






Abstract
The full spectrum of the disease phenotype and viral genotype of coronavirus disease 2019 (COVID-19) have yet to be thoroughly explored in children. Here, we analyze the relationships between viral genetic variants and clinical characteristics in children.
Whole-genome sequencing was performed on respiratory specimens collected for all SARS-CoV-2-positive children (n = 141) between March 13 and June 16, 2020. Viral genetic variations across the SARS-CoV-2 genome were identified and investigated to evaluate genomic correlates of disease severity.
Higher viral load was detected in symptomatic patients (P = .0007) and in children <5 years old (P = .0004). Genomic analysis revealed a mean pairwise difference of 10.8 single nucleotide variants (SNVs), and the majority (55.4%) of SNVs led to an amino acid change in the viral proteins. The D614G mutation in the spike protein was present in 99.3% of the isolates. The calculated viral mutational rate of 22.2 substitutions/year contrasts the 13.5 substitutions/year observed in California isolates without the D614G mutation. Phylogenetic clade 20C was associated with severe cases of COVID-19 (odds ratio, 6.95; P = .0467). Epidemiological investigation revealed major representation of 3 of 5 major Nextstrain clades (20A, 20B, and 20C) consistent with multiple introductions of SARS-CoV-2 in Southern California.
Genomic evaluation demonstrated greater than expected genetic diversity, presence of the D614G mutation, increased mutation rate, and evidence of multiple introductions of SARS-CoV-2 into Southern California. Our findings suggest a possible association of phylogenetic clade 20C with severe disease, but small sample size precludes a definitive conclusion. Our study warrants larger and multi-institutional genomic evaluation and has implications for infection control practices.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), was first reported in December 2019 in Wuhan, Hubei Province, China [1]. As of September 12, 2020, >28 million COVID-19 cases and 916 992 deaths have been reported worldwide [2]. The growing significance of COVID-19 in the pediatric population has become more evident in recent months, as cases have grown from a reported 1.7% in April 2020 to 7.7% of total reported US cases in August 2020 [3]. Between July and August 2020 alone, 179 990 new cases were reported in children, a 90% increase [4]. While COVID-19 in children is often reported to be much less severe, analysis of pediatric COVID-19 hospitalization data from 14 states in the United States found that 1 in 3 hospitalized children were admitted to an intensive care unit (ICU) [5]. Awareness of the risk of serious long-term sequelae of COVID-19 in children such as multisystem inflammatory syndrome in children (MIS-C) has increased [6, 7]. Hence, even among children with asymptomatic or mild disease manifestations, diagnosis and monitoring of disease course have become a key public health imperative as we seek to safely re-open schools and restart other public activities.
Evaluation of the viral genome during a pandemic can not only aid in understanding the scale and epidemiology of the outbreak, but also provide valuable insights into viral evolution, including potential links to transmissibility and disease severity [8, 9]. Studies investigating SARS-CoV-2 genomes have identified mutations that could potentially alter viral pathogenicity and transmissibility [10, 11]; however, few have focused on how this may impact pediatric COVID-19 patients, and these were limited in scope and sample size. As of September 1, 2020, California has 756 181 confirmed COVID-19 cases and 14 273 deaths [4]. The vast majority of these cases are in Southern California. Los Angeles County has the highest number of confirmed cases and second-highest reported deaths in the United States [2]. However, genomic data from these cases are currently limited, with the majority of the genomes deposited in the GISAID database originating in Northern California [12].
Using viral whole-genome sequencing (WGS) of clinical specimens, we evaluated the associations between disease severity and viral genetic variations in children. This is the largest single-institution pediatric COVID-19 case series with integrated clinical and genomic epidemiology reported to date. As such, it represents a significant contribution to the body of knowledge regarding the clinical, genomic, and epidemiological features of COVID-19 in children.
METHODS
Study Population
Specimens from 141 COVID-19 patients were obtained from Children’s Hospital Los Angeles (CHLA) between March 19 and June 16, 2020. Patients were tested for a variety of reasons including COVID-19-related symptoms, hospital admission unrelated to COVID-19, and asymptomatic preprocedural screening. Clinical data including age, gender, ethnicity, hospitalization status, coexisting conditions, antimicrobial therapy, modes of oxygen supplementation, history of travel and contacts, clinical signs and symptoms, laboratory test results, and radiographic findings were obtained from the electronic medical record (EMR). Patients were classified based on the following clinical signs, symptoms, and radiographic findings using the Centers for Disease Control and Prevention’s (CDC’s) classification: (a) asymptomatic or presymptomatic infections: positive SARS-CoV-2 test in the absence of symptoms; (b) mild illness: signs and symptoms associated with upper respiratory tract infections (URIs; eg, fever, cough, malaise, sore throat, headache, vomiting, diarrhea) but without shortness of breath, dyspnea, or abnormal chest imaging; (c) moderate illness: evidence of lower respiratory tract infection (LRTI) or radiographic findings and having saturation of oxygen (SpO2) ≥94% on room air; (d) severe illness: SpO2 <94% on room air, a ratio of arterial partial pressure of oxygen to fraction of inspired oxygen (PaO2/FiO2) <300 mmHg, respiratory frequency >30 breaths per minute, or lung infiltrates >50%; (e) critical illness: individuals who have respiratory failure, septic shock, and/or multiple organ dysfunction [13].
Detection of SARS-CoV-2 RNA
Nasopharyngeal swabs were sent to the Clinical Virology Laboratory at Children’s Hospital Los Angeles for testing: (1) total nucleic acid was extracted using the NucliSENS easyMag (bioMérieux, Durham, NC, USA) followed by real-time reverse transcription polymerase chain reaction (RT-PCR) using the CDC 2019-Novel Coronavirus Real-Time RT-PCR assay; (2) total nucleic acid was extracted using the Thermo Fisher KingFisher Flex (Thermo Fisher Scientiftic, Waltham, MA, USA) with the MagMAX Viral and Pathogen Nucleic Acid Isolation Kits followed by RT-PCR using the TaqPath COVID-19 Combo Kit. Viral loads (copies/mL) were calculated based on a standard curve generated by testing samples with known viral copy numbers. Briefly, viral samples consisting of a gradient of known viral copy numbers (2.0×107 copies/mL to 2×103 copies/mL) were processed followed by RT-PCR in triplicate. A standard curve was generated by plotting a linear line on a graph with Ct values plotted on the y-axis and viral load in log (copies/mL) on the x-axis.
Viral Genome Library Construction and Sequencing
WGS of extracted viral RNA was performed as previously described using Paragon Genomics CleanPlex SARS-CoV-2 Research and Surveillance NGS Panel [14]. Libraries were quantified and normalized to ~7 nM and pooled to a final concentration of 4 nM; libraries were denatured and diluted according to Illumina protocols and loaded on the MiSeq at 10 pM. Paired-end and dual-indexed 2×150-bp sequencing was done using Micro Kit, version 2 (300 cycles). Sample performance was selected based on the following metrics: average depth ≥1000× and variant calling ≥100×.
Consensus Genome Assembly
Nucleotide sequences were aligned with NovoAlign (Novocraft Technologies, Selengor, Malaysia). Coverage profiles, variant calls, and consensus genomes were generated using Lightweight Utility tools of Bioinformatics Analysis (LUBA), an in-house proprietary software system [15]. Consensus sequences were built by adjusting the reference genome at high allele frequency (≥0.5) single nucleotide variants (SNVs) and indel loci. Base quality-adjusted pileup was generated, and the reference sequence was modified accordingly at positions with alternative bases and IN/DELS that accounted for >50% of the aligned reads.
Consensus Genome Comparison, Phylogenetic Analysis, and Clade Analysis
Consensus genomes obtained for the 141 CHLA isolates were compared with the Wuhan isolate (NC_045512.2) using the SARS-CoV-2 Genome App, version 1.1 (https://cov2annot.cpmbiodev.net), and CHLA COVID-19 Analysis Research Database (CARD) [16] to identify synonymous, nonsynonymous, and intergenic variations. CHLA isolates were also compared against the global collection of 77 966 available SARS-CoV-2 sequences in CARD to identify Southern California–specific haplotypes.
Phylogenetic analysis and evolutionary rate estimation were performed using packages available through Nextstrain command-line interface (version 2.0.0.) [17]. Consensus sequences for 141 CHLA isolates were combined with full-length SARS-CoV-2 sequences of 436 additional isolates from GISAID that carried the identical, ancestral, or descendent haplotypes as the 141 isolates (Supplementary Table 1) to generate a multiple sequence alignment with MAFFT (version 7.460) using speed-oriented option—FFT-NS-i (iterative refinement method, 2 cycles) optimized for large data sets [18]. A maximum likelihood tree using Bayesian information criteria was generated with IQ-TREE (version 2.0.3) [19] using the GTR substitution model. The resulting rate estimation and phylogeny was then time-resolved using TreeTime (version 0.7.6) [20] and visualized using auspice [17]. Phylogenetic clade analysis was performed using Nextclade (version 0.3.7). Similar procedures were also followed for evolutionary rate estimation of 577 SARS-CoV-2 genomes without the D614G mutation; however, analysis was restricted to isolates from California.
Statistical Analysis
Differences of genome copies/mL were compared using the Mann-Whitney test. The association between viral genotypes and disease severity was investigated using the Fisher exact test and odds ratio calculation via a 2×2 contingency table. The groupings compared were the investigated phylogenetic clades (eg, number of patients infected with clade 20C vs number of patients not infected with clade 20C) vs the different categories of disease severity (eg, number of patients with severe disease vs number of patients without severe disease).
Data Availability
The genomic data and associated metadata necessary for phylogenetic analysis are now part of CHLA CARD [16]. Other de-identified clinical data shown in the manuscript are available upon request from the corresponding author.
Patient Consent Statement
The design of the study, conducted at Children’s Hospital Los Angeles, was approved by the Institutional Review Board under IRB CHLA-16-00429; a waiver of consent was approved.
RESULTS
Patient Demographics
During a 13-week period (March 19 to June 16, 2020), we identified 141 pediatric patients with confirmed COVID-19. All patients resided in Southern California. Demographics and clinical presentation are summarized in Figure 1 and Supplementary Table 2. Most children in our cohort were of Hispanic ethnicity (95/141, 67.4%). The median age of the patients (range) was 8.3 years (10 days–21 years), 53.2% were male, and 13.4% (19) had underlying conditions. Sixteen (11.4 %) patients were admitted to the hospital, of whom 14 (87.5%) were admitted due to COVID-19-related symptoms. Of the remaining 125 patients not requiring hospital admission, 48 (38.4%) were screened for preoperative procedures, 48 (38.4%) presented to the emergency department (ED), and 29 (23.2%) presented at outpatient clinics. Thirty-three (23.2%) patients had direct contact with individuals positive for COVID-19 or URI symptoms suspicious for COVID-19, suggesting that community transmission of the virus plays an important role in pediatric cases. No cases were associated with a positive travel history. The median time to discharge of hospitalized patients was 3.0 days, and no deaths were reported during this period.
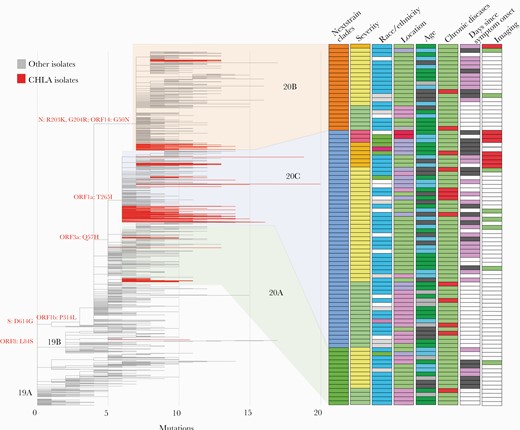
Viral genotypes and clinical features of CHLA patients. Maximum likelihood tree showing phylogenetic clades of CHLA isolates in relation to a subsample of global isolates. CHLA isolates are shown in red. Clade-defining mutations are shown with various clinical metadata indicated on the right. Each row represents a single patient. Nextstrain clades: orange = 20B, blue = 20C, green = 20A. Disease severity: pink = severe, orange = moderate, yellow = mild, green = asymptomatic. Race/ethnicity: blue = Hispanic, white = unknown, purple = Black, gray = other, red = Asian, green = White. Location: red = ICU, blue = inpatient, green = ED, pink = outpatient. Age: green = ≥10 years, gray = ≥5 to <10 years, black = ≥1 to <5 years, blue = <1 year. Chronic diseases: red = present, green = absent. Days since symptom onset: purple = 1–1.5 days, black = >2 days, white = unknown. Imaging: red = chest imaging with significant findings, green = clear, white = unknown. Abbreviations: CHLA, Children’s Hospital Los Angeles; ED, emergency department.
Clinical Characteristics
Extensive chart review was conducted on 88 patients with available inpatient and outpatient EMRs to capture disease severity, comorbidities, and clinical course. The majority of patients were symptomatic (62/88, 70.5%), including 2 patients who were presymptomatic. While the majority (52/62, 83.9%) of symptomatic patients experienced mild infection, 16.1% (10/62) had moderate or severe COVID-19. Three (4.8%) suffered from severe COVID-19 and were admitted to the ICU (Figure 1). Six (4.2%) patients required oxygen supplementation. Six of 16 patients (37.5%) with chest imaging had opacities in the lungs, and 2 had massive lung infiltrations (Figure 1). Of the 7 moderate and 3 severe cases, 4 patients total were <1 year old (2 moderate and 2 severe cases). All 3 severe cases were acute with no progression to MIS-C. There were also no critical cases in this cohort.
The median viral load obtained from all positive results (range) was 2.3×107 (81–1.0×1012) copies/mL, with higher viral loads detected in symptomatic compared with asymptomatic patients (2.7×108 vs 9.9×105 copies/mL; P = .0007) (Figure 2A). Patients tested within 2 days of symptom onset also had a higher viral load compared with samples collected >2 days from symptom onset (9.2×108 vs 2.4×107 copies/mL; P = .001) (Figure 2B). All patients <5 years old were symptomatic and had higher viral loads (2.3×108 vs 1.9×106 copies/mL; P = .0004) (Figure 2C). No difference in viral load was observed in association with chronic underlying conditions, gender, or disease severity.
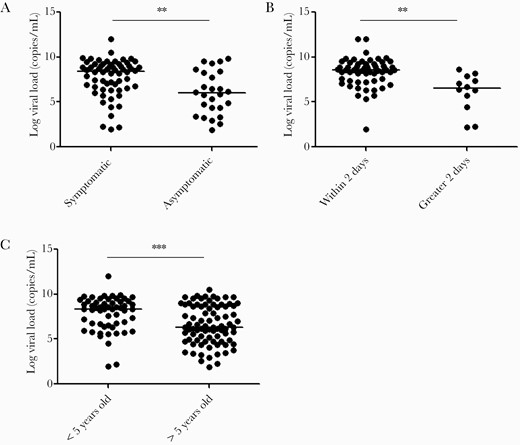
Viral load comparison among patient groups. Increased viral loads were observed in (A) symptomatic individuals, (B) patients tested within 2 days of symptoms, and (C) patients who were <5 years old. The line depicts the median viral load. Statistical analysis was performed using the Mann-Whitney test (**P < .005; ***P < .0005). Abbreviation: CHLA, Children’s Hospital Los Angeles.
Viral Characteristics and Genomic Diversity
WGS of SARS-CoV-2 isolates from our 141 patients revealed 355 unique SNVs and 15 insertions/deletions (IN/DELs) compared with the Wuhan isolate (NC-045512.2) [21], with an average of 10.8 (0–43) variations per isolate (Figure 3; Supplementary Table 3). Of the 355 unique SNVs, 205 were nonsynonymous, 133 were synonymous, and 17 were intergenic (Figure 3A; Supplementary Table 3). SNVs were located mostly in open reading frames (ORFs), with the highest numbers in ORF1ab (n = 231) and S (n = 42) (Figure 3A; Supplementary Table 3). The majority (10/15) of the insertion/deletions caused a frame-shift mutation in ORF1ab. Notably, the recently described D614G mutation in the spike protein [10, 22], caused by nucleotide G-to-A substitution at position 23 403 in the Wuhan reference strain NC_045512.2 (Figure 3B; Supplementary Table 3), was present in all but 1 of our isolates (140/141, 99.3%). There were 2 other mutations that coexist with the D614G mutation in all isolates located in ORF1ab: F924F (c.2772C > T) is a synonymous mutation, while P4715L (c.14144C > T) is a nonsynonymous mutation (Figure 3B). Using CHLA CARD [16] to assess the incidence of these variations globally, we compared our isolates with a global database of 77 966 SARS-CoV-2 isolates worldwide and found 41 variations unique to the isolates present in our cohort, while the remaining 329 variations were present in at least 1 other global isolate (Supplementary Table 3). These 41 variants unique to our cohort include 18 missense, 10 synonymous, 10 frameshift, 1 inframe, 1 stop loss, and 1 start loss. Interestingly, each of the 10 frameshift mutations was detected in 1 isolate. The only other frameshift mutation found in the cohort was the c.361delA mutation in ORF8, which was also found in only 1 isolate in our cohort but reported 25 times previously according to CHLA CARD. While the clinical significance of these variations remains unclear, they point toward the genetic heterogeneity of SARS-CoV-2 isolates circulating in Southern California.
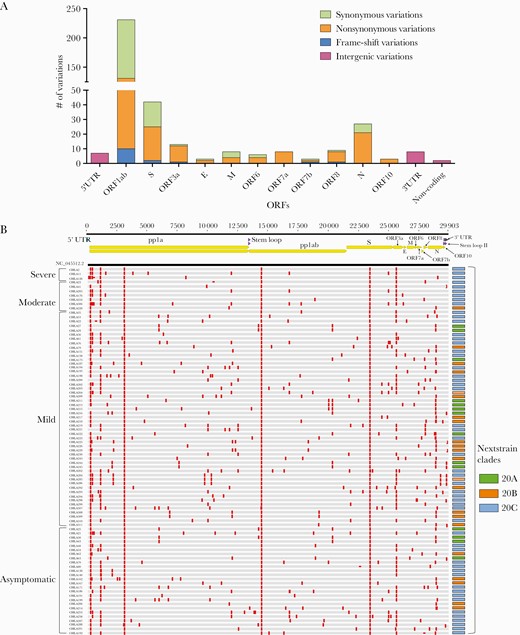
Single nucleotide variations in genomes obtained from 88 CHLA patients with available medical records. A, Summary of characteristics of SNVs in open reading frames and noncoding regions. B, Spread of SNVs across the genome in 88 CHLA isolates as compared with the Wuhan isolate (NC_045512.2). Vertical red lines on the genomes indicate presence of an SNV relative to the reference genome. Genomes are grouped based on disease severity seen in patients. The panel on the right shows the Nextstrain clade for each isolate. Upper panel shows the structure of the severe acute respiratory syndrome coronavirus 2 genome. Abbreviations: CHLA, Children’s Hospital Los Angeles; ORF, open reading frame; SNV, single nucleotide variant.
Phylogenetic analysis of the 141 SARS-CoV-2 genomes demonstrated clustering with other isolates from the United States and across the world (Supplementary Figure 1). All but 1 of the isolates from our cohort belonged to 3 of the 5 phylogenetic clades (20A, 20B, 20C) defined by Nextstrain (Figure 1; Supplementary Figure 1) [17, 23]. The majority (61.0%, 86/141) of isolates belong to phylogenetic clade 20C, a largely North American clade (Figure 1). There was limited evidence of early emerging genotypes of SARS-CoV-2 in this cohort. Only 1 isolate without the D614G variation in the spike protein belonging to phylogenetic clade 19B was identified, and none of the isolates belonging to phylogenetic clade 19A, which consists of the very first isolates from Asia (Figure 1).
The estimated evolutionary rate was calculated to be 7.4×10–4 substitutions per site per year, yielding, based on the SARS-CoV-2 30-kb genome, an overall mutation rate of 22.2 substitutions per year (Supplementary Figure 2A). The inferred time to most recent common ancestor (TMRCA) based on the molecular clock analysis of these isolates was 2019-12-24 (CI: 2019-11-25–2019-12-26). To further investigate the association between viral isolates with the D614G mutation and the rate of viral evolution, we analyzed 577 viral genomes from California at different time points without the D614G mutation. The mutation rate of these isolates was 13.5 substitutions per year (Supplementary Figure 2B), substantially lower than what was observed for our isolates (Supplementary Figure 2A).
Correlation Between Disease and Viral Genetic Variations
Comparison of viral sequences revealed significant variations across the viral genome, a subset of which is illustrated in Figure 2B. In fact, only 23 of 370 SNVs across the viral genome were present in >5% (7/141) of the isolates regardless of the disease phenotype of the patient (Supplementary Table 3), which made attempts to examine potential associations of individual variants with disease severity significantly underpowered. There was some notable clustering for disease severity and phylogenetic clade, although the overall numbers were small. While the viral isolates detected from asymptomatic patients and patients with mild disease span all 3 phylogenetic clades (20A, 20B, and 20C), LRTI was only seen in patients with phylogenetic clades 20B and 20C, and more severe clinical course was only encountered in patients with phylogenetic clade 20C (Figures 1 and 3B). In fact, 9 of 10 patients with either moderate or severe forms of COVID-19 were infected by virus from phylogenetic clade 20C, which is significantly higher (OR, 6.95; P = .0467) than the fraction of patients with mild or asymptomatic infections (44/78) (Figure 3B). No other segregation of clinical and demographic features including age, race/ethnicity, and presence of comorbidities based on viral genotypes was observed (Figure 1).
DISCUSSION
Pediatric patients can present with a broad spectrum of clinical manifestations associated with COVID-19, ranging from asymptomatic infections to severe illness. Over half of our 141-person pediatric cohort was symptomatic, with a subset requiring hospitalization. Four of 10 cases classified as moderate to severe cases were in patients aged <1 year, consistent with reports of this age group accounting for the highest percentage of hospitalization in pediatric COVID-19 cases [3, 24]. When compared with a recent MMWR summarizing signs and symptoms in 291 pediatric patients, shortness of breath (14.3%) or cough (49.2%) in our patient cohort was comparable, but fever was more prevalent in our patients at 71.4% compared with 56% [25]. On the other hand, the hospitalization rate and ICU admission reported in our study were consistent with the estimated ranges reported of 5.7%–20% and 0.58%–2.0%, respectively [25]. However, only a small number of patients in our study experienced moderate illness with LRTI compared with ~65% of 171 children in China with confirmed COVID-19 [25]. These variabilities in disease severity emphasize the critical need for pediatric-specific studies to identify and/or confirm the possible contribution of host factors including age, ethnicity, and immune status.
We observed significantly higher viral loads in symptomatic patients and in children <5 years of age, corroborating findings from a recent study that demonstrated a significantly higher amount of viral nucleic acid in young children [26, 27]. The association between viral load and days from symptom onset is consistent with findings from other studies that reported detection of viral RNA as early as 1 day post–symptom onset and highest viral loads in the first 2 days of symptoms onset [28–31]. These findings have direct implications for infection control within the community and hospital settings.
Genomic evaluation of SARS-CoV-2 in this cohort demonstrated that nearly all of our isolates carried the D614G mutation in the spike protein. The prevalence of this mutation in our patients earlier in the pandemic (95% in March/April) was particularly striking when compared with other California isolates in the GISAID database, which showed a 10% prevalence in February, 65% in March, and not achieving a comparable prevalence of 96% until June. The D614G mutation has been shown to have been rapidly fixed in isolates from Europe and North America and has been associated with lower Ct values in vitro and in vivo, but not with disease severity or case fatality rates [10, 31]. The role of the D614G mutation in SARS-CoV-2 pathogenicity continues to be a focus of active investigation. While some studies suggest that this mutation may increase transmissibility, a clear consensus awaits further investigation [32–34]. In this pediatric cohort, the mutation does not appear to be a primary driver of disease severity, as it was present in all spectra of disease, ranging from asymptomatic to severe infection. We also identified the 2 common mutations (F924F and P4715L) alongside the D614G mutation in our isolates [35]. ORF1ab P4715L is located in Nsp12, which is important for viral replication. A comprehensive analysis of >12 300 SARS-CoV-2 genome sequences across 28 countries reported that mutation at P4715L and D614G may correlate with higher fatality rates [36]. Nevertheless, the functional significance of these mutations still needs to be fully investigated. Confirmation of these observed mutations and better understanding of their potential significance will require sequencing and correlative studies of larger numbers of pediatric COVID-19 cases.
We also observed an unexpectedly increased mutation rate (22.5 substitutions per year) in this cohort when compared with other SARS-CoV-2 cases from California without the D614G mutation during the same period (13.5 substitutions per year). The degree to which this difference reflects an underlying biologic difference vs the temporal and geographic dynamics of the pandemic in California remains to be determined. However, the effect of this higher-than-expected rate of mutation is readily apparent in the unexpected diversity of variation across the viral genome as illustrated in Figure 2B. We postulate that the increased transmissibility caused by D614G may have also led to this increased mutation rate. Nonetheless, the clinical and biological implications remain to be carefully ascertained.
From a genomic epidemiology perspective, the isolates in our study almost exclusively belonged to the 3 most recent SARS-CoV-2 phylogenetic clades (20A, 20B, 20C) as defined in Nextstrain [17]. This is consistent with previously described patterns of multiple introduction and community transmission of SARS-CoV-2 lineages in California [37, 38]. Analysis of the genome sequences from the 22 pediatric patients in California not in this study that are currently available for evaluation confirmed the same 3 phylogenetic clades, but with a difference in clade distribution. In these cases, phylogenetic clade 20A was most prominent (data not shown). The genetic diversity among the isolates points to multiple potential introductions of the virus in Southern California from across the United States and the world. An unexpected finding in our cohort was an apparent association between disease severity and clade 20C. Recognizing the limitations of small case numbers, it is notable that all severe cases and all but 1 moderately severe case fell into the phylogenetic clade 20C. Two additional mutations (C1059T and G25563T) differentiate phylogenetic clade 20C from 20A and 20B, and at least 1 of these may potentially be associated with increased disease severity [39]. G25563T leads to the Q57H missense mutation ORF3a that is associated with host cell apoptosis and contributes to increased infectivity and virulence [40, 41]. Confirmation of the effect of these mutations, as well as further exploration of the genotype–phenotype correlation of phylogenetic clade 20C, warrants further examination in larger-scale pediatric populations.
There are some limitations that warrant discussion. First, this is a single-center study on 141 pediatric patients in Southern California with clinical characteristics available on 88 patients. Although we observed interesting associations between viral genotypes and disease severity, this study was not sufficiently powered to establish a statistically significant correlation between individual viral genetic variations and disease severity. Finally, due to the over-representation of the Hispanic population (67.3%) in our pediatric cohort, the clinical and genomic findings from this study may not reflect all ethnic groups. Notwithstanding the predominant Hispanic population in this cohort, 6 of the 10 moderate or severe cases occurred in non-Hispanic patients. Data from a larger and geographically diverse pediatric population will be required to better characterize these potential associations between disease severity and viral genetic variations.
This study represents the largest single-institution pediatric COVID-19 case series with integrated clinical, genomic, and epidemiological characteristics in the United States. While we observed limited shared variations between the isolates, our findings demonstrate unique variation in each isolate at the cohort or population level, including observation of moderate and severe COVID-19 cases corresponding almost exclusively to phylogenetic clade 20C. Furthermore, the majority of these variants led to amino acid changes in the viral proteins, possibly indicating an ongoing adaptation of the virus in the human population. This genetic diversity of SARS-CoV-2 at the population level has not been well appreciated in previous descriptions of SAR-CoV-2 in children; substantial collaborative efforts will be required across multiple institutions to confidently establish an association between genetic variations and any specific disease manifestation in pediatric COVID-19 patients.
Supplementary Data
Supplementary materials are available at Open Forum Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Acknowledgments
We would like to acknowledge the staff members of the Clinical Microbiology and Virology laboratory and the Center for Personalized Medicine at Children’s Hospital Los Angeles for dedication to providing excellent patient care. We thank Emily Gai for her assistance in editing and formatting of viral sequences. We would like to acknowledge the frontline health care workers who remain devoted in the fight against COVID-19. We would also like to acknowledge NCBI, GISAID, and Nextstrain for providing valuable resources for SARS-CoV-2 genomics.
Financial support. This study was partly supported by the Children’s Hospital Los Angeles, the Saban Research Institute.
Potential conflicts of interest. All authors: no reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
Johns Hopkins Coronavirus Resource Center.
Nextstain.org.
Author notes
Equal contribution


{kind=link}
{kind=link}
{kind=link}
Comments