E) Mutational spectrum for each tumor sector for each patient in the cohort.
Heterogeneity in Clonal Architecture Between Tumor Regions
Given our observation of mutational heterogeneity between sectors, we next determined the clonal architecture of each tumor and reconstructed its evolution. Using the SciClone16 algorithm, the VAFs of SNVs in copy-number neutral, loss of heterozygosity–free genomic regions were clustered in both one dimension (per sector) and 2 dimensions (per tumor, incorporating information from all sectors). In one representative sample, tumor B65 (Fig. 4), a subset of subclones was detectable in only one of the 2 regions. Furthermore, subclones 2 and 3 represent “cryptic” subclones that would not be distinguishable from the founding clone without additional information from spatially distinct samples. Eight of 10 (80%) cases contained subclonal populations that were private to one of the tumor sectors, consistent with the genetic heterogeneity observed between tumor regions (Supplementary Figure S3).
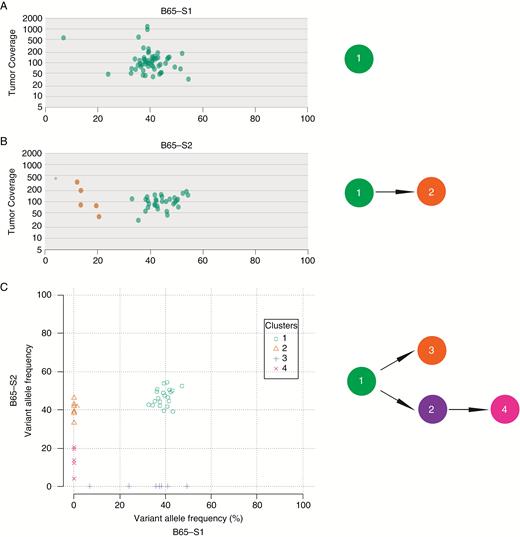
Regional heterogeneity in subclonal architecture. (A, B) SciClone-generated tumor coverage versus VAF plots for tumor B65 sectors 1 (A) and 2 (B) (left). Reconstructed probable phylogenies between subclones in each respective tumor sector are shown in the right panel. (C) SciClone-generated 2-dimensional plot of sector 2 versus sector 1 VAFs for patient B65 (left) and a corresponding example of the ClonEvol-generated probable branching phylogenetic trees based on VAF information from both tumor sectors (right).
To understand how multisector sequencing can aid in determining tumor evolution, we imported mutation clusters and VAF information into ClonEvol, which reconstructs possible phylogenetic trees. For tumor B65, using individual sectors in isolation resulted in a linear clonal evolution model (Fig. 4A, B), but incorporating both sectors revealed a branching structure (Fig. 4C), linking subclone phylogenetic trees in the multiple sectors. In 80% of cases, multisector information added complexity to the inferred phylogenies (Supplementary Figure S4).
Heterogeneity in Putative Targetable Somatic Variants Between Sectors
We next explored the potential therapeutic implications of mutational heterogeneity in these multisector tumor samples. Using DGIdb,17 we identified potentially druggable mutations in at least one of the distinct tumor sectors for all patients (Supplementary Tables S8 and S9). Eight of 10 (80%) cases had potentially targetable mutations that were not shared between sectors (Fig. 5A). Even when detected in both tumor sectors, potentially druggable mutations occurred at different VAFs between sectors in a significant number (75%) of cases. This finding is exemplified by the divergent VAFs of potential druggable gene variants between 2 sectors (S1 and S2) for tumors B58, B65, and B42 (Fig. 5B, Supplementary Figure S5). In 4 of the 10 cases (40%), private, druggable mutations were more numerous than those shared between regions, suggesting that GBM tumors can harbor heterogeneous, spatially restricted subclones bearing actionable mutations.
Regional heterogeneity in potentially therapeutic gene variants and in the predicted neoantigen landscape. (A) Chart showing the numbers of private, clonal-shared, and subclonal-shared mutations in potentially therapeutic genes for each patient. (B) Mutation versus VAF plots highlighting heterogeneity in potentially therapeutic gene variants for patient B58 (left) and B65 (right) for 2 distinct tumor sectors. (C) Chart depicting the numbers of private, clonal-shared, and subclonal-shared predicted neoantigens for each tumor. (D) Mutation versus VAF plot of predicted neoantigens for patient B58 (left) and B65 (right) for 2 tumor sectors. Mutations along the vertical axis are grouped into 3 classes: shared; sector 1 (S1) specific; and sector 2 (S2) specific.
To further crystallize the clinical implications of single versus multisite sequencing, we focused on somatic mutations in the glioblastoma driver genes EGFR and phosphatase and tensin homolog (PTEN) as demonstrative examples. Of the 4 tumors with somatic mutations in EGFR, only one variant, A289V in tumor W016, was clonal and detected in both sectors. All other EGFR SNVs were subclonal and private to one tumor sector. Although the private EGFR mutation in case B42 (E320Q) has not been previously described, the other private EGFR SNVs (T363I in B72 and M567I in B103) are listed in the COSMIC database and are predicted to be potentially pathogenic (score ≥0.89 for all 3 variants) based on the Functional Analysis Through Hidden Markov Models in silico algorithmic approach.25,26 Somatic mutations in the tumor suppressor PTEN were detected in 2 patients in our cohort. For patient B42, the potentially pathogenic PTEN L112V mutation was clonal and shared between tumor sectors. In contrast, the PTEN truncation mutation E7* in tumor B65 was subclonal in one region but absent in the second.
Heterogeneity in the Neoantigen Landscape
To determine the immunotherapeutic implications of the observed spatial mutational heterogeneity, we applied a cancer immunogenomics approach to predict the candidate neoantigen landscape in each tumor sector.11,18 In this approach, multiple computational algorithms are used to determine the affinity with which translated mutant peptides determined from identified missense variants bind to a patient’s human leukocyte antigen molecules. In each tumor examined, both shared and private neoantigens were identified (Fig. 5C, Supplementary Table S10). This observation is exemplified by the neoantigen landscapes of 2 sectors for tumors B58 and B65 (Fig. 5D). Within both tumors, although a subset of mostly high VAF neoantigens was shared between 2 spatially distinct areas, lower VAF neoantigens were identified in one of the 2 regions. Moreover, W016, the tumor with the highest mutational burden, harbored a commensurately high number of predicted subclonal rather than clonal neoantigens, largely specific to the hypermutated sector S1 (Fig. 5C). Together, these data show that spatial mutational heterogeneity correlates with heterogeneity of the candidate neoantigen landscape and that a hypermutator state leads to an elevated number of subclonal neoepitopes.
Discussion
In this study, we examined regional genetic heterogeneity among multiple sectors of primary GBM tumors by whole-exome sequencing and targeted sequencing of the TERT promoter locus. Sottoriva et al provided early evidence of intratumoral heterogeneity in GBM using genome-wide CNA analyses, although we did not detect such CNAs using exome-derived analyses.27 Verhaak and colleagues performed multisector whole-exome sequencing of GBM tumors and focused on evolutionary patterns that lead to disease recurrence.28 Another study examined transcriptional heterogeneity and utilized targeted sequencing to demonstrate regional heterogeneity of mutations in DNA-repair genes.29 From a precision oncology standpoint, Lee et al recently found that patient-derived GBM cells from multisector samples were more sensitive to drugs that target truncal rather than private genetic alterations.30 Here we focused on the mutational burden in primary GBM tumors by whole-exome sequencing of multiple regions to examine the impact of regional heterogeneity on our understanding of pretreatment tumor biology as well as the potential clinical implications for targeted- and immune-based therapies.
Within our cohort, 2 of 10 patients exhibited a putative hypermutator phenotype. A majority of the high mutational load in both hypermutator tumors was specific to one region. To our knowledge, the concomitant existence of non-hypermutated and hypermutated clones within distinct anatomic regions of GBM has not been previously described. Also, while prior studies28,31–33 have reported a 20%–30% incidence of a hypermutated state in recurrent GBM after treatment with temozolomide, its occurrence in primary treatment-naïve adult and pediatric GBM has only been reported in the setting of polymerase epsilon gene (POLE) deficiency34,35 and biallelic mismatch repair deficiency,36 respectively. Although POLE mutations were not observed in our patients, it is possible that mutations in base excision repair or mismatch repair genes, which were recently described in treatment-naïve GBM,29 could contribute to the development of a hypermutator phenotype. Given the spatial heterogeneity in mutational burden, a single sector biopsy could potentially miss the hypermutated tumor region, and therefore incorrectly classify the mutational burden of the tumor. It is also possible that the high incidence of hypermutation in recurrent, posttreatment GBM31–33 may actually represent an enrichment of pretreatment hypermutated subclones that are potentially not observed due to undersampling. Because hypermutated clones are likely insensitive to temozolomide,37 the occurrence of these populations within the tumor may also correlate with resistance to alkylating therapy, especially in the setting of subtotal resection. Further multisector sampling studies combined with outcome data will be needed to explore this possibility.
From an immunotherapy standpoint, elevated mutational burden—the engine for neoantigen production—correlates with response to checkpoint blockade immunotherapy in solid tumors.38–40 Although it is not the only parameter that influences responses to this drug class, it appears to be an important one that extends across cancer types. Indeed, several groups have reported compelling case studies of newly diagnosed hypermutated GBM that exhibits responses to checkpoint blockade immunotherapy.35,36 Especially in the MGMT promoter unmethylated setting, in which temozolomide is not as efficacious, the finding of hypermutated subsets distributed in newly diagnosed tumors may suggest that immunotherapy could be a consideration. Finally, each tumor harbored both private and shared neoantigens in each region, suggesting that consideration of spatial diversity in neoantigen-targeting approaches, such as polyvalent vaccines, may be important.
Across our study cohort, we observed regional heterogeneity in both the occurrence and relative frequencies of several cancer driver genes, such as EGFR, TP53, and PIK3CA. Additionally, clonality analysis revealed that in 90% of tumors sequenced, subclonal mutations were more likely to be private than shared between sectors, with the hypermutator cases representing extremes of regional restriction of subclonal mutations. These findings, along with convergent results on the substantial regional heterogeneity of other solid tumors, highlight the potential limitations of current single-site diagnostic biopsy in capturing the full spectrum of mutations in an individual tumor, which may be critical to a more comprehensive understanding of a tumor’s biology and response to therapy.12,13,41 In accordance with the observed regional mutational heterogeneity, subclonal inference using Bayesian clustering methods16 demonstrated the existence of spatially restricted subclones in 8 out of the 10 (80%) tumors in this cohort. We recognize that percentage of tumor cellularity, which differed by no more than 25% between paired patient samples in our study, can impact the overall number of subclones detected using our model assumptions. However, we would emphasize that the evidence for pervasive subclonal heterogeneity between regions under specific invariant model assumptions is the major biologically meaningful finding in our analysis. We found that integration of information from spatially distinct tumor samples in these cases allowed us to distinguish cryptic subclones from the founding clone, which was not achievable using only one tumor sample. Our analysis may, however, underestimate the extent of tumor evolution and heterogeneity, since we did not assess extra chromosomal DNA, which has been shown to harbor oncogenes and allow for more rapid tumor evolution than can be achieved by chromosomal amplification.42
We also observed significant heterogeneity in potentially druggable mutations between tumor sectors. Eighty percent of the tumors sequenced had private targetable mutations, and these were more frequent than shared targetable mutations in 40% of the patients. Recently, Morrissy et al found a predominance of mutations that were clonal in one biopsy specimen but were subclonal or undetected in additional biopsies in medulloblastomas and a limited number of high-grade gliomas. They estimated that to attain an 80% probability of detecting at least 80% of all potential actionable mutations, at least 5 biopsies would be required.43 This spatially distinct expression of druggable genes could, in part, explain the failure of prior clinical trials that employed targeted mutation approaches to treat GBM. Multisector sampling may therefore be required to select the appropriate “personalized” drug regimen that would engage multiple, spatially separated, targetable mutations for broader treatment coverage of the existing subclonal landscape.
Interestingly, all patients in our cohort harbored one of the 2 TERT promoter mutations (G228A or G250A). This finding is in alignment with recent studies which have reported a high incidence (54%–84%) of TERT promoter mutations in GBM patients.8,44–49 Importantly, the TERT promoter mutation was present in the founding clone in all tumor sectors examined. This is the first report of TERT promoter mutations being clonal in IDH1 wildtype GBM, consistent with recent studies which have suggested that TERT promoter mutation is an early event in GBM pathogenesis. Intriguingly, this also suggests that TERT might represent a strategic therapeutic target in GBM patients.
Although single-site biopsies may be sufficient for the detection of known clonal mutations, including TERT (as described herein) and IDH1, in GBM, our study suggests that inclusion of multisite biopsy specimens may be critical in the design of future clinical trials that test the efficacies of small-molecule or immune-based therapies due to the striking spatial divergence of most mutations. From a practical standpoint, it will be important to determine how many sites need to be sampled per tumor to capture the majority of biologically relevant, spatially restricted subclones.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
Funding
This work was supported by the Barnes-Jewish Hospital Foundation and Washington University Institute of Clinical and Translational Sciences (A.H.K.), the Christopher Davidson and Knight Family Fund (A.H.K., G.P.D.), NIH R01NS066905 (D.S.M., S.J.F.), and Washington University Medical Scientist Training Program grant NIH T32 GM07200 (T.M.).
Conflict of interest statement. G.P.D. is a co-founder of Immunovalent Therapeutics. The other authors declare no conflicts of interest.
Acknowledgment
We would like to thank Dr. Timothy Ley for thoughtful discussion.
References
Author notes
Contributed equally.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}