





Abstract
This report provides data for the extent of B cell depletion and recovery, efficacy, safety and immunogenicity of Sandoz rituximab (SDZ-RTX; GP2013; Rixathon®) compared with reference rituximab (Ref-RTX) up to week 52 of the ASSIST-RA study.
Patients were randomized to SDZ-RTX or Ref-RTX in combination with methotrexate according to the RTX label. The primary endpoint was analysed at week 24. Responders (28-joint DAS [DAS28] decrease from baseline >1.2) at week 24 with residual disease activity (DAS28 ≥2.6) were eligible for a second treatment course between week 24 and 52. Endpoints after week 24 included change from baseline in peripheral B cells, DAS28, ACR 20% response rate (ACR20), Clinical and Simplified Disease Activity Indexes (CDAI, SDAI) and HAQ disability index (HAQ-DI). Safety and immunogenicity were assessed by the incidence of adverse events and antidrug antibodies.
Primary and secondary endpoints up to week 24 were met. Overall, 260/312 randomized patients completed treatment up to week 52. SDZ-RTX resulted in B cell concentrations over time similar to Ref-RTX. The efficacy of SDZ-RTX was similar to Ref-RTX up to week 52, as measured by DAS28, ACR20/50/70, CDAI, SDAI and HAQ-DI. Safety of SDZ-RTX was similar to Ref-RTX regarding frequency, type and severity of adverse events, which were consistent with the known Ref-RTX safety profile. The incidence of antidrug antibodies was low and transient similarly across treatment groups.
SDZ-RTX demonstrated similar B cell concentrations over time, efficacy, safety and immunogenicity to Ref-RTX over 52 weeks of the ASSIST-RA study.
SDZ-RTX demonstrated similar efficacy to reference rituximab (RTX-EU/RTX-US) across multiple measures of rheumatoid arthritis activity.
SDZ-RTX maintained similar efficacy/safety to RTX-EU/RTX-US in patients receiving one and two treatment courses.
No differences were observed between treatments in B cell depletion/recovery, and SDZ-RTX immunogenicity was low.
Introduction
Rituximab (RTX) has been approved since 2006 for the treatment of patients with RA. Treatment guidelines recommend RTX as therapy for patients with inadequate response to conventional synthetic DMARDs (csDMARDs) or TNF inhibitors (TNFis), based on direct and indirect evidence of similar efficacy to other biologic DMARDs (bDMARDs) [1–3].
Biosimilar medicines have enabled more patients to access biologic therapies when needed by bringing savings to individual patients and/or societies [4, 5]. Sandoz rituximab (SDZ-RTX; GP2013) is approved in the European Union (EU) and other highly regulated markets for use in all indications of reference rituximab (Ref-RTX) [6]. Approval was granted based on the totality of evidence for biosimilarity, including analytical and preclinical studies [7, 8], and two double-blind, randomized, controlled trials [9, 10]. The ASSIST-RA study (GP13-201; NCT01274182) and the ASSIST-FL study (GP13-301; NCT01419665) established similar pharmacokinetics/pharmacodynamics (PK/PD), efficacy and safety profiles for SDZ-RTX and Ref-RTX in patients with RA [9] and follicular lymphoma [10].
The primary objective of the ASSIST-RA study was PK equivalence between SDZ-RTX and EU- and US-sourced Ref-RTX (Ref-RTX-EU or Ref-RTX-US). The main efficacy objective was to show non-inferiority of SDZ-RTX vs Ref-RTX (both EU and US) in terms of the change from baseline in the 28-joint DAS (DAS28) at week 24. Results of the primary and key secondary endpoints through week 24 were previously reported [9]. Here we report the long-term results up to week 52.
Methods
The study was approved by institution ethics committees and was conducted in accordance with the principles set out in the Declaration of Helsinki. Written informed consent was obtained from all patients.
Patients were ≥18 years of age and had active RA that was refractory to csDMARDs and ≥1 TNFi. Full inclusion and exclusion criteria were published previously [9]. Patients were randomized via interactive response technology: in study Part 1, to receive SDZ-RTX or Ref-RTX-EU (ratio 1:1), and in study Part 2, to receive SDZ-RTX or Ref-RTX-US (ratio 1:2). Randomization was stratified by the number of prior anti-TNF treatments or other biologics. After the primary (PK) and secondary (PD, efficacy) endpoints were assessed at week 24, patients were eligible for a second treatment course in accordance with initial randomization. The second treatment was administered at the investigator’s discretion at any time between weeks 24 and 52, as long as there had been an initial response (decrease from baseline >1.2 points on the DAS28 using ESR or CRP) but residual disease activity remained at week 24 (DAS28 score ≥2.6). The second treatment course was administered in the same way as the first course: 1000 mg of study drug given on two separate occasions, 2 weeks apart. All patients were followed up to week 52 and patients receiving a second treatment course had a final safety, efficacy and PD assessment 26 weeks after the first infusion of the second course. Study patients, investigators and sponsor staff involved in data monitoring and analysis were blinded until the end of the study.
At week 24, non-responders requiring alternative treatment with study-prohibited medicines (intra-articular or intramuscular glucocorticoid injection, analgesics [in addition to prescribed RA therapy and excluding paracetamol and low-strength opioids], non-biologic DMARDs [except methotrexate, chloroquine, hydroxychloroquine, sulfasalazine], other bDMARDs) were withdrawn from the study.
The primary endpoint was the area under the serum concentration–time curve from infusion to time infinity (AUCinf) assessed at week 24. The key secondary endpoints were peripheral B cell concentration, assessed as the percentage change from baseline, maximal drug concentration (Cmax) after infusion and efficacy as assessed by the change from baseline in DAS28 at week 24. The proportion of patients achieving a 20%/50%/70% improvement in the ACR disease criteria (ACR20/50/70), Clinical Disease Activity Index (CDAI), Simplified Disease Activity Index (SDAI) and HAQ disability index (HAQ-DI) served as further secondary endpoints.
Safety assessments included adverse event (AE) reporting and infusion-related reactions (IRRs) after each infusion. Immunogenicity was evaluated following a multitiered approach comprising screening and confirmation of antidrug antibodies (ADAs). Samples positive for ADAs were further analysed in terms of titre determination and characterized in a cell-based assay to assess the neutralizing capacity of ADAs. Further methodology details are shown in the supplementary material, Methods section, available at Rheumatology online.
The study was powered to assess the equivalence in the PK primary endpoint and non-inferiority in the change from baseline in DAS28 at week 24 (with a non-inferiority margin of 0.6). Data to week 24 for the primary and secondary endpoints have already been published [9]. In this report we present data summarized descriptively to week 52 (end of the study), which include the results of the second treatment course.
The change from baseline in DAS28 used a mixed model for repeated measures, adjusted for treatment, time and interaction between time and treatment as categorical variables and baseline DAS28 as a continuous variable. Patients for whom a baseline value or all post-baseline values were missing were excluded from the analysis and no imputation was made for missing data (total DAS28 score or any component). DAS28 and other efficacy variables were also analysed descriptively by visit. The proportion of patients achieving an ACR20/50/70 improvement and the proportion of patients achieving remission or low disease activity (LDA) based on the CDAI were calculated based on completer and per-protocol analyses. For efficacy and safety analyses presented here, both Ref-RTX groups were pooled. Further details of statistical methods can be found in the supplementary material, Methods section, available at Rheumatology online.
Results
Patients
Patient disposition is presented in Supplementary Fig. S1, available at Rheumatology online. In total, 312 patients were randomized to receive treatment (SDZ-RTX: n = 133; Ref-RTX-EU: n = 87; Ref-RTX-US: n = 92); 290 (92.9%) patients completed week 24 and continued in the study (123 receiving SDZ-RTX and 167 receiving Ref-RTX) and 260 (83.3% of the initial population) completed week 52. Overall, 218 patients (69.9%) completed week 52 having received a second treatment course: 91 patients (68.4%) receiving SDZ-RTX and 127 patients (70.9%) receiving Ref-RTX. A total of 43 patients completed week 52 without a second course (Supplementary Fig. S1, available at Rheumatology online).
Patient baseline demographics and disease characteristics were reported previously [9]. Demographics and characteristics were well balanced between the treatment groups and as expected for patients with active RA. There were no meaningful differences between the groups with regard to prior RA treatments, baseline DAS28 (5.83 for SDZ-RTX; 5.88 for Ref-RTX) or baseline HAQ-DI (1.95 for SDZ-RTX; 1.89 for Ref-RTX). Approximately 98% of the study patients tested positive for either RF or cyclic citrullinated protein antibodies.
Week 24 results were reported previously [9]. Briefly, equivalent PK and PD at week 24 were demonstrated between SDZ-RTX and both reference medicine comparators (Ref-RTX-EU and Ref-RTX-US). The decrease in disease activity was similar across treatment arms: least square mean (SE) of the DAS28 change from baseline was −2.07 (0.108) for SDZ-RTX and −2.11 (0.095) for Ref-RTX. The median time from study start to the second treatment course was 197 days (minimum 168, maximum 379) for SDZ-RTX and 197 days (minimum 163, maximum 378) for Ref-RTX.
B cell kinetics
Among patients who did not receive a second treatment course, B cell levels continued to recover from week 12 to the end of observation at week 52 in all treatment groups (Fig. 1a;Supplementary Table S1, available at Rheumatology online). B cell recovery was similar for SDZ-RTX and Ref-RTX-EU/Ref-RTX-US, although interpatient variability was highlighted by large error bars at later time points. For patients who received a second treatment course, B cell levels remained low to week 52, with a similar extent of depletion for SDZ-RTX and Ref-RTX-EU/Ref-RTX-US (Fig. 1b). The timing of the administration of the second treatment course from week 24 contributed to the observed higher interindividual variability in B cell counts between week 16 and 38. This variability decreased by week 52, by which time all patients receiving a second treatment course had received that treatment (Fig. 1b).
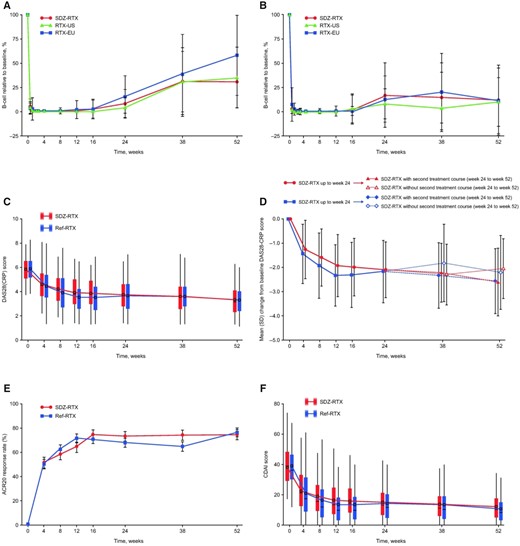
Pharmacodynamic and efficacy results to week 52 of the ASSIST-RA study
(A) B cell count as the mean percentage (s.d.) of baseline level for patients not receiving a second treatment course. (B) B cell count as the mean percentage (s.d.) of baseline level for patients receiving a second treatment course. (C) Absolute DAS28-CRP scores for all patients. (D) Mean (s.d.) DAS28-CRP scores for patients receiving and not receiving a second treatment course. (E) ACR20 response rates (completer analysis). (F) Absolute CDAI scores.
Efficacy
Efficacy endpoints assessed to week 52 are presented in Fig. 1c–f. Following the initial decrease in DAS28 up to week 24, disease activity remained well controlled to the end of the study at week 52 in both treatment groups (Fig. 1c;Supplementary Tables S2 and S3, available at Rheumatology online). At week 52, the mean DAS28 scores were 3.3 (s.d. 1.2) in the SDZ-RTX group and 3.3 (s.d. 1.2) in the Ref-RTX group. There was wide variation in DAS28 scores within each treatment group at all time points (Fig. 1c). Compared with baseline, the decrease in DAS28 at week 52 was a least square mean of −2.41 (SE 0.12) in the SDZ-RTX group and −2.47 (SE 0.10) in the Ref-RTX group. Between week 38 and week 52, a trend toward lower disease activity was observed for patients who received a second treatment course compared with those who did not; disease activity was similar for SDZ-RTX and Ref-RTX in either subgroup (Fig. 1d). Changes in disease activity according to the DAS28 were supported by the other efficacy endpoints. ACR20 response rates increased similarly following the first treatment course to week 24 in all treatment groups and were maintained up to week 52 (completer analysis: 74.5% in the SDZ-RTX group and 76.5% in the Ref-RTX group [Fig. 1e;Supplementary Table S4, available at Rheumatology online]; per-protocol [PP] analysis: 59.8% in the SDZ-RTX group and 62.7% in the Ref-RTX group). ACR50 response rates at week 52 were 42.2% for SDZ-RTX vs 41.2% for Ref-RTX in the completer analysis and 33.9% for SDZ-RTX vs 33.7% for Ref-RTX in the PP analysis. ACR70 response rates were 20.6% for SDZ-RTX and 25.0% for Ref-RTX in the completer analysis and 16.5% for SDZ-RTX vs 20.5% for Ref-RTX in the PP analysis.
The CDAI (Fig. 1f;Supplementary Table S5, available at Rheumatology online) and SDAI scores had a similar profile to the DAS28, with initial rapid decreases after the first treatment course that were maintained to week 24 and further decreases to week 52 in both treatment groups (week 52 mean CDAI scores: 12.8 [s.d. 10.7] for SDZ-RTX, 12.2 [s.d. 9.8] for Ref-RTX; SDAI scores: 13.7 [s.d. 11.2] for SDZ-RTX, 13.2 [s.d. 10.0] for Ref-RTX). Similar numbers of patients had LDA and were in remission in both treatment groups at week 52. For the CDAI, LDA was observed in 33.7% of patients for SDZ-RTX and 36.8% of patients for Ref-RTX (completer analysis; corresponding data from PP analysis were 26.0% and 30.1%, respectively). CDAI remission was observed in 14.3% of patients for SDZ-RTX and 13.2% of patients for Ref-RTX (completer analysis; corresponding data from PP analysis were 11.0% and 10.8%, respectively).
For the SDAI, LDA was observed in 37.9% of patients for SDZ-RTX and 33.3% of patients for Ref-RTX (completer analysis; corresponding data from PP analysis were 28.3% and 27.1%, respectively). SDAI remission was observed in 13.7% of patients for SDZ-RTX and 14.1% of patients for Ref-RTX (completer analysis; corresponding data from PP analysis were 10.2% and 11.4%, respectively).
Improvements in HAQ-DI scores from high baseline values were noted for both SDZ-RTX and Ref-RTX, reflecting the decreased disease activity. The mean change from baseline to week 52 in HAQ-DI score was −0.52 (s.d. 0.65) for SDZ-RTX and −0.47 (s.d. 0.65) for Ref-RTX.
Safety
Overall, the proportions of patients reporting AEs was similar between SDZ-RTX and Ref-RTX with no clinically meaningful differences between the groups (Table 1). Most AEs were mild or moderate in severity and mild, moderate and severe AEs were reported by similar proportions of patients in each treatment group. There was a low incidence of serious AEs without clinically meaningful differences for SDZ-RTX or Ref-RTX. IRRs were reported with similar frequency in both treatment groups and the incidence decreased after each subsequent infusion (including second treatment course; Table 1). Three patients died during the first 24 weeks of the study (one from breast cancer during the screening period, before study treatment; one from multi-organ failure, in the SDZ-RTX arm, suspected by the investigator to be related to an accidental MTX overdose by the patient; and one from purulent pericarditis, in the RTX-US arm); none of the deaths was considered related to the study drug [9].
Summary of safety (adverse events) and immunogenicity to end of studya
| Characteristics | SDZ-RTX (n = 133) | Ref-RTX (n = 179) |
|---|---|---|
| Safety summary, n (%) | ||
| AEs | 88 (66.2) | 119 (66.5) |
| Leading to study drug discontinuation | 7 (5.3) | 13 (7.3) |
| Suspected by the investigator to be study drug-related | 43 (32.3) | 57 (31.8) |
| Serious AEs | 16 (12.0) | 23 (12.8) |
| AEs ≥4% regardless of study drug relationship (MedDRA Preferred Term) | ||
| Urinary tract infection | 11 (8.3) | 7 (3.9) |
| Nasopharyngitis | 9 (6.8) | 14 (7.8) |
| Nausea | 9 (6.8) | 8 (4.5) |
| Fatigue | 6 (4.5) | 1 (0.6) |
| Headache | 6 (4.5) | 11 (6.1) |
| Infusion-related reaction | 6 (4.5) | 8 (4.5) |
| RA | 6 (4.5) | 13 (7.3) |
| Upper respiratory tract infection | 6 (4.5) | 11 (6.1) |
| Cough | 5 (3.8) | 11 (6.1) |
| Hypertension | 5 (3.8) | 8 (4.5) |
| IRR for first treatment course | ||
| On day or day after first infusion | 21 (15.8) | 27 (15.1) |
| On day or day after second infusion | 10 (7.5) | 9 (5.0) |
| IRR for second treatment course | ||
| On day or day after first infusion | 2 (1.5) | 7 (3.9) |
| On day or day after second infusion | 4 (3.0) | 4 (2.2) |
| Immunogenicity, n/N (%) | ||
| Overall post-baseline | ||
| ADA negative | 106/127 (83.5) | 137/166 (82.5) |
| ADA positive | 21/127 (16.5) | 29/166 (17.5) |
| Neutralizing ADA | 5/127 (3.9) | 1/166 (0.6) |
| Transient ADA | 13/127 (10.2) | 19/166 (11.4) |
| Characteristics | SDZ-RTX (n = 133) | Ref-RTX (n = 179) |
|---|---|---|
| Safety summary, n (%) | ||
| AEs | 88 (66.2) | 119 (66.5) |
| Leading to study drug discontinuation | 7 (5.3) | 13 (7.3) |
| Suspected by the investigator to be study drug-related | 43 (32.3) | 57 (31.8) |
| Serious AEs | 16 (12.0) | 23 (12.8) |
| AEs ≥4% regardless of study drug relationship (MedDRA Preferred Term) | ||
| Urinary tract infection | 11 (8.3) | 7 (3.9) |
| Nasopharyngitis | 9 (6.8) | 14 (7.8) |
| Nausea | 9 (6.8) | 8 (4.5) |
| Fatigue | 6 (4.5) | 1 (0.6) |
| Headache | 6 (4.5) | 11 (6.1) |
| Infusion-related reaction | 6 (4.5) | 8 (4.5) |
| RA | 6 (4.5) | 13 (7.3) |
| Upper respiratory tract infection | 6 (4.5) | 11 (6.1) |
| Cough | 5 (3.8) | 11 (6.1) |
| Hypertension | 5 (3.8) | 8 (4.5) |
| IRR for first treatment course | ||
| On day or day after first infusion | 21 (15.8) | 27 (15.1) |
| On day or day after second infusion | 10 (7.5) | 9 (5.0) |
| IRR for second treatment course | ||
| On day or day after first infusion | 2 (1.5) | 7 (3.9) |
| On day or day after second infusion | 4 (3.0) | 4 (2.2) |
| Immunogenicity, n/N (%) | ||
| Overall post-baseline | ||
| ADA negative | 106/127 (83.5) | 137/166 (82.5) |
| ADA positive | 21/127 (16.5) | 29/166 (17.5) |
| Neutralizing ADA | 5/127 (3.9) | 1/166 (0.6) |
| Transient ADA | 13/127 (10.2) | 19/166 (11.4) |
End of study was week 52 or 26 weeks after the first infusion of the second treatment course, whichever was later. ADA: antidrug antibody; AE: adverse event; IRR: infusion related reaction; Ref-RTX: reference rituximab; SDZ-RTX: Sandoz rituximab; SOC: system organ class.
Summary of safety (adverse events) and immunogenicity to end of studya
| Characteristics | SDZ-RTX (n = 133) | Ref-RTX (n = 179) |
|---|---|---|
| Safety summary, n (%) | ||
| AEs | 88 (66.2) | 119 (66.5) |
| Leading to study drug discontinuation | 7 (5.3) | 13 (7.3) |
| Suspected by the investigator to be study drug-related | 43 (32.3) | 57 (31.8) |
| Serious AEs | 16 (12.0) | 23 (12.8) |
| AEs ≥4% regardless of study drug relationship (MedDRA Preferred Term) | ||
| Urinary tract infection | 11 (8.3) | 7 (3.9) |
| Nasopharyngitis | 9 (6.8) | 14 (7.8) |
| Nausea | 9 (6.8) | 8 (4.5) |
| Fatigue | 6 (4.5) | 1 (0.6) |
| Headache | 6 (4.5) | 11 (6.1) |
| Infusion-related reaction | 6 (4.5) | 8 (4.5) |
| RA | 6 (4.5) | 13 (7.3) |
| Upper respiratory tract infection | 6 (4.5) | 11 (6.1) |
| Cough | 5 (3.8) | 11 (6.1) |
| Hypertension | 5 (3.8) | 8 (4.5) |
| IRR for first treatment course | ||
| On day or day after first infusion | 21 (15.8) | 27 (15.1) |
| On day or day after second infusion | 10 (7.5) | 9 (5.0) |
| IRR for second treatment course | ||
| On day or day after first infusion | 2 (1.5) | 7 (3.9) |
| On day or day after second infusion | 4 (3.0) | 4 (2.2) |
| Immunogenicity, n/N (%) | ||
| Overall post-baseline | ||
| ADA negative | 106/127 (83.5) | 137/166 (82.5) |
| ADA positive | 21/127 (16.5) | 29/166 (17.5) |
| Neutralizing ADA | 5/127 (3.9) | 1/166 (0.6) |
| Transient ADA | 13/127 (10.2) | 19/166 (11.4) |
| Characteristics | SDZ-RTX (n = 133) | Ref-RTX (n = 179) |
|---|---|---|
| Safety summary, n (%) | ||
| AEs | 88 (66.2) | 119 (66.5) |
| Leading to study drug discontinuation | 7 (5.3) | 13 (7.3) |
| Suspected by the investigator to be study drug-related | 43 (32.3) | 57 (31.8) |
| Serious AEs | 16 (12.0) | 23 (12.8) |
| AEs ≥4% regardless of study drug relationship (MedDRA Preferred Term) | ||
| Urinary tract infection | 11 (8.3) | 7 (3.9) |
| Nasopharyngitis | 9 (6.8) | 14 (7.8) |
| Nausea | 9 (6.8) | 8 (4.5) |
| Fatigue | 6 (4.5) | 1 (0.6) |
| Headache | 6 (4.5) | 11 (6.1) |
| Infusion-related reaction | 6 (4.5) | 8 (4.5) |
| RA | 6 (4.5) | 13 (7.3) |
| Upper respiratory tract infection | 6 (4.5) | 11 (6.1) |
| Cough | 5 (3.8) | 11 (6.1) |
| Hypertension | 5 (3.8) | 8 (4.5) |
| IRR for first treatment course | ||
| On day or day after first infusion | 21 (15.8) | 27 (15.1) |
| On day or day after second infusion | 10 (7.5) | 9 (5.0) |
| IRR for second treatment course | ||
| On day or day after first infusion | 2 (1.5) | 7 (3.9) |
| On day or day after second infusion | 4 (3.0) | 4 (2.2) |
| Immunogenicity, n/N (%) | ||
| Overall post-baseline | ||
| ADA negative | 106/127 (83.5) | 137/166 (82.5) |
| ADA positive | 21/127 (16.5) | 29/166 (17.5) |
| Neutralizing ADA | 5/127 (3.9) | 1/166 (0.6) |
| Transient ADA | 13/127 (10.2) | 19/166 (11.4) |
End of study was week 52 or 26 weeks after the first infusion of the second treatment course, whichever was later. ADA: antidrug antibody; AE: adverse event; IRR: infusion related reaction; Ref-RTX: reference rituximab; SDZ-RTX: Sandoz rituximab; SOC: system organ class.
Immunogenicity
Similar proportions of patients reported ADAs in the SDZ-RTX (16.5%) and Ref-RTX groups (17.5%; Table 1). Most ADAs were of low titre and transient in nature. There was no increase in the proportion of patients with ADAs between week 24 and 52, following the second treatment course, in any treatment group.
Discussion
The current analysis confirms the similarity of SDZ-RTX with both Ref-RTX-EU and Ref-RTX-US in terms of peripheral B cell concentrations, efficacy, safety and immunogenicity over up to 52 weeks of follow-up in patients with active RA. These results provide additional evidence for biosimilarity over a longer treatment period, extending the previous report with additional data on outcomes following a second treatment course. Previous studies have shown that RTX re-treatment is associated with a prolonged treatment response in patients with RA, enabling consideration of dose reduction over the longer term with no adverse effect on safety outcomes [11, 12]. In our study, efficacy responses with SDZ-RTX remained stable over time and were consistent with those for Ref-RTX. B cell recovery is another sensitive parameter to evaluate potential differences between the two compounds. In this analysis, B cell recovery occurred at a similar rate for SDZ-RTX and Ref-RTX and started at around week 12, which is consistent with the known B cell recovery profile of Ref-RTX [13]. Depletion of B cells continued throughout the study period for patients who received a second treatment course. SDZ-RTX demonstrated similar safety and immunogenicity profiles to Ref-RTX, indicating good tolerability of SDZ-RTX in this patient population.
Despite some differences in methodologies and reporting of endpoints, our efficacy and safety results are generally consistent with long-term experience with Ref-RTX treatment in clinical trials [14] and in clinical practice [15]. Our results are also generally consistent with short- and long-term studies of the similarities of efficacy and safety for other proposed and approved RTX biosimilars [16, 17]. IRRs to RTX most commonly occur following the first infusion and decrease with subsequent infusions [18]. In this study, we observed a similar pattern of IRRs for both treatments, with fewer IRRs reported after each subsequent infusion. Most ADAs were low titre and the incidence of ADAs was consistent with the known rate of ADAs for RTX use in patients with RA [18]. A single ADA assay was used, which is capable of detecting ADAs against SDZ-RTX as well as against either Ref-RTX-EU or Ref-RTX-US.
Limitations of this study include the relatively small sample size and lack of statistical hypothesis testing beyond week 24. Aspects of the study design may also have influenced the observed results. First, the two-part design meant that two separate comparisons were included in the study, namely SDZ-RTX vs Ref-RTX-EU and SDZ-RTX vs Ref-RTX-US, with data on the two reference medicines pooled for the final efficacy, safety and immunogenicity analysis. Second, the decision to administer a second treatment course was at the investigator’s discretion. Two approaches are generally used for maintenance RTX treatment in RA: prescribe regular treatment courses approximately every 6 months, or monitor for recurrence of B cells or clinical signs of relapse before prescribing an additional treatment course [19]. In this study, the timing of administration of the second treatment course depended on the investigator’s preference, so it was not possible to control for different approaches and both scenarios may have been captured. Third, only a small proportion of patients did not receive a second treatment course, limiting the interpretability of comparisons between patients who did or did not receive a second treatment course. However, it is worth noting that there was no imbalance in the timing of administration of the second treatment course between patients who received SDZ-RTX or those who received Ref-RTX. Finally, comparisons with other studies are limited due to differences in patient populations, study designs and endpoint assessments. This trial did not include a switch component where patients receiving Ref-RTX transition to the biosimilar. Data for a switch from Ref-RTX-EU or Ref-RTX-US to SDZ-RTX from another study have recently been published [20].
Overall, our results confirm that the clinical response to SDZ-RTX (in terms of PK, efficacy, safety and immunogenicity) is similar to that of Ref-RTX and remains stable over time. Treatment with SDZ-RTX is well tolerated and no unexpected safety reactions were observed over 52 weeks in the ASSIST-RA study.
Funding: This study was funded by Hexal AG, a Sandoz Company, in all countries except the USA, and by Sandoz in the USA. Medical writing support under the guidance of the authors was provided by Amanda Hatton and Ben Caldwell of Spirit, a division of Spirit Medical Communications Group Ltd, Manchester, UK and funded by Sandoz. Sandoz is a Novartis division.
Disclosure statement: JSS reports grants and honoraria from AbbVie, AstraZeneca, Lilly, Novartis, Sandoz (a Novartis division), Pfizer, Roche and Chugai, and honoraria from Amgen, Astro, Bristol-Myers Squibb, Celgene, Celltrion, Gilead, ILTOO, MSD, Samsung Bioepis, Sanofi and UCB. SBC, HPT, MS, AK, AB and JG-R have received investigator fees from Sandoz (a Novartis division). LC, JP and DK are employees of Sandoz/Hexal AG (a Novartis division). TS is an employee of Novartis.
Supplementary data
Supplementary data are available at Rheumatology online.
References
European Medicines Agency. Assessment Report: Rixathon [updated 7 August
European Medicines Agency. Assessment Report: MabThera [updated 22 August

{kind=link}
Comments