





Scope and purpose of the guideline
Background

NICE has accredited the process used by the BSR to produce its guidance on the management of systemic lupus erythematosus in adults. Accreditation is valid for 5 years from 10 June 2013. More information on accreditation can be viewed at www.nice.org.uk/accreditation. For full details on our accreditation visit: www.nice.org.uk/accreditation.
SLE (or lupus for short) is a multisystem, autoimmune disease, involving complex pathogenetic mechanisms that can present at any age. It most commonly presents in women in the reproductive age group, although lupus is increasingly recognized after the age of 40 years, particularly in Europeans [1–3]. Lupus affected nearly 1 in 1000 of the population in the UK in 2012 [4] and was most frequently observed in people of African-Caribbean and South Asian descent [4–6]. The age-standardized incidence in the UK according to the Clinical Practice Research Datalink is 8.3/100 000/year for females and 1.4/100 000/year for males [4], and the highest incidence rates are seen in those of African-Caribbean descent: 31.4/100 000/year, compared with 6.7/100 000/year for those of white European descent. The mean age at diagnosis is 48.9 years [4], but it is lower in those of African ancestry in the UK [4–6] and North America [2, 7].
The disease is prone to relapses and remissions, resulting in considerable morbidity due to flares of disease activity and accumulated damage, and an increased risk of premature death, mostly due to infection or cardiovascular disease [2, 8–14]. Death from active lupus is rare in the UK [15, 16]; however, a 10% mortality over 20 years and a mean age of death of 53.7 years was recently reported [16]. About one-third of SLE patients in the UK develop LN [16–18]. Patients of African ancestry tend to present young with LN in the UK, as in the USA and elsewhere [2, 17, 19], and are at considerable risk of developing end-stage renal disease (ESRD) and of dying prematurely. In another UK cohort, ESRD occurred in 20% of LN patients within 10 years of diagnosis, and the mean age at death in LN patients was 40.3 years, with an average of 7.5 years between development of LN and death [18].
The mainstay of therapy for active lupus until recently has been NSAIDs, CSs, antimalarials such as HCQ, and immunosuppressants such as AZA and CYC, although only prednisolone and HCQ are licensed for lupus [8, 20]. With the exception of LN, there were relatively few trials until the last 15 years, and in 2011, belimumab became the first drug to be licensed for the treatment of active lupus for over 50 years [20]. New therapies that will reduce the need for CSs to control lupus activity and to reduce the development of damage and infection are needed to improve outcome [10–12, 16, 21]. In the meantime it is important to manage patients optimally with the treatment strategies that are available.
Need for the guideline
Despite some improvement in survival data over the last 40 years [2, 13], lupus patients still die on average 25 years earlier than the mean for women and men in the UK [16]. The disease can present with slowly or rapidly progressive active disease at any age and can be associated with the rapid accumulation of damage if not promptly diagnosed, appropriately treated and regularly monitored [2, 8, 14, 19, 20]. An up-to-date comprehensive guideline to optimize these aspects of management that is consistent with current evidence and National Health Service (NHS) practice is warranted to improve the outcome of this variable and potentially life-threatening disease that causes considerable morbidity. There have been no previous UK-based guidelines for lupus. The European (EULAR) recommendations for the management of lupus in general were not very detailed and were published in 2008 [22], although more specific recommendations were published for neuropsychiatric lupus in 2010 [23], and joint EULAR and European Renal Association–European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for LN were published in 2012 [24], as well as ACR guidelines for the management of LN in 2012 [25].
Objectives of the guideline
The aim of this guideline was to produce recommendations for the management of adult lupus patients in the UK that cover the diagnosis, assessment and monitoring of lupus and the treatment of mild, moderate and severe active lupus disease, but which do not imply a legal obligation. The resulting recommendations are based on an extensive review of the literature up to June 2015 to produce evidence-based guidelines, particularly for the treatment of non-renal lupus, supplemented as necessary by expert opinion and consensus agreement (Tables 1 and 2). The guideline development group recommended that patients with LN are managed according to the EULAR/ERA-EDTA recommendations for LN [24] and provide their strengths of agreement (SOAs) with a summary of the most important items in those recommendations (Table 3).
Levels of evidence and grades of recommendation for diagnosis, assessment and monitoring of non-renal SLE
| Statement/item | Number of studies | Overall SIGN level of evidence | Grade of recommendation | Selected references covering items discussed in text |
|---|---|---|---|---|
| Diagnosis from clinical and serological features | ||||
| Prognostic value of: | ||||
Clinical features
|
|
|
| |
| Assessment and monitoring of SLE disease activity and damage | ||||
| Clinical flare | 6 | 2+ | C | [48, 49] |
| Good diagnostic utility of: | ||||
|
|
|
| |
| ESR correlates with active lupus | 2 | 2+ | C | [69, 70] |
| Prognostic value of lupus disease activity and damage indices | >60 | 2 ++ | B | Reviewed in [12, 71] [11, 14–16, 32, 72, 73] |
| Monitoring and treating cardiovascular risk factors in SLE patients | 6 | 2+ | C | Reviewed in [22, 71, 74–76] |
| Frequency of monitoring SLE: | ||||
|
|
|
| |
| Monitoring for drug toxicity/levels | 2 | 2+ | C | [78, 79] |
| Statement/item | Number of studies | Overall SIGN level of evidence | Grade of recommendation | Selected references covering items discussed in text |
|---|---|---|---|---|
| Diagnosis from clinical and serological features | ||||
| Prognostic value of: | ||||
Clinical features
|
|
|
| |
| Assessment and monitoring of SLE disease activity and damage | ||||
| Clinical flare | 6 | 2+ | C | [48, 49] |
| Good diagnostic utility of: | ||||
|
|
|
| |
| ESR correlates with active lupus | 2 | 2+ | C | [69, 70] |
| Prognostic value of lupus disease activity and damage indices | >60 | 2 ++ | B | Reviewed in [12, 71] [11, 14–16, 32, 72, 73] |
| Monitoring and treating cardiovascular risk factors in SLE patients | 6 | 2+ | C | Reviewed in [22, 71, 74–76] |
| Frequency of monitoring SLE: | ||||
|
|
|
| |
| Monitoring for drug toxicity/levels | 2 | 2+ | C | [78, 79] |
SIGN: Scottish Intercollegiate Guidelines Network
Levels of evidence and grades of recommendation for diagnosis, assessment and monitoring of non-renal SLE
| Statement/item | Number of studies | Overall SIGN level of evidence | Grade of recommendation | Selected references covering items discussed in text |
|---|---|---|---|---|
| Diagnosis from clinical and serological features | ||||
| Prognostic value of: | ||||
Clinical features
|
|
|
| |
| Assessment and monitoring of SLE disease activity and damage | ||||
| Clinical flare | 6 | 2+ | C | [48, 49] |
| Good diagnostic utility of: | ||||
|
|
|
| |
| ESR correlates with active lupus | 2 | 2+ | C | [69, 70] |
| Prognostic value of lupus disease activity and damage indices | >60 | 2 ++ | B | Reviewed in [12, 71] [11, 14–16, 32, 72, 73] |
| Monitoring and treating cardiovascular risk factors in SLE patients | 6 | 2+ | C | Reviewed in [22, 71, 74–76] |
| Frequency of monitoring SLE: | ||||
|
|
|
| |
| Monitoring for drug toxicity/levels | 2 | 2+ | C | [78, 79] |
| Statement/item | Number of studies | Overall SIGN level of evidence | Grade of recommendation | Selected references covering items discussed in text |
|---|---|---|---|---|
| Diagnosis from clinical and serological features | ||||
| Prognostic value of: | ||||
Clinical features
|
|
|
| |
| Assessment and monitoring of SLE disease activity and damage | ||||
| Clinical flare | 6 | 2+ | C | [48, 49] |
| Good diagnostic utility of: | ||||
|
|
|
| |
| ESR correlates with active lupus | 2 | 2+ | C | [69, 70] |
| Prognostic value of lupus disease activity and damage indices | >60 | 2 ++ | B | Reviewed in [12, 71] [11, 14–16, 32, 72, 73] |
| Monitoring and treating cardiovascular risk factors in SLE patients | 6 | 2+ | C | Reviewed in [22, 71, 74–76] |
| Frequency of monitoring SLE: | ||||
|
|
|
| |
| Monitoring for drug toxicity/levels | 2 | 2+ | C | [78, 79] |
SIGN: Scottish Intercollegiate Guidelines Network
Levels of evidence and grades of recommendation for medications used in the treatment of non-renal SLE
| Treatment (recommended target dosage) | Main uses (unless contra-indications) | Total number of papers | Overall SIGN level of evidence | Grade of recommendation | Comments: including number of reports and references for RCTs, cohort studies and systematic reviews/meta-analyses (SRs) |
|---|---|---|---|---|---|
| Antimalarials: HCQ ≤6.5 mg/kg/day | Mild lupus, prevent flare in all patients, prevent damage, steroid-sparing | 45 | 1 ++ | A | 7 RCTs [80–86]; 36 cohort studies [87–120]; 2 SRs [121, 122] |
| MTX ≤25 mg/week | Mild and moderate lupus, prevent flare, steroid sparing | 12 | 1+ | A | |
| NSAIDs | Symptom control in mild non-renal lupus only | 1 | 3 | D | 1 SR covers case series/reports [134] |
| Sunscreen (high-SPF UV-A and UV-B) | Prevents UV-induced rashes and other manifestations | 7 | 2 ++ | B | 1 blind RCT [135]; 5 cohort studies [136–140]; 1 case series [141] |
| Low-dose oral prednisolone (≤7.5 mg) | Mild lupus and to prevent flares | 0 | 4 | D | Expert opinion |
| Higher doses of oral prednisolone ≤0.5 mg/kg/day | Moderate lupus and prevention of flares | 0 2 | 4 2+ | D C | To prevent flare: 1 blind RCT [46] and 1 open-label RCT [60] |
| I.m. triamcinolone | Moderate lupus | 1 | 2+ | C | 1 open-label RCT [142] |
| I.m. methylprednisolone (80–120 mg) | Moderate lupus | 0 | 4 | D | Expert opinion |
| I.v. methylprednisolone (100–250 mg) | Moderate lupus | 1 | 2+ | C/D | 1 blind RCT for 100 mg vs 1000 mg [143] |
| I.v. methylprednisolone (500 mg–1 g) × 1–3 pulses | Moderate and severe lupus | 6 | 2+ | C | 2 small blind RCTs [143, 144]; 1 open-label trial [145]; 3 cohort studies [146–148] |
| AZA (if TPMT normal) 2–3 mg/kg/day | Moderate lupus, prevent flare, steroid sparing | 10 | 2+ | C | 4 open-label RCTs [149–152]; 5 cohort studies [153–157]; 1 case series [158] |
| MMF 2–3 g/day | Moderate/severe lupus, prevent flare, steroid-sparing | 13 | 2 ++ | B | 3 open-label RCTs [159–161]; 7 cohort studies [162–168]; 1 case series [169]; 2 SRs [133, 170] |
| Mycophenolic acid/sodium 1.44–2.16 g/day | For patients intolerant of MMF | 2 | 3 | D | 1 open-label RCT [171]; 1 cohort study [172] |
| Ciclosporin ≤2.5 mg/kg/day | Moderate/severe lupus including cytopenias, prevent flare, steroid-sparing | 11 | 2+ | C | 2 open-label RCTs [152, 173]; 8 cohort studies [174–181]; 1 SR [133] |
| Tacrolimus 1–3 mg/day (assess drug levels) | Moderate/severe lupus, steroid-sparing | 3 | 3 | D | 2 cohort studies [182, 183]; 1 SR [133] |
| LEF (20 mg/day) | Moderate lupus without subacute rash | 3 | 3 | D | 1 small blind RCT [184]; 1 cohort study [185]; 1 SR [133] |
| CYC (see text for dosing) | Severe lupus, including NPSLE, prevent flare, steroid-sparing | 30 | 2 ++ | B | 4 open-label RCTs [186–189]; 25 cohort studies covered by 1 SR [133] |
| Rituximab 1000 mg × 2 | Refractory severe and moderate lupus; steroid-sparing | 33 | 2+ | C | 1 blind RCT [190, 191]; 3 open-label RCTs [192–194]; 24 cohort studies [195–198 not in SRs]; 2 case series [194, 199]; 2 SRs, including 1 meta-analysis [200, 201]; 1 SR with 26 extra case reports/series [202] |
| Belimumab 10 mg/kg/4 weeks | Refractory moderate/severe lupus; prevent flare and steroid-sparing (not NPSLE) | 5 | 1+ | B | 2 phase III blind RCTs [203, 204]; 1 phase II blind RCT [205]; post hoc combined analysis [206]; 1 open-label extension [207, 208]; 1 meta-analysis [209] |
| IVIG (see text) | Refractory severe lupus (including catastrophic APS) | 19 | 2− | D | Rarely indicated: 3 open-label trials [210–212]; 10 cohort studies [213–222]; 4 case series [223–226]; 2 SRs with 1 meta-analysis [227, 228] |
| Plasmapharesis | TTP; refractory severe SLE | 10 | 2 ++ for TTP; 3 otherwise | B for TTP; D otherwise | Rarely indicated: 9 cohort/case series [229–237]; 1SR [238] |
| Treatment (recommended target dosage) | Main uses (unless contra-indications) | Total number of papers | Overall SIGN level of evidence | Grade of recommendation | Comments: including number of reports and references for RCTs, cohort studies and systematic reviews/meta-analyses (SRs) |
|---|---|---|---|---|---|
| Antimalarials: HCQ ≤6.5 mg/kg/day | Mild lupus, prevent flare in all patients, prevent damage, steroid-sparing | 45 | 1 ++ | A | 7 RCTs [80–86]; 36 cohort studies [87–120]; 2 SRs [121, 122] |
| MTX ≤25 mg/week | Mild and moderate lupus, prevent flare, steroid sparing | 12 | 1+ | A | |
| NSAIDs | Symptom control in mild non-renal lupus only | 1 | 3 | D | 1 SR covers case series/reports [134] |
| Sunscreen (high-SPF UV-A and UV-B) | Prevents UV-induced rashes and other manifestations | 7 | 2 ++ | B | 1 blind RCT [135]; 5 cohort studies [136–140]; 1 case series [141] |
| Low-dose oral prednisolone (≤7.5 mg) | Mild lupus and to prevent flares | 0 | 4 | D | Expert opinion |
| Higher doses of oral prednisolone ≤0.5 mg/kg/day | Moderate lupus and prevention of flares | 0 2 | 4 2+ | D C | To prevent flare: 1 blind RCT [46] and 1 open-label RCT [60] |
| I.m. triamcinolone | Moderate lupus | 1 | 2+ | C | 1 open-label RCT [142] |
| I.m. methylprednisolone (80–120 mg) | Moderate lupus | 0 | 4 | D | Expert opinion |
| I.v. methylprednisolone (100–250 mg) | Moderate lupus | 1 | 2+ | C/D | 1 blind RCT for 100 mg vs 1000 mg [143] |
| I.v. methylprednisolone (500 mg–1 g) × 1–3 pulses | Moderate and severe lupus | 6 | 2+ | C | 2 small blind RCTs [143, 144]; 1 open-label trial [145]; 3 cohort studies [146–148] |
| AZA (if TPMT normal) 2–3 mg/kg/day | Moderate lupus, prevent flare, steroid sparing | 10 | 2+ | C | 4 open-label RCTs [149–152]; 5 cohort studies [153–157]; 1 case series [158] |
| MMF 2–3 g/day | Moderate/severe lupus, prevent flare, steroid-sparing | 13 | 2 ++ | B | 3 open-label RCTs [159–161]; 7 cohort studies [162–168]; 1 case series [169]; 2 SRs [133, 170] |
| Mycophenolic acid/sodium 1.44–2.16 g/day | For patients intolerant of MMF | 2 | 3 | D | 1 open-label RCT [171]; 1 cohort study [172] |
| Ciclosporin ≤2.5 mg/kg/day | Moderate/severe lupus including cytopenias, prevent flare, steroid-sparing | 11 | 2+ | C | 2 open-label RCTs [152, 173]; 8 cohort studies [174–181]; 1 SR [133] |
| Tacrolimus 1–3 mg/day (assess drug levels) | Moderate/severe lupus, steroid-sparing | 3 | 3 | D | 2 cohort studies [182, 183]; 1 SR [133] |
| LEF (20 mg/day) | Moderate lupus without subacute rash | 3 | 3 | D | 1 small blind RCT [184]; 1 cohort study [185]; 1 SR [133] |
| CYC (see text for dosing) | Severe lupus, including NPSLE, prevent flare, steroid-sparing | 30 | 2 ++ | B | 4 open-label RCTs [186–189]; 25 cohort studies covered by 1 SR [133] |
| Rituximab 1000 mg × 2 | Refractory severe and moderate lupus; steroid-sparing | 33 | 2+ | C | 1 blind RCT [190, 191]; 3 open-label RCTs [192–194]; 24 cohort studies [195–198 not in SRs]; 2 case series [194, 199]; 2 SRs, including 1 meta-analysis [200, 201]; 1 SR with 26 extra case reports/series [202] |
| Belimumab 10 mg/kg/4 weeks | Refractory moderate/severe lupus; prevent flare and steroid-sparing (not NPSLE) | 5 | 1+ | B | 2 phase III blind RCTs [203, 204]; 1 phase II blind RCT [205]; post hoc combined analysis [206]; 1 open-label extension [207, 208]; 1 meta-analysis [209] |
| IVIG (see text) | Refractory severe lupus (including catastrophic APS) | 19 | 2− | D | Rarely indicated: 3 open-label trials [210–212]; 10 cohort studies [213–222]; 4 case series [223–226]; 2 SRs with 1 meta-analysis [227, 228] |
| Plasmapharesis | TTP; refractory severe SLE | 10 | 2 ++ for TTP; 3 otherwise | B for TTP; D otherwise | Rarely indicated: 9 cohort/case series [229–237]; 1SR [238] |
TPMT: thiopurine S-methyltransferase (see text); TTP: thrombocytopaenic purpura.
Levels of evidence and grades of recommendation for medications used in the treatment of non-renal SLE
| Treatment (recommended target dosage) | Main uses (unless contra-indications) | Total number of papers | Overall SIGN level of evidence | Grade of recommendation | Comments: including number of reports and references for RCTs, cohort studies and systematic reviews/meta-analyses (SRs) |
|---|---|---|---|---|---|
| Antimalarials: HCQ ≤6.5 mg/kg/day | Mild lupus, prevent flare in all patients, prevent damage, steroid-sparing | 45 | 1 ++ | A | 7 RCTs [80–86]; 36 cohort studies [87–120]; 2 SRs [121, 122] |
| MTX ≤25 mg/week | Mild and moderate lupus, prevent flare, steroid sparing | 12 | 1+ | A | |
| NSAIDs | Symptom control in mild non-renal lupus only | 1 | 3 | D | 1 SR covers case series/reports [134] |
| Sunscreen (high-SPF UV-A and UV-B) | Prevents UV-induced rashes and other manifestations | 7 | 2 ++ | B | 1 blind RCT [135]; 5 cohort studies [136–140]; 1 case series [141] |
| Low-dose oral prednisolone (≤7.5 mg) | Mild lupus and to prevent flares | 0 | 4 | D | Expert opinion |
| Higher doses of oral prednisolone ≤0.5 mg/kg/day | Moderate lupus and prevention of flares | 0 2 | 4 2+ | D C | To prevent flare: 1 blind RCT [46] and 1 open-label RCT [60] |
| I.m. triamcinolone | Moderate lupus | 1 | 2+ | C | 1 open-label RCT [142] |
| I.m. methylprednisolone (80–120 mg) | Moderate lupus | 0 | 4 | D | Expert opinion |
| I.v. methylprednisolone (100–250 mg) | Moderate lupus | 1 | 2+ | C/D | 1 blind RCT for 100 mg vs 1000 mg [143] |
| I.v. methylprednisolone (500 mg–1 g) × 1–3 pulses | Moderate and severe lupus | 6 | 2+ | C | 2 small blind RCTs [143, 144]; 1 open-label trial [145]; 3 cohort studies [146–148] |
| AZA (if TPMT normal) 2–3 mg/kg/day | Moderate lupus, prevent flare, steroid sparing | 10 | 2+ | C | 4 open-label RCTs [149–152]; 5 cohort studies [153–157]; 1 case series [158] |
| MMF 2–3 g/day | Moderate/severe lupus, prevent flare, steroid-sparing | 13 | 2 ++ | B | 3 open-label RCTs [159–161]; 7 cohort studies [162–168]; 1 case series [169]; 2 SRs [133, 170] |
| Mycophenolic acid/sodium 1.44–2.16 g/day | For patients intolerant of MMF | 2 | 3 | D | 1 open-label RCT [171]; 1 cohort study [172] |
| Ciclosporin ≤2.5 mg/kg/day | Moderate/severe lupus including cytopenias, prevent flare, steroid-sparing | 11 | 2+ | C | 2 open-label RCTs [152, 173]; 8 cohort studies [174–181]; 1 SR [133] |
| Tacrolimus 1–3 mg/day (assess drug levels) | Moderate/severe lupus, steroid-sparing | 3 | 3 | D | 2 cohort studies [182, 183]; 1 SR [133] |
| LEF (20 mg/day) | Moderate lupus without subacute rash | 3 | 3 | D | 1 small blind RCT [184]; 1 cohort study [185]; 1 SR [133] |
| CYC (see text for dosing) | Severe lupus, including NPSLE, prevent flare, steroid-sparing | 30 | 2 ++ | B | 4 open-label RCTs [186–189]; 25 cohort studies covered by 1 SR [133] |
| Rituximab 1000 mg × 2 | Refractory severe and moderate lupus; steroid-sparing | 33 | 2+ | C | 1 blind RCT [190, 191]; 3 open-label RCTs [192–194]; 24 cohort studies [195–198 not in SRs]; 2 case series [194, 199]; 2 SRs, including 1 meta-analysis [200, 201]; 1 SR with 26 extra case reports/series [202] |
| Belimumab 10 mg/kg/4 weeks | Refractory moderate/severe lupus; prevent flare and steroid-sparing (not NPSLE) | 5 | 1+ | B | 2 phase III blind RCTs [203, 204]; 1 phase II blind RCT [205]; post hoc combined analysis [206]; 1 open-label extension [207, 208]; 1 meta-analysis [209] |
| IVIG (see text) | Refractory severe lupus (including catastrophic APS) | 19 | 2− | D | Rarely indicated: 3 open-label trials [210–212]; 10 cohort studies [213–222]; 4 case series [223–226]; 2 SRs with 1 meta-analysis [227, 228] |
| Plasmapharesis | TTP; refractory severe SLE | 10 | 2 ++ for TTP; 3 otherwise | B for TTP; D otherwise | Rarely indicated: 9 cohort/case series [229–237]; 1SR [238] |
| Treatment (recommended target dosage) | Main uses (unless contra-indications) | Total number of papers | Overall SIGN level of evidence | Grade of recommendation | Comments: including number of reports and references for RCTs, cohort studies and systematic reviews/meta-analyses (SRs) |
|---|---|---|---|---|---|
| Antimalarials: HCQ ≤6.5 mg/kg/day | Mild lupus, prevent flare in all patients, prevent damage, steroid-sparing | 45 | 1 ++ | A | 7 RCTs [80–86]; 36 cohort studies [87–120]; 2 SRs [121, 122] |
| MTX ≤25 mg/week | Mild and moderate lupus, prevent flare, steroid sparing | 12 | 1+ | A | |
| NSAIDs | Symptom control in mild non-renal lupus only | 1 | 3 | D | 1 SR covers case series/reports [134] |
| Sunscreen (high-SPF UV-A and UV-B) | Prevents UV-induced rashes and other manifestations | 7 | 2 ++ | B | 1 blind RCT [135]; 5 cohort studies [136–140]; 1 case series [141] |
| Low-dose oral prednisolone (≤7.5 mg) | Mild lupus and to prevent flares | 0 | 4 | D | Expert opinion |
| Higher doses of oral prednisolone ≤0.5 mg/kg/day | Moderate lupus and prevention of flares | 0 2 | 4 2+ | D C | To prevent flare: 1 blind RCT [46] and 1 open-label RCT [60] |
| I.m. triamcinolone | Moderate lupus | 1 | 2+ | C | 1 open-label RCT [142] |
| I.m. methylprednisolone (80–120 mg) | Moderate lupus | 0 | 4 | D | Expert opinion |
| I.v. methylprednisolone (100–250 mg) | Moderate lupus | 1 | 2+ | C/D | 1 blind RCT for 100 mg vs 1000 mg [143] |
| I.v. methylprednisolone (500 mg–1 g) × 1–3 pulses | Moderate and severe lupus | 6 | 2+ | C | 2 small blind RCTs [143, 144]; 1 open-label trial [145]; 3 cohort studies [146–148] |
| AZA (if TPMT normal) 2–3 mg/kg/day | Moderate lupus, prevent flare, steroid sparing | 10 | 2+ | C | 4 open-label RCTs [149–152]; 5 cohort studies [153–157]; 1 case series [158] |
| MMF 2–3 g/day | Moderate/severe lupus, prevent flare, steroid-sparing | 13 | 2 ++ | B | 3 open-label RCTs [159–161]; 7 cohort studies [162–168]; 1 case series [169]; 2 SRs [133, 170] |
| Mycophenolic acid/sodium 1.44–2.16 g/day | For patients intolerant of MMF | 2 | 3 | D | 1 open-label RCT [171]; 1 cohort study [172] |
| Ciclosporin ≤2.5 mg/kg/day | Moderate/severe lupus including cytopenias, prevent flare, steroid-sparing | 11 | 2+ | C | 2 open-label RCTs [152, 173]; 8 cohort studies [174–181]; 1 SR [133] |
| Tacrolimus 1–3 mg/day (assess drug levels) | Moderate/severe lupus, steroid-sparing | 3 | 3 | D | 2 cohort studies [182, 183]; 1 SR [133] |
| LEF (20 mg/day) | Moderate lupus without subacute rash | 3 | 3 | D | 1 small blind RCT [184]; 1 cohort study [185]; 1 SR [133] |
| CYC (see text for dosing) | Severe lupus, including NPSLE, prevent flare, steroid-sparing | 30 | 2 ++ | B | 4 open-label RCTs [186–189]; 25 cohort studies covered by 1 SR [133] |
| Rituximab 1000 mg × 2 | Refractory severe and moderate lupus; steroid-sparing | 33 | 2+ | C | 1 blind RCT [190, 191]; 3 open-label RCTs [192–194]; 24 cohort studies [195–198 not in SRs]; 2 case series [194, 199]; 2 SRs, including 1 meta-analysis [200, 201]; 1 SR with 26 extra case reports/series [202] |
| Belimumab 10 mg/kg/4 weeks | Refractory moderate/severe lupus; prevent flare and steroid-sparing (not NPSLE) | 5 | 1+ | B | 2 phase III blind RCTs [203, 204]; 1 phase II blind RCT [205]; post hoc combined analysis [206]; 1 open-label extension [207, 208]; 1 meta-analysis [209] |
| IVIG (see text) | Refractory severe lupus (including catastrophic APS) | 19 | 2− | D | Rarely indicated: 3 open-label trials [210–212]; 10 cohort studies [213–222]; 4 case series [223–226]; 2 SRs with 1 meta-analysis [227, 228] |
| Plasmapharesis | TTP; refractory severe SLE | 10 | 2 ++ for TTP; 3 otherwise | B for TTP; D otherwise | Rarely indicated: 9 cohort/case series [229–237]; 1SR [238] |
TPMT: thiopurine S-methyltransferase (see text); TTP: thrombocytopaenic purpura.
Strength of agreement of authors with the main EULAR/ERA-EDTA recommendations for the management of LN
| Management of SLE patients with renal involvement | SOAa |
|---|---|
| Assessment of renal involvement | |
| 1. Indications for first renal biopsy in SLE | 97 |
| Any sign of renal involvement—in particular, urinary findings such as reproducible proteinuria ≥0.5 g/24 h, especially with glomerular haematuria and/or cellular casts—should be an indication for renal biopsy. Renal biopsy is indispensable since, in most cases, clinical, serologic and laboratory tests cannot accurately predict renal biopsy findings. | |
| 2. Pathological assessment of kidney biopsy | 98 |
| The use of the International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 classification system is recommended, with assessment not only of active and chronic glomerular and tubulointerstitial changes, but also of vascular lesions associated with aPLs/APS. | |
| Treatment of renal involvement | |
| 3. Indications and goals of immunosuppressive treatment in LN | 98 |
| 3.1 Initiation of immunosuppressive treatment should be guided by a diagnostic renal biopsy. Immunosuppressive agents are recommended in class IIIA or IIIA/C (±V) and IVA or IVA/C (±V) nephritis, and also in pure class V nephritis if proteinuria exceeds 1 g/24 h despite the optimal use of renin–angiotensin–aldosterone system blockers. | |
| 3.2 The ultimate goals of treatment in LN are long-term preservation of renal function, prevention of disease flares, avoidance of treatment-related harms and improved quality of life and survival. Treatment should aim for complete renal response with UPCR <50 mg/mol and normal or near-normal (within 10% of normal GFR if previously abnormal) renal function. Partial renal response, defined as ≥ 50% reduction in proteinuria to subnephrotic levels and normal or near-normal renal function, should be achieved preferably by 6 months but no later than 12 months following initiation of treatment. | 98 |
| 4. Treatment of adult LN—initial treatment | |
| 4.1 For patients with class IIIA or IIIA/C (±V) and class IVA or IVA/C (±V) LN, mycophenolic acid (MPA) (MMF target dose: 3 g/day for 6 months, or MPA sodium at equivalent dose) or low-dose i.v. CYC (total dose 3 g over 3 months), in combination with glucocorticoids, are recommended as initial treatment as they have the best efficacy/toxicity ratio. | 93 |
| 4.2 In patients with adverse prognostic factors (acute deterioration in renal function, substantial cellular crescents and/or fibrinoid necrosis), similar regimens may be used, but CYC can also be prescribed monthly at higher doses (0.75–1 g/m2) for 6 months or orally (2–2.5 mg/kg/day) for 3 months. | 92 |
| 4.3 To increase efficacy and reduce cumulative glucocorticoid doses, treatment regimens should be combined initially with three consecutive pulses of i.v. methylprednisolone 500–750 mg, followed by oral prednisone 0.5 mg/kg/day for 4 weeks, reducing to ≤ 10 mg/day by 4–6 months | 98 |
| 4.4 In pure class V nephritis with nephrotic-range proteinuria, MPA (MMF target dose 3 g/day for 6 months) in combination with oral prednisone (0.5 mg/kg/day) may be used as initial treatment based on better efficacy/toxicity ratio. CYC or calcineurin inhibitors (ciclosporin, tacrolimus) or rituximab are recommended as alternative options or for non-responders. | 95 |
| 4.5 AZA (2 mg/kg/day) may be considered as an alternative to MPA or CYC in selected patients without adverse prognostic factors (as defined 4.2), or when these drugs are contraindicated, not tolerated or unavailable. AZA use is associated with a higher flare risk. | 96 |
| Subsequent treatment | |
| 4.6 In patients improving after initial treatment, subsequent immunosuppression is recommended with either MPA at lower doses (initial target MMF dose 2 g/day) or AZA (2 mg/kg/day) for at least 3 years, in combination with low-dose prednisone (5–7.5 mg/day). Gradual drug withdrawal, glucocorticoids first, can then be attempted. | 97 |
| 4.7 Patients who responded to initial treatment with MPA should remain on MPA unless pregnancy is contemplated, in which case they should switch to AZA at least 3 months prior to conception. | 98 |
| 4.8 Calcineurin inhibitors can be considered in pure class V nephritis. | 93 |
| Refractory disease | |
| 4.9 For patients who fail treatment with MPA or CYC, either because of lack of effect (as defined above) or due to adverse events, we recommend that the treatment is switched from MPA to CYC, or CYC to MPA, or that rituximab be given. | 95 |
| 5. Adjunct treatment in patients with LN | |
| 5.1 Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are indicated for patients with proteinuria (UPCR >50 mg/mmol) or hypertension. | 98 |
| 6. Management of end-stage renal disease in LN | |
| 6.1 All methods of renal replacement treatment can be used in lupus patients, but there may be increased risk of infections in peritoneal dialysis patients still on immunosuppressive agents, and vascular access thrombosis in patients with aPLs. | 98 |
| 6.2 Transplantation should be performed when lupus activity has been absent, or at a low level, for at least 3–6 months, with superior results obtained with living donor and pre-emptive transplantation. aPLs should be sought during transplant preparation because they are associated with an increased risk of vascular events in the transplanted kidney. | 96 |
| 7. APS-associated nephropathy in SLE | |
| 7.1 In patients with lupus and APS-associated nephropathy (APSN), HCQ and/or antiplatelet/anticoagulant treatment should be considered. | 91 |
| Management of SLE patients with renal involvement | SOAa |
|---|---|
| Assessment of renal involvement | |
| 1. Indications for first renal biopsy in SLE | 97 |
| Any sign of renal involvement—in particular, urinary findings such as reproducible proteinuria ≥0.5 g/24 h, especially with glomerular haematuria and/or cellular casts—should be an indication for renal biopsy. Renal biopsy is indispensable since, in most cases, clinical, serologic and laboratory tests cannot accurately predict renal biopsy findings. | |
| 2. Pathological assessment of kidney biopsy | 98 |
| The use of the International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 classification system is recommended, with assessment not only of active and chronic glomerular and tubulointerstitial changes, but also of vascular lesions associated with aPLs/APS. | |
| Treatment of renal involvement | |
| 3. Indications and goals of immunosuppressive treatment in LN | 98 |
| 3.1 Initiation of immunosuppressive treatment should be guided by a diagnostic renal biopsy. Immunosuppressive agents are recommended in class IIIA or IIIA/C (±V) and IVA or IVA/C (±V) nephritis, and also in pure class V nephritis if proteinuria exceeds 1 g/24 h despite the optimal use of renin–angiotensin–aldosterone system blockers. | |
| 3.2 The ultimate goals of treatment in LN are long-term preservation of renal function, prevention of disease flares, avoidance of treatment-related harms and improved quality of life and survival. Treatment should aim for complete renal response with UPCR <50 mg/mol and normal or near-normal (within 10% of normal GFR if previously abnormal) renal function. Partial renal response, defined as ≥ 50% reduction in proteinuria to subnephrotic levels and normal or near-normal renal function, should be achieved preferably by 6 months but no later than 12 months following initiation of treatment. | 98 |
| 4. Treatment of adult LN—initial treatment | |
| 4.1 For patients with class IIIA or IIIA/C (±V) and class IVA or IVA/C (±V) LN, mycophenolic acid (MPA) (MMF target dose: 3 g/day for 6 months, or MPA sodium at equivalent dose) or low-dose i.v. CYC (total dose 3 g over 3 months), in combination with glucocorticoids, are recommended as initial treatment as they have the best efficacy/toxicity ratio. | 93 |
| 4.2 In patients with adverse prognostic factors (acute deterioration in renal function, substantial cellular crescents and/or fibrinoid necrosis), similar regimens may be used, but CYC can also be prescribed monthly at higher doses (0.75–1 g/m2) for 6 months or orally (2–2.5 mg/kg/day) for 3 months. | 92 |
| 4.3 To increase efficacy and reduce cumulative glucocorticoid doses, treatment regimens should be combined initially with three consecutive pulses of i.v. methylprednisolone 500–750 mg, followed by oral prednisone 0.5 mg/kg/day for 4 weeks, reducing to ≤ 10 mg/day by 4–6 months | 98 |
| 4.4 In pure class V nephritis with nephrotic-range proteinuria, MPA (MMF target dose 3 g/day for 6 months) in combination with oral prednisone (0.5 mg/kg/day) may be used as initial treatment based on better efficacy/toxicity ratio. CYC or calcineurin inhibitors (ciclosporin, tacrolimus) or rituximab are recommended as alternative options or for non-responders. | 95 |
| 4.5 AZA (2 mg/kg/day) may be considered as an alternative to MPA or CYC in selected patients without adverse prognostic factors (as defined 4.2), or when these drugs are contraindicated, not tolerated or unavailable. AZA use is associated with a higher flare risk. | 96 |
| Subsequent treatment | |
| 4.6 In patients improving after initial treatment, subsequent immunosuppression is recommended with either MPA at lower doses (initial target MMF dose 2 g/day) or AZA (2 mg/kg/day) for at least 3 years, in combination with low-dose prednisone (5–7.5 mg/day). Gradual drug withdrawal, glucocorticoids first, can then be attempted. | 97 |
| 4.7 Patients who responded to initial treatment with MPA should remain on MPA unless pregnancy is contemplated, in which case they should switch to AZA at least 3 months prior to conception. | 98 |
| 4.8 Calcineurin inhibitors can be considered in pure class V nephritis. | 93 |
| Refractory disease | |
| 4.9 For patients who fail treatment with MPA or CYC, either because of lack of effect (as defined above) or due to adverse events, we recommend that the treatment is switched from MPA to CYC, or CYC to MPA, or that rituximab be given. | 95 |
| 5. Adjunct treatment in patients with LN | |
| 5.1 Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are indicated for patients with proteinuria (UPCR >50 mg/mmol) or hypertension. | 98 |
| 6. Management of end-stage renal disease in LN | |
| 6.1 All methods of renal replacement treatment can be used in lupus patients, but there may be increased risk of infections in peritoneal dialysis patients still on immunosuppressive agents, and vascular access thrombosis in patients with aPLs. | 98 |
| 6.2 Transplantation should be performed when lupus activity has been absent, or at a low level, for at least 3–6 months, with superior results obtained with living donor and pre-emptive transplantation. aPLs should be sought during transplant preparation because they are associated with an increased risk of vascular events in the transplanted kidney. | 96 |
| 7. APS-associated nephropathy in SLE | |
| 7.1 In patients with lupus and APS-associated nephropathy (APSN), HCQ and/or antiplatelet/anticoagulant treatment should be considered. | 91 |
Reproduced from Bertsias et al. Joint European League Against Rheumatism and European Renal Association–European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis 71: 771–82. Copyright 2012, with permission from BMJ Publishing Group Ltd [24]. Numbers are mean (s.d.) and median (IQR) agreement level among authors. A score of 10 represents the highest SOA. GFR: glomerular filtration rate; SOA: strength of agreement; UPCR: urine protein:creatinine ratio.
Strength of agreement of authors with the main EULAR/ERA-EDTA recommendations for the management of LN
| Management of SLE patients with renal involvement | SOAa |
|---|---|
| Assessment of renal involvement | |
| 1. Indications for first renal biopsy in SLE | 97 |
| Any sign of renal involvement—in particular, urinary findings such as reproducible proteinuria ≥0.5 g/24 h, especially with glomerular haematuria and/or cellular casts—should be an indication for renal biopsy. Renal biopsy is indispensable since, in most cases, clinical, serologic and laboratory tests cannot accurately predict renal biopsy findings. | |
| 2. Pathological assessment of kidney biopsy | 98 |
| The use of the International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 classification system is recommended, with assessment not only of active and chronic glomerular and tubulointerstitial changes, but also of vascular lesions associated with aPLs/APS. | |
| Treatment of renal involvement | |
| 3. Indications and goals of immunosuppressive treatment in LN | 98 |
| 3.1 Initiation of immunosuppressive treatment should be guided by a diagnostic renal biopsy. Immunosuppressive agents are recommended in class IIIA or IIIA/C (±V) and IVA or IVA/C (±V) nephritis, and also in pure class V nephritis if proteinuria exceeds 1 g/24 h despite the optimal use of renin–angiotensin–aldosterone system blockers. | |
| 3.2 The ultimate goals of treatment in LN are long-term preservation of renal function, prevention of disease flares, avoidance of treatment-related harms and improved quality of life and survival. Treatment should aim for complete renal response with UPCR <50 mg/mol and normal or near-normal (within 10% of normal GFR if previously abnormal) renal function. Partial renal response, defined as ≥ 50% reduction in proteinuria to subnephrotic levels and normal or near-normal renal function, should be achieved preferably by 6 months but no later than 12 months following initiation of treatment. | 98 |
| 4. Treatment of adult LN—initial treatment | |
| 4.1 For patients with class IIIA or IIIA/C (±V) and class IVA or IVA/C (±V) LN, mycophenolic acid (MPA) (MMF target dose: 3 g/day for 6 months, or MPA sodium at equivalent dose) or low-dose i.v. CYC (total dose 3 g over 3 months), in combination with glucocorticoids, are recommended as initial treatment as they have the best efficacy/toxicity ratio. | 93 |
| 4.2 In patients with adverse prognostic factors (acute deterioration in renal function, substantial cellular crescents and/or fibrinoid necrosis), similar regimens may be used, but CYC can also be prescribed monthly at higher doses (0.75–1 g/m2) for 6 months or orally (2–2.5 mg/kg/day) for 3 months. | 92 |
| 4.3 To increase efficacy and reduce cumulative glucocorticoid doses, treatment regimens should be combined initially with three consecutive pulses of i.v. methylprednisolone 500–750 mg, followed by oral prednisone 0.5 mg/kg/day for 4 weeks, reducing to ≤ 10 mg/day by 4–6 months | 98 |
| 4.4 In pure class V nephritis with nephrotic-range proteinuria, MPA (MMF target dose 3 g/day for 6 months) in combination with oral prednisone (0.5 mg/kg/day) may be used as initial treatment based on better efficacy/toxicity ratio. CYC or calcineurin inhibitors (ciclosporin, tacrolimus) or rituximab are recommended as alternative options or for non-responders. | 95 |
| 4.5 AZA (2 mg/kg/day) may be considered as an alternative to MPA or CYC in selected patients without adverse prognostic factors (as defined 4.2), or when these drugs are contraindicated, not tolerated or unavailable. AZA use is associated with a higher flare risk. | 96 |
| Subsequent treatment | |
| 4.6 In patients improving after initial treatment, subsequent immunosuppression is recommended with either MPA at lower doses (initial target MMF dose 2 g/day) or AZA (2 mg/kg/day) for at least 3 years, in combination with low-dose prednisone (5–7.5 mg/day). Gradual drug withdrawal, glucocorticoids first, can then be attempted. | 97 |
| 4.7 Patients who responded to initial treatment with MPA should remain on MPA unless pregnancy is contemplated, in which case they should switch to AZA at least 3 months prior to conception. | 98 |
| 4.8 Calcineurin inhibitors can be considered in pure class V nephritis. | 93 |
| Refractory disease | |
| 4.9 For patients who fail treatment with MPA or CYC, either because of lack of effect (as defined above) or due to adverse events, we recommend that the treatment is switched from MPA to CYC, or CYC to MPA, or that rituximab be given. | 95 |
| 5. Adjunct treatment in patients with LN | |
| 5.1 Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are indicated for patients with proteinuria (UPCR >50 mg/mmol) or hypertension. | 98 |
| 6. Management of end-stage renal disease in LN | |
| 6.1 All methods of renal replacement treatment can be used in lupus patients, but there may be increased risk of infections in peritoneal dialysis patients still on immunosuppressive agents, and vascular access thrombosis in patients with aPLs. | 98 |
| 6.2 Transplantation should be performed when lupus activity has been absent, or at a low level, for at least 3–6 months, with superior results obtained with living donor and pre-emptive transplantation. aPLs should be sought during transplant preparation because they are associated with an increased risk of vascular events in the transplanted kidney. | 96 |
| 7. APS-associated nephropathy in SLE | |
| 7.1 In patients with lupus and APS-associated nephropathy (APSN), HCQ and/or antiplatelet/anticoagulant treatment should be considered. | 91 |
| Management of SLE patients with renal involvement | SOAa |
|---|---|
| Assessment of renal involvement | |
| 1. Indications for first renal biopsy in SLE | 97 |
| Any sign of renal involvement—in particular, urinary findings such as reproducible proteinuria ≥0.5 g/24 h, especially with glomerular haematuria and/or cellular casts—should be an indication for renal biopsy. Renal biopsy is indispensable since, in most cases, clinical, serologic and laboratory tests cannot accurately predict renal biopsy findings. | |
| 2. Pathological assessment of kidney biopsy | 98 |
| The use of the International Society of Nephrology/Renal Pathology Society (ISN/RPS) 2003 classification system is recommended, with assessment not only of active and chronic glomerular and tubulointerstitial changes, but also of vascular lesions associated with aPLs/APS. | |
| Treatment of renal involvement | |
| 3. Indications and goals of immunosuppressive treatment in LN | 98 |
| 3.1 Initiation of immunosuppressive treatment should be guided by a diagnostic renal biopsy. Immunosuppressive agents are recommended in class IIIA or IIIA/C (±V) and IVA or IVA/C (±V) nephritis, and also in pure class V nephritis if proteinuria exceeds 1 g/24 h despite the optimal use of renin–angiotensin–aldosterone system blockers. | |
| 3.2 The ultimate goals of treatment in LN are long-term preservation of renal function, prevention of disease flares, avoidance of treatment-related harms and improved quality of life and survival. Treatment should aim for complete renal response with UPCR <50 mg/mol and normal or near-normal (within 10% of normal GFR if previously abnormal) renal function. Partial renal response, defined as ≥ 50% reduction in proteinuria to subnephrotic levels and normal or near-normal renal function, should be achieved preferably by 6 months but no later than 12 months following initiation of treatment. | 98 |
| 4. Treatment of adult LN—initial treatment | |
| 4.1 For patients with class IIIA or IIIA/C (±V) and class IVA or IVA/C (±V) LN, mycophenolic acid (MPA) (MMF target dose: 3 g/day for 6 months, or MPA sodium at equivalent dose) or low-dose i.v. CYC (total dose 3 g over 3 months), in combination with glucocorticoids, are recommended as initial treatment as they have the best efficacy/toxicity ratio. | 93 |
| 4.2 In patients with adverse prognostic factors (acute deterioration in renal function, substantial cellular crescents and/or fibrinoid necrosis), similar regimens may be used, but CYC can also be prescribed monthly at higher doses (0.75–1 g/m2) for 6 months or orally (2–2.5 mg/kg/day) for 3 months. | 92 |
| 4.3 To increase efficacy and reduce cumulative glucocorticoid doses, treatment regimens should be combined initially with three consecutive pulses of i.v. methylprednisolone 500–750 mg, followed by oral prednisone 0.5 mg/kg/day for 4 weeks, reducing to ≤ 10 mg/day by 4–6 months | 98 |
| 4.4 In pure class V nephritis with nephrotic-range proteinuria, MPA (MMF target dose 3 g/day for 6 months) in combination with oral prednisone (0.5 mg/kg/day) may be used as initial treatment based on better efficacy/toxicity ratio. CYC or calcineurin inhibitors (ciclosporin, tacrolimus) or rituximab are recommended as alternative options or for non-responders. | 95 |
| 4.5 AZA (2 mg/kg/day) may be considered as an alternative to MPA or CYC in selected patients without adverse prognostic factors (as defined 4.2), or when these drugs are contraindicated, not tolerated or unavailable. AZA use is associated with a higher flare risk. | 96 |
| Subsequent treatment | |
| 4.6 In patients improving after initial treatment, subsequent immunosuppression is recommended with either MPA at lower doses (initial target MMF dose 2 g/day) or AZA (2 mg/kg/day) for at least 3 years, in combination with low-dose prednisone (5–7.5 mg/day). Gradual drug withdrawal, glucocorticoids first, can then be attempted. | 97 |
| 4.7 Patients who responded to initial treatment with MPA should remain on MPA unless pregnancy is contemplated, in which case they should switch to AZA at least 3 months prior to conception. | 98 |
| 4.8 Calcineurin inhibitors can be considered in pure class V nephritis. | 93 |
| Refractory disease | |
| 4.9 For patients who fail treatment with MPA or CYC, either because of lack of effect (as defined above) or due to adverse events, we recommend that the treatment is switched from MPA to CYC, or CYC to MPA, or that rituximab be given. | 95 |
| 5. Adjunct treatment in patients with LN | |
| 5.1 Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers are indicated for patients with proteinuria (UPCR >50 mg/mmol) or hypertension. | 98 |
| 6. Management of end-stage renal disease in LN | |
| 6.1 All methods of renal replacement treatment can be used in lupus patients, but there may be increased risk of infections in peritoneal dialysis patients still on immunosuppressive agents, and vascular access thrombosis in patients with aPLs. | 98 |
| 6.2 Transplantation should be performed when lupus activity has been absent, or at a low level, for at least 3–6 months, with superior results obtained with living donor and pre-emptive transplantation. aPLs should be sought during transplant preparation because they are associated with an increased risk of vascular events in the transplanted kidney. | 96 |
| 7. APS-associated nephropathy in SLE | |
| 7.1 In patients with lupus and APS-associated nephropathy (APSN), HCQ and/or antiplatelet/anticoagulant treatment should be considered. | 91 |
Reproduced from Bertsias et al. Joint European League Against Rheumatism and European Renal Association–European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis 71: 771–82. Copyright 2012, with permission from BMJ Publishing Group Ltd [24]. Numbers are mean (s.d.) and median (IQR) agreement level among authors. A score of 10 represents the highest SOA. GFR: glomerular filtration rate; SOA: strength of agreement; UPCR: urine protein:creatinine ratio.
Target population, target audience and stakeholder involvement
The guidelines address the management of adult patients only and have been developed by a multidisciplinary guideline development group set up by the British Society for Rheumatology (BSR) and led by C.G., consisting of academic (C.G., I.N.B., D.D.C., M.K., D.I.) and NHS consultants in rheumatology (M.A., B.G.) and nephrology (D.J., L.L.), rheumatology trainees (M.G., K.S.), a GP (B.E.), a clinical nurse specialist (S.B.), a patient representative (Y.N.) and a lay member (P.N.). All participants declared any conflicts of interest and these are listed at the end of this article. The target audience includes rheumatologists and other clinicians such as nephrologists, immunologists and dermatologists, trainees in these specialties and emergency medicine, GPs, clinical nurse specialists and other allied health professionals involved in the care of adult lupus patients. Opinions of other key stakeholders such as other consultant members of the BSR, additional trainees, podiatrists, nurse specialists and representatives of Lupus UK were sought during the preparation of these guidelines.
Areas that the guideline does not cover
This guideline does not cover the evidence for topical or systemic therapy for isolated cutaneous lupus, nor does it discuss paediatric lupus, as there is relatively little literature on paediatric lupus. As the disease tends to come on after puberty, most of the recommendations are likely to be appropriate for children/adolescents, with suitable dose modifications. We provide only summary advice about the use of drugs in the management of pregnant lupus patients, and refer to the extensive review of drugs used in pregnancy and breast-feeding that have been recently published [239, 240]. The management of complications of lupus, including chronic fatigue, cardiovascular risk, osteoporosis, infection and cancer risk are not discussed in detail, as these issues should be managed as for other patients with similar risk factors according to national and international guidelines. Management of thrombosis will depend on whether or not the criteria for APS are met [241].
Rigor of development
Selection of questions for the literature review, and statement of extent of previous National Institute for Health and Care Excellence, Royal College of Physicians, and Scottish Intercollegiate Guidelines Network guidelines
A multidisciplinary guideline development group was formed and followed the BSR Protocol for Guidelines and EULAR standardized operating procedures to define the focus of the work, the target population and the target audience. Discussions were supplemented by consensus-building strategies, including a modified Delphi technique, in order to reduce and clearly define the list of research questions to be addressed by the literature search (see supplementary data section Search strategy, available at Rheumatology Online). There are no BSR, Royal College of Physicians (RCP), National Institute for Health and Care Excellence (NICE) or Scottish Intercollegiate Guidelines Network (SIGN) guidelines or recommendations for the management of lupus in the UK to help improve the outcome of this variable and potentially life-threatening disease, but lupus has been included in the on-line resource Map of Medicine.
Literature review: eligibility criteria and limitations of the search
A systematic search of MEDLINE (PubMed) and the Cochrane Database of Systematic Reviews was performed, and all publications in peer-reviewed English language journals up to June 2015 were considered. A detailed search was performed using an array of relevant terms (see supplementary data section Search strategy and supplementary Table S1, available at Rheumatology Online), and papers were screened for eligibility based on their title, abstract and/or full content. Studies were eligible if they had studied at least 50 patients for prevalence and prognosis of manifestations, 10 patients for diagnosis and monitoring, or 5 patients for therapy.
Studies on animals, children, review articles, commentaries, conference abstracts or statements, and expert opinion statements were excluded. Narrative review articles and existing guidelines were checked for references, but only meta-analyses and systematic reviews were included, together with original research articles, in the analysis. Over 8000 articles were identified during the literature search, and over 600 were deemed eligible for detailed review by at least two members of the group. There was considerable overlap in the topics covered by the papers, which were reviewed by various members of the group.
Development of the guideline: levels of evidence and consensus agreement
The recommendations were developed in line with the BSR’s Guidelines Protocol, using RCP, SIGN and AGREE II methodology to assess the level of evidence (LOE) and grade of recommendation (GOR). Papers selected for review and the evidence obtained from them were categorized by at least two members of the group, according to the study design, using the SIGN methodology (supplementary Table S2, available at Rheumatology Online), and the level of the evidence was graded by combining information on the design and validity of the available research studies to provide the GOR for each component of each statement. The results of the literature search were summarized, aggregated and distributed to the expert committee by three of us (C.G., M.G., M.A.), and the GOR for each item was ratified by the expert committee. Draft recommendations were discussed and rephrased at a face-to-face meeting and subsequently by email, following an updated literature review. The LOEs and the GORs for the data supporting the guideline recommendations are shown in Tables 1 and 2. Finally, the six recommendations for the management of SLE and the main items in the EULAR/ERA-EDTA recommendations for LN [24] (Table 3) were voted on by clinical members of the guideline development group. For each recommendation, the SOA of all clinical members of the group was sought on a scale of 1 (no agreement) to 10 (complete agreement); the mean percentage agreement was calculated and is shown after each recommendation (all >90% and supported by other members of the group). The guideline will be reviewed in 5 years’ time.
The guideline
Eligibility criteria
This guideline is designed to cover the management of adult patients with SLE by healthcare professionals. These recommendations are based on the literature review covering the diagnosis, assessment, monitoring and treatment of mild, moderate and severe lupus, including neuropsychiatric (NP) disease. The focus of the literature review was on non-renal disease, as the EULAR/ERA-EDTA recommendations for LN (see below) were published [24] close to the time that we started work on this guideline.
Exclusion criteria
Management of paediatric lupus, renal lupus, topical treatment for cutaneous lupus, and drug treatment in pregnancy have been excluded from our literature search and guideline development. BSR guidelines on the use of drugs in pregnant patients with rheumatic diseases (including lupus) have been developed in parallel with this guideline.
Introduction to the recommendations and supporting evidence
For each question addressed by the literature review (supplementary data section Search strategy, available at Rheumatology Online), we provide first the recommendations and the overall LOE, GOR and SOA for each, followed by the rationale. The rationale consists of a summary of the evidence supporting the statements (including cautions in the case of drug therapy). It is organized by topic and includes some key points about the studies leading to the recommendations and a conclusion for each topic discussed. The number of studies and types of studies (with references) leading to the LOE and GOR are summarized in Table 1 for the items contributing to the recommendations on diagnosis, assessment and monitoring of lupus, and in Table 2 for those relating to the treatment and prevention of mild, moderate and severe non-renal lupus. In Table 3 we provide our SOA with key points of the EULAR/ERA-EDTA recommendations for the management of LN [24], so that the management of the most important aspects of lupus are covered by this guideline in a single document.
Recommendations for clinical and serological features prompting consideration of a diagnosis of SLE
SLE is a multisystem autoimmune disorder. The diagnosis requires a combination of clinical features and the presence of at least one relevant immunological abnormality. If there is a clinical suspicion of lupus, blood tests (including serological marker tests) should be checked (LOE 2 ++, GOR B, SOA 98%).
ANAs are present in ∼95% of SLE patients. If the test is negative, there is a low clinical probability of the patient having SLE. A positive ANA test occurs in ∼5% of the adult population, and alone it has poor diagnostic value in the absence of clinical features of autoimmune rheumatic disease (2 ++/B, SOA 96%).
The presence of anti-dsDNA antibodies (2 ++/B), low complement levels (2+/C) or anti-Smith (Sm) antibodies (2+/C) are highly predictive of a diagnosis of SLE in patients with relevant clinical features. Anti-Ro/La and anti-RNP antibodies are less-specific markers of SLE (2+/C) as they are found in other autoimmune rheumatic disorders as well as SLE (2+/C) (SOA 95%).
aPLs should be tested in all lupus patients at baseline, especially in those with an adverse pregnancy history or arterial/venous thrombotic events (2 ++/B). Confirmatory tests for APS are positive LA, aCL (IgG, IgM) and/or anti-beta-2 glycoprotein-1 (IgG, IgM) on two occasions at least 12 weeks apart (2 ++/B) (SOA 97%).
Rationale
Clinical manifestations
SLE is a multisystem autoimmune disease [1, 8] with considerable heterogeneity. This makes the diagnosis, assessment and monitoring a challenging process [10, 26–28, 41]. Delays in diagnosis are well recognized and remain a concern [242]. Some of the most typical features and their cumulative incidence are shown in supplementary Table S3, available at Rheumatology Online [7, 10, 26–29]. It is important to ensure that the diagnosis of lupus is appropriate before considering treatment [41, 243]. Given the variety of clinical manifestations that can occur, lupus should be considered in the differential diagnosis of many acute and sub-acute presentations, particularly, but not exclusively, in individuals at increased risk of the disease, such as women from African, South Asian or Chinese backgrounds [2, 244]. Lupus can also affect men, resulting in severe disease, including renal involvement and greater risk of damage compared with women in some but not all reports [15, 16, 30, 31].
Renal and neurological involvement are major causes for morbidity and mortality in SLE [2, 7, 15, 16, 32, 33]. Renal disease is clinically silent and must be actively sought to prevent renal damage as discussed below. A working party of the ACR distinguished 19 NP manifestations that may occur in SLE patients [245]. Not all are directly attributable to the SLE disease process, and the true incidence of these manifestations is hard to ascertain as most of them are uncommon [23, 246]. Gastrointestinal and hepatic features occur in 39–67% of patients [42, 247] and are often not recognized as being due to lupus. As with cardiorespiratory features, they must be distinguished carefully from infection, adverse events from drugs and co-morbid conditions. Ophthalmic manifestations of lupus are rare, but potentially sight-threatening, and need careful evaluation by an experienced ophthalmologist [248–250].
Serological (immunological) manifestations
The clinical features of acute lupus are mostly due to inflammatory processes triggered by the formation of immune complexes involving autoantibodies and complement consumption, although thrombosis associated with aPLs may contribute to the pathogenesis in some patients [1, 8, 10]. With a clinical suspicion of SLE, an initial autoantibody screen should be performed. Approximately 95% of lupus patients are ANA positive, and 98% of patients will have positive ANA and/or anti-dsDNA antibodies [26, 36, 37]. ANA tests, although sensitive, are not specific for the diagnosis of lupus, and ANAs can occur in a variety of other conditions, including SS, SSc, DM, viral infections (e.g. infectious mononucleosis) and malignancy [36, 41]. The ANA test can increase in titre over time or can become negative in treated patients, and the results can vary with different assays [34, 37].
If patients have a strong clinical likelihood of having lupus, anti-dsDNA antibody testing should be done [38]. Anti-dsDNA and anti-Sm antibodies are much more specific for lupus, being very rare in other conditions [36] but they are less sensitive than ANA (supplementary Table S3, available at Rheumatology Online) [10, 26–29, 251]. Both the Farr and the ELISA methods are acceptable for measuring anti-dsDNA antibodies, with the former yielding higher sensitivity and specificity rates [24, 39, 40]. The Crithidia luciliae immunofluorescence test also has a high specificity for SLE. Additional routine serological tests are the complement C3 and C4 levels [43]. C3 generally has a higher sensitivity than serum C4 for active LN, but both tests have modest specificity and their clinical utility lies in their high negative predictive value (>90%) to exclude active disease, especially renal disease [24, 44–46].
Anti-Ro (SSA), anti-La (SSB) and anti-RNP antibodies are less specific markers for the presence of SLE, as they are found in other autoimmune rheumatic disorders [41]. Anti-Ro and anti-La are most strongly associated with primary SS but do occur in lupus patients, especially those with photosensitivity and subacute cutaneous lupus. Anti-Ro and anti-La antibodies can cause neonatal lupus syndrome including congenital heart block (CHB) in children born to mothers with these antibodies (see Recommendations for monitoring of SLE section) [64, 65]. Anti-RNP antibodies are found in overlap conditions such as MCTD [41].
All lupus patients should be tested for aPLs because their presence indicates a group at increased risk of arterial/venous thrombotic events and adverse pregnancy outcomes [241, 252, 253]. As APS and SLE often overlap, and APS sometimes evolves in to SLE, the presence of APS should also prompt assessment for lupus. Confirmatory tests for APS are positive LA, aCL (IgG, IgM), and/or anti-beta-2 glycoprotein-1(IgG, IgM) antibodies on two occasions at least 12 weeks apart [241, 252]. The LA test is the most specific of the three tests and is associated with a higher positive predictive value. The most high-risk aPL profile (triple positivity including positive LA, aCL and anti-β2-glycoprotein-I antibody) is associated with a cumulative incidence of thrombosis after 10 years of 37.1% [254].
Classification criteria for lupus
Based on the ACR (previously the American Rheumatism Association) revised criteria for SLE published in 1982 [255] and the 1997 modification [256], a patient may be classified as having SLE if they have 4 or more of 11 criteria present (Table 4). However, not all patients who meet these criteria have lupus, and not all patients diagnosed clinically with lupus have four or more of these criteria, which may appear or disappear over time [7, 33, 35, 257]. There has been a tendency to consider patients who meet the ACR classification criteria for lupus to have the disease, even if they only have certain clinical features without evidence of one or more of the immunological abnormalities that are the hallmark of this autoimmune disease. Conversely, sometimes the disease has been diagnosed on the basis of auto-antibodies and haematological features, without consideration of whether the whole clinical and serological picture is consistent with lupus being the most likely diagnosis.
The ACR criteria for classification of SLEa
| Criterion | Definition |
|---|---|
| Malar rash | Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds |
| Discoid rash | Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions |
| Photosensitivity | Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation |
| Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by a physician |
| Arthritis | Non-erosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling or effusion |
| Serositis | Pleuritis: convincing history of pleuritic pain or rub heard by a physician or evidence of pleural effusion OR Pericarditis: documented by ECG or rub or evidence of pericardial effusion |
| Renal disorder | Persistent proteinuria >0.5 g/day or > 3+ if quantitation not performed OR |
| Cellular casts: may be red cell, haemoglobin, granular, tubular or mixed | |
| Neurologic disorder | Seizures: in the absence of offending drugs or known metabolic derangements; e.g. uremia, ketoacidosis or electrolyte imbalance OR |
| Psychosis: in the absence of offending drugs or known metabolic derangements, e.g. uremia, ketoacidosis or electrolyte imbalance | |
| Haematologic disorder | Haemolytic anaemia with reticulocytosis OR |
| Leukopenia <4000/mm3 total on two or more occasions OR | |
| Lymphopenia <1500/mm3 on two or more occasions OR | |
| Thrombocytopenia <100 000/mm3 in the absence of offending drugs | |
| Immunologic disorder | Anti-DNA: antibody to native DNA in abnormal titre OR |
| Anti-Sm: presence of antibody to Sm nuclear antigen OR | |
Positive finding of aPLs on:
| |
| ANA | An abnormal titre of ANA by immunofluorescence, or an equivalent assay at any point in time and in the absence of drugs known to be associated with drug-induced lupus syndrome |
| Criterion | Definition |
|---|---|
| Malar rash | Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds |
| Discoid rash | Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions |
| Photosensitivity | Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation |
| Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by a physician |
| Arthritis | Non-erosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling or effusion |
| Serositis | Pleuritis: convincing history of pleuritic pain or rub heard by a physician or evidence of pleural effusion OR Pericarditis: documented by ECG or rub or evidence of pericardial effusion |
| Renal disorder | Persistent proteinuria >0.5 g/day or > 3+ if quantitation not performed OR |
| Cellular casts: may be red cell, haemoglobin, granular, tubular or mixed | |
| Neurologic disorder | Seizures: in the absence of offending drugs or known metabolic derangements; e.g. uremia, ketoacidosis or electrolyte imbalance OR |
| Psychosis: in the absence of offending drugs or known metabolic derangements, e.g. uremia, ketoacidosis or electrolyte imbalance | |
| Haematologic disorder | Haemolytic anaemia with reticulocytosis OR |
| Leukopenia <4000/mm3 total on two or more occasions OR | |
| Lymphopenia <1500/mm3 on two or more occasions OR | |
| Thrombocytopenia <100 000/mm3 in the absence of offending drugs | |
| Immunologic disorder | Anti-DNA: antibody to native DNA in abnormal titre OR |
| Anti-Sm: presence of antibody to Sm nuclear antigen OR | |
Positive finding of aPLs on:
| |
| ANA | An abnormal titre of ANA by immunofluorescence, or an equivalent assay at any point in time and in the absence of drugs known to be associated with drug-induced lupus syndrome |
The proposed classification is based on 11 criteria. For the purpose of identifying patients in clinical studies, a person shall be said to have SLE if any 4 or more of the 11 criteria are present, serially or simultaneously, during any interval of observation. Adapted from Tan EM et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271–7, copyright 1982 [255]; and Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725, copyright 1997 [256], with permission from John Wiley & Sons. Anti-Sm: anti-Smith antibody.
The ACR criteria for classification of SLEa
| Criterion | Definition |
|---|---|
| Malar rash | Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds |
| Discoid rash | Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions |
| Photosensitivity | Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation |
| Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by a physician |
| Arthritis | Non-erosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling or effusion |
| Serositis | Pleuritis: convincing history of pleuritic pain or rub heard by a physician or evidence of pleural effusion OR Pericarditis: documented by ECG or rub or evidence of pericardial effusion |
| Renal disorder | Persistent proteinuria >0.5 g/day or > 3+ if quantitation not performed OR |
| Cellular casts: may be red cell, haemoglobin, granular, tubular or mixed | |
| Neurologic disorder | Seizures: in the absence of offending drugs or known metabolic derangements; e.g. uremia, ketoacidosis or electrolyte imbalance OR |
| Psychosis: in the absence of offending drugs or known metabolic derangements, e.g. uremia, ketoacidosis or electrolyte imbalance | |
| Haematologic disorder | Haemolytic anaemia with reticulocytosis OR |
| Leukopenia <4000/mm3 total on two or more occasions OR | |
| Lymphopenia <1500/mm3 on two or more occasions OR | |
| Thrombocytopenia <100 000/mm3 in the absence of offending drugs | |
| Immunologic disorder | Anti-DNA: antibody to native DNA in abnormal titre OR |
| Anti-Sm: presence of antibody to Sm nuclear antigen OR | |
Positive finding of aPLs on:
| |
| ANA | An abnormal titre of ANA by immunofluorescence, or an equivalent assay at any point in time and in the absence of drugs known to be associated with drug-induced lupus syndrome |
| Criterion | Definition |
|---|---|
| Malar rash | Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds |
| Discoid rash | Erythematous raised patches with adherent keratotic scaling and follicular plugging; atrophic scarring may occur in older lesions |
| Photosensitivity | Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation |
| Oral ulcers | Oral or nasopharyngeal ulceration, usually painless, observed by a physician |
| Arthritis | Non-erosive arthritis involving two or more peripheral joints, characterized by tenderness, swelling or effusion |
| Serositis | Pleuritis: convincing history of pleuritic pain or rub heard by a physician or evidence of pleural effusion OR Pericarditis: documented by ECG or rub or evidence of pericardial effusion |
| Renal disorder | Persistent proteinuria >0.5 g/day or > 3+ if quantitation not performed OR |
| Cellular casts: may be red cell, haemoglobin, granular, tubular or mixed | |
| Neurologic disorder | Seizures: in the absence of offending drugs or known metabolic derangements; e.g. uremia, ketoacidosis or electrolyte imbalance OR |
| Psychosis: in the absence of offending drugs or known metabolic derangements, e.g. uremia, ketoacidosis or electrolyte imbalance | |
| Haematologic disorder | Haemolytic anaemia with reticulocytosis OR |
| Leukopenia <4000/mm3 total on two or more occasions OR | |
| Lymphopenia <1500/mm3 on two or more occasions OR | |
| Thrombocytopenia <100 000/mm3 in the absence of offending drugs | |
| Immunologic disorder | Anti-DNA: antibody to native DNA in abnormal titre OR |
| Anti-Sm: presence of antibody to Sm nuclear antigen OR | |
Positive finding of aPLs on:
| |
| ANA | An abnormal titre of ANA by immunofluorescence, or an equivalent assay at any point in time and in the absence of drugs known to be associated with drug-induced lupus syndrome |
The proposed classification is based on 11 criteria. For the purpose of identifying patients in clinical studies, a person shall be said to have SLE if any 4 or more of the 11 criteria are present, serially or simultaneously, during any interval of observation. Adapted from Tan EM et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271–7, copyright 1982 [255]; and Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725, copyright 1997 [256], with permission from John Wiley & Sons. Anti-Sm: anti-Smith antibody.
To address these and some other issues, the SLICC group devised alternative classification criteria for lupus [258]. These criteria introduced a requirement for at least one clinical and one immunological criterion and two others from an expanded list of items (Table 5) compared with the ACR criteria (Table 4) [256]. They also allowed biopsy-proven LN in the presence of ANA or anti-dsDNA antibodies to be classified as lupus, without the need for other criteria [258]. The serological criteria include low complement (C3 and/or C4), as this item reflects complement consumption due to the formation of immune complexes in active lupus disease.
Clinical and Immunologic Criteria Used in the SLICC Classification Criteria for SLEa
| Clinical Criteria |
|---|
| Acute cutaneous lupus including: |
| lupus malar rash (do not count if malar discoid), bullous lupus, toxic epidermal necrolysis variant of SLE, maculopapular lupus rash, photosensitive lupus rash, (in the absence of dermatomyositis), or subacute cutaneous lupus, nonindurated psoriaform and/or annular polycyclic lesions that resolve without scarring, although occasionally with postinflammatory dyspigmentation or telangiectasias) |
| Chronic cutaneous lupus including: |
| classical discoid rash, localized (above the neck), generalized (above and below the neck), hypertrophic, (verrucous) lupus, lupus panniculitis (profundus), mucosal lupus, lupus erythematosus tumidus, chilblains lupus, discoid lupus/lichen planus overlap |
| Oral ulcers: |
| Palate, buccal, tongue, or nasal ulcers (in the absence of other causes, such as vasculitis, Behcet’s disease, infection (herpes viruses), inflammatory bowel disease, reactive arthritis, acidic foods) |
| Nonscarring alopecia: |
| diffuse thinning or hair fragility with visible broken hairs (in the absence of other causes such as alopecia areata, drugs, iron deficiency and androgenic alopecia) |
| Synovitis involving two or more joints: |
| characterized by swelling or effusion or tenderness in 2 or more joints and thirty minutes or more of morning stiffness. |
| Serositis: |
| typical pleurisy for > 1 day or pleural effusions or pleural rub or typical pericardial pain (pain with recumbency improved by sitting forward) for > 1 day or pericardial effusion or pericardial rub or pericarditis by EKG (in the absence of other causes, such as infection, uremia, and Dressler’s pericarditis) |
| Renal: |
| Urine protein:creatinine ratio (or 24 hr urine protein) representing 500 mg of protein/24 hr or red blood cell casts |
| Neurologic: |
| seizures, psychosis, mononeuritis multiplex (in the absence of other known causes such as primary vasculitis), myelitis, peripheral or cranial neuropathy (in the absence of other known causes such as primary vasculitis, infection, and diabetes mellitus), acute confusional state (in the absence of other causes, including toxic-metabolic, uremia, drugs) |
| Hemolytic anemia |
| Leukopenia: < 4000/mm3 at least once (in the absence of other known causes such as Felty’s, drugs, portal hypertension) |
| OR |
| Lymphopenia: < 1000/mm3 at least once (in the absence of other known causes such as corticosteroids, drugs and infection) |
| Thrombocytopenia: <100,000/mm3 at least once (in the absence of other known causes such as drugs, portal hypertension, TTP) |
| Clinical Criteria |
|---|
| Acute cutaneous lupus including: |
| lupus malar rash (do not count if malar discoid), bullous lupus, toxic epidermal necrolysis variant of SLE, maculopapular lupus rash, photosensitive lupus rash, (in the absence of dermatomyositis), or subacute cutaneous lupus, nonindurated psoriaform and/or annular polycyclic lesions that resolve without scarring, although occasionally with postinflammatory dyspigmentation or telangiectasias) |
| Chronic cutaneous lupus including: |
| classical discoid rash, localized (above the neck), generalized (above and below the neck), hypertrophic, (verrucous) lupus, lupus panniculitis (profundus), mucosal lupus, lupus erythematosus tumidus, chilblains lupus, discoid lupus/lichen planus overlap |
| Oral ulcers: |
| Palate, buccal, tongue, or nasal ulcers (in the absence of other causes, such as vasculitis, Behcet’s disease, infection (herpes viruses), inflammatory bowel disease, reactive arthritis, acidic foods) |
| Nonscarring alopecia: |
| diffuse thinning or hair fragility with visible broken hairs (in the absence of other causes such as alopecia areata, drugs, iron deficiency and androgenic alopecia) |
| Synovitis involving two or more joints: |
| characterized by swelling or effusion or tenderness in 2 or more joints and thirty minutes or more of morning stiffness. |
| Serositis: |
| typical pleurisy for > 1 day or pleural effusions or pleural rub or typical pericardial pain (pain with recumbency improved by sitting forward) for > 1 day or pericardial effusion or pericardial rub or pericarditis by EKG (in the absence of other causes, such as infection, uremia, and Dressler’s pericarditis) |
| Renal: |
| Urine protein:creatinine ratio (or 24 hr urine protein) representing 500 mg of protein/24 hr or red blood cell casts |
| Neurologic: |
| seizures, psychosis, mononeuritis multiplex (in the absence of other known causes such as primary vasculitis), myelitis, peripheral or cranial neuropathy (in the absence of other known causes such as primary vasculitis, infection, and diabetes mellitus), acute confusional state (in the absence of other causes, including toxic-metabolic, uremia, drugs) |
| Hemolytic anemia |
| Leukopenia: < 4000/mm3 at least once (in the absence of other known causes such as Felty’s, drugs, portal hypertension) |
| OR |
| Lymphopenia: < 1000/mm3 at least once (in the absence of other known causes such as corticosteroids, drugs and infection) |
| Thrombocytopenia: <100,000/mm3 at least once (in the absence of other known causes such as drugs, portal hypertension, TTP) |
| Immunologic Criteria |
|---|
| ANA level above laboratory reference range |
| Anti-dsDNA antibody level above laboratory reference range (or > 2 fold the laboratory reference range if tested by ELISA) |
| Anti-Sm |
| Antiphospholipid antibody: |
| any of the following: lupus anticoagulant, false-positive rapid plasma regain (RPR), medium or high titer, nticardiolipin antibody level (IgG, IgM or IgA), anti-β2 glycoprotein I (IgG, IgM or IgA) |
| Low complement: |
| low C3, low C4, low CH50 |
| Direct Coombs’ test (in the absence of hemolytic anemia) |
| Immunologic Criteria |
|---|
| ANA level above laboratory reference range |
| Anti-dsDNA antibody level above laboratory reference range (or > 2 fold the laboratory reference range if tested by ELISA) |
| Anti-Sm |
| Antiphospholipid antibody: |
| any of the following: lupus anticoagulant, false-positive rapid plasma regain (RPR), medium or high titer, nticardiolipin antibody level (IgG, IgM or IgA), anti-β2 glycoprotein I (IgG, IgM or IgA) |
| Low complement: |
| low C3, low C4, low CH50 |
| Direct Coombs’ test (in the absence of hemolytic anemia) |
Patients can be classified as having SLE if they satisfy four of the clinical and immunological criteria, including at least one clinical criterion and one immunologic criterion, OR if they have biopsy-proven nephritis compatible with SLE in the presence of ANAs or anti-dsDNA antibodies. Reproduced from Petri M et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 64:2677–86. Copyright 2012. With permission from John Wiley & Sons [258]. TTP: thrombocytopaenic purpura; anti-Sm: anti-Smith antibodies.
Clinical and Immunologic Criteria Used in the SLICC Classification Criteria for SLEa
| Clinical Criteria |
|---|
| Acute cutaneous lupus including: |
| lupus malar rash (do not count if malar discoid), bullous lupus, toxic epidermal necrolysis variant of SLE, maculopapular lupus rash, photosensitive lupus rash, (in the absence of dermatomyositis), or subacute cutaneous lupus, nonindurated psoriaform and/or annular polycyclic lesions that resolve without scarring, although occasionally with postinflammatory dyspigmentation or telangiectasias) |
| Chronic cutaneous lupus including: |
| classical discoid rash, localized (above the neck), generalized (above and below the neck), hypertrophic, (verrucous) lupus, lupus panniculitis (profundus), mucosal lupus, lupus erythematosus tumidus, chilblains lupus, discoid lupus/lichen planus overlap |
| Oral ulcers: |
| Palate, buccal, tongue, or nasal ulcers (in the absence of other causes, such as vasculitis, Behcet’s disease, infection (herpes viruses), inflammatory bowel disease, reactive arthritis, acidic foods) |
| Nonscarring alopecia: |
| diffuse thinning or hair fragility with visible broken hairs (in the absence of other causes such as alopecia areata, drugs, iron deficiency and androgenic alopecia) |
| Synovitis involving two or more joints: |
| characterized by swelling or effusion or tenderness in 2 or more joints and thirty minutes or more of morning stiffness. |
| Serositis: |
| typical pleurisy for > 1 day or pleural effusions or pleural rub or typical pericardial pain (pain with recumbency improved by sitting forward) for > 1 day or pericardial effusion or pericardial rub or pericarditis by EKG (in the absence of other causes, such as infection, uremia, and Dressler’s pericarditis) |
| Renal: |
| Urine protein:creatinine ratio (or 24 hr urine protein) representing 500 mg of protein/24 hr or red blood cell casts |
| Neurologic: |
| seizures, psychosis, mononeuritis multiplex (in the absence of other known causes such as primary vasculitis), myelitis, peripheral or cranial neuropathy (in the absence of other known causes such as primary vasculitis, infection, and diabetes mellitus), acute confusional state (in the absence of other causes, including toxic-metabolic, uremia, drugs) |
| Hemolytic anemia |
| Leukopenia: < 4000/mm3 at least once (in the absence of other known causes such as Felty’s, drugs, portal hypertension) |
| OR |
| Lymphopenia: < 1000/mm3 at least once (in the absence of other known causes such as corticosteroids, drugs and infection) |
| Thrombocytopenia: <100,000/mm3 at least once (in the absence of other known causes such as drugs, portal hypertension, TTP) |
| Clinical Criteria |
|---|
| Acute cutaneous lupus including: |
| lupus malar rash (do not count if malar discoid), bullous lupus, toxic epidermal necrolysis variant of SLE, maculopapular lupus rash, photosensitive lupus rash, (in the absence of dermatomyositis), or subacute cutaneous lupus, nonindurated psoriaform and/or annular polycyclic lesions that resolve without scarring, although occasionally with postinflammatory dyspigmentation or telangiectasias) |
| Chronic cutaneous lupus including: |
| classical discoid rash, localized (above the neck), generalized (above and below the neck), hypertrophic, (verrucous) lupus, lupus panniculitis (profundus), mucosal lupus, lupus erythematosus tumidus, chilblains lupus, discoid lupus/lichen planus overlap |
| Oral ulcers: |
| Palate, buccal, tongue, or nasal ulcers (in the absence of other causes, such as vasculitis, Behcet’s disease, infection (herpes viruses), inflammatory bowel disease, reactive arthritis, acidic foods) |
| Nonscarring alopecia: |
| diffuse thinning or hair fragility with visible broken hairs (in the absence of other causes such as alopecia areata, drugs, iron deficiency and androgenic alopecia) |
| Synovitis involving two or more joints: |
| characterized by swelling or effusion or tenderness in 2 or more joints and thirty minutes or more of morning stiffness. |
| Serositis: |
| typical pleurisy for > 1 day or pleural effusions or pleural rub or typical pericardial pain (pain with recumbency improved by sitting forward) for > 1 day or pericardial effusion or pericardial rub or pericarditis by EKG (in the absence of other causes, such as infection, uremia, and Dressler’s pericarditis) |
| Renal: |
| Urine protein:creatinine ratio (or 24 hr urine protein) representing 500 mg of protein/24 hr or red blood cell casts |
| Neurologic: |
| seizures, psychosis, mononeuritis multiplex (in the absence of other known causes such as primary vasculitis), myelitis, peripheral or cranial neuropathy (in the absence of other known causes such as primary vasculitis, infection, and diabetes mellitus), acute confusional state (in the absence of other causes, including toxic-metabolic, uremia, drugs) |
| Hemolytic anemia |
| Leukopenia: < 4000/mm3 at least once (in the absence of other known causes such as Felty’s, drugs, portal hypertension) |
| OR |
| Lymphopenia: < 1000/mm3 at least once (in the absence of other known causes such as corticosteroids, drugs and infection) |
| Thrombocytopenia: <100,000/mm3 at least once (in the absence of other known causes such as drugs, portal hypertension, TTP) |
| Immunologic Criteria |
|---|
| ANA level above laboratory reference range |
| Anti-dsDNA antibody level above laboratory reference range (or > 2 fold the laboratory reference range if tested by ELISA) |
| Anti-Sm |
| Antiphospholipid antibody: |
| any of the following: lupus anticoagulant, false-positive rapid plasma regain (RPR), medium or high titer, nticardiolipin antibody level (IgG, IgM or IgA), anti-β2 glycoprotein I (IgG, IgM or IgA) |
| Low complement: |
| low C3, low C4, low CH50 |
| Direct Coombs’ test (in the absence of hemolytic anemia) |
| Immunologic Criteria |
|---|
| ANA level above laboratory reference range |
| Anti-dsDNA antibody level above laboratory reference range (or > 2 fold the laboratory reference range if tested by ELISA) |
| Anti-Sm |
| Antiphospholipid antibody: |
| any of the following: lupus anticoagulant, false-positive rapid plasma regain (RPR), medium or high titer, nticardiolipin antibody level (IgG, IgM or IgA), anti-β2 glycoprotein I (IgG, IgM or IgA) |
| Low complement: |
| low C3, low C4, low CH50 |
| Direct Coombs’ test (in the absence of hemolytic anemia) |
Patients can be classified as having SLE if they satisfy four of the clinical and immunological criteria, including at least one clinical criterion and one immunologic criterion, OR if they have biopsy-proven nephritis compatible with SLE in the presence of ANAs or anti-dsDNA antibodies. Reproduced from Petri M et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 64:2677–86. Copyright 2012. With permission from John Wiley & Sons [258]. TTP: thrombocytopaenic purpura; anti-Sm: anti-Smith antibodies.
These revised SLICC lupus criteria have been accepted by the European Medicines Agency, the US Food and Drug Administration and NHS England as being suitable for the inclusion of patients in clinical trials and in the commissioning policy for rituximab. They are more intuitive than the previous ACR classification criteria when considering a diagnosis of lupus, and allow a larger number of patients to meet criteria; however, diagnosis should not be restricted to patients who meet the classification criteria, as they can encompass other manifestations in the appropriate serological context [259]. The SLICC criteria have been tested in a number of cohorts and in most studies have shown an increase in sensitivity and reduced specificity, so care is needed if features are better explained by an alternative diagnosis [260–263].
Conclusions
When considering a patient with a possible diagnosis of lupus, a detailed clinical history and examination is required in order to identify relevant clinical features, including assessment of haematological and renal parameters. The diagnosis should not be made without evidence of at least one autoantibody or low complement levels to support the diagnosis of this autoimmune disease, consistent with the SLICC classification criteria. The ACR (Table 4) and SLICC (Table 5) classification criteria are not diagnostic criteria but may be helpful when considering the diagnosis; however, they do not cover all the clinical manifestations of lupus. The LOEs and GORs for parameters supporting the diagnosis of lupus are shown in Table 1.
Recommendations for the assessment of SLE patients
Clinical manifestations in SLE patients may be due to disease activity, damage, drug toxicity or the presence of co-morbidity. In the case of disease activity, it is important to ascertain whether this is due to active inflammation or thrombosis, as this will define treatment strategies (LOE 2 ++, GOR B, SOA 97%).
Clinical assessment of a lupus patient should include a thorough history and review of systems, full clinical examination and monitoring of vital signs, urinalysis, laboratory tests, assessment of health status and quality of life, and measurement of disease activity and damage using standardized SLE assessment tools (2 ++/B). Imaging (4/D), renal (2 ++/B) and other biopsies (4/D) should be performed where indicated (SOA 100%).
Disease activity is categorized into mild, moderate and severe, with the occurrence of flares (2+/C). Mild disease activity is clinically stable lupus with no life-threatening organ involvement, mainly manifesting as arthritis, mucocutaneous lesions and mild pleuritis. Patients with moderate disease activity have more serious manifestations, and severe disease is defined as organ- or life-threatening (4/D) (SOA 93%).
Rationale
Assessment of lupus
A systematic approach should be taken because of the diversity and complexity of clinical and laboratory manifestations (supplementary Table S3, available at Rheumatology Online) [264–266]. Clinical manifestations may be due to one or any combination of the following: disease activity from active inflammation or thrombosis, acute drug toxicity, chronic damage due to the effects of the disease or its treatment (such as lung fibrosis or atherosclerosis), or co-morbidity (e.g. infection). It is important to take a detailed history and to perform a clinical examination, including vital signs and urinalysis, to establish the likely differential diagnoses and then to organize the relevant investigations as suggested in Table 6, depending on the circumstances. In addition, when assessing disease activity with a view to planning treatment, it is necessary to determine the circumstances that may have led to a lupus flare (such as exposure to sunlight, concurrent or recent infection, hormonal changes, or timing of previous disease-related therapeutic change) as this will guide further investigation, treatment change (including non-drug measures) and disease monitoring required thereafter.
Assessment and monitoring of SLE in lupus patients
| Item | Initial assessment | Assessment (active disease) | Monitoring (stable disease) | Pregnancy |
|---|---|---|---|---|
| Patients with active disease should be reviewed at least every 1–3 months | Patients with stable/low disease activity should be reviewed every 6–12 months | Pregnancy counselling and follow-up | ||
| History and examination | ||||
| Detailed history | X | focused history | focused history | obstetric history |
| Clinical examination | X | X | Xa | X |
| Vital signs (Blood pressure, heart rate, weight) | X | X | X | X |
| Drug review including vaccination status | X | X | X | X |
| Bloods | ||||
| Full blood count | X | X | X | X |
| Other tests for anaemia | Xa | Xa | Xa | Xa |
| Renal function | X | X | X | X |
| Bone profile | X | Xa | Xa | X |
| Liver function tests | X | Xa | Xa | X |
| Creatine kinase | X | Xa | Xa | Xa |
| CRP | X | Xa | Xa | Xa |
| Vitamin D3 | X | Xa | Annually | X |
| Thyroid function | X | Xa | Xa | X |
| Immunology | ||||
| ANA | X | – | – | Xa |
| Anti-dsDNA titre, C3/C4 level | X | X | X | X |
| aPL (LA, aCL, anti-beta2-glycoptroteinI) | X | Xa | Xa | Repeat if negative in the past |
| Anti-Ro/La, anti-RNP and anti-Sm antibodies | X |
| – | Repeat if negative in the past |
| Immunoglobulins | X | Xa | Annuallya | Xa |
| Direct Coombs' test | X | Xa | Xa | Xa |
| Urine | ||||
| Urinalysis (screen for proteinuria, haematuria, leucocyturia and nitrites to exclude infection) | X | X | X | X |
| Urine random protein:creatinine ratio Or 24-h urine collection for protein | Xa | Xa | Xa | Xa |
| Urine microscopy (and culture) | Xa | Xa | Xa | Xa |
| Other investigations | ||||
| Microbiology (other) | Xa | Xa | Xa | Xa |
| Biopsy (e.g. skin, kidney) | Xa | Xa | Xa | Xb |
| Lung function tests | Xa | Xa | Xa | Xa |
| Neurophysiology | Xa | Xa | Xa | Xa |
| ECG | X | Xa | Xa | Xa |
| Imaging | ||||
| Chest X-ray | X | Xa | Xa | Xb |
| Other imaging (US, CT, MRI) | Xa | Xa | Xa | Xb |
| Modifiable cardiovascular risk factors | ||||
| Hypertension | X | Xa | Annually | X |
| Dyslipidaemia | X | Xa | Annually | Xa |
| Diabetes mellitus | X | Xa | Annually | X |
| High BMI | X | Xa | Annually | X |
| Smoking | X | Xa | Annually | X |
| Disease activity and damage scores | ||||
| BILAG (BILAG 2004 index) or | X | Xa | Annually | BILAG2004Pc |
| SLEDAI (SLEDAI–2K or SELENA SLEDAI) | X | Xa | Annually | SLEPDAId |
| SLICC/ACR Damage Index | X | Xa | Annually | X |
| Quality of life questionnaires | ||||
| Short-form 36 or LupusQoL | X | Xa | Annually | Xa |
| Item | Initial assessment | Assessment (active disease) | Monitoring (stable disease) | Pregnancy |
|---|---|---|---|---|
| Patients with active disease should be reviewed at least every 1–3 months | Patients with stable/low disease activity should be reviewed every 6–12 months | Pregnancy counselling and follow-up | ||
| History and examination | ||||
| Detailed history | X | focused history | focused history | obstetric history |
| Clinical examination | X | X | Xa | X |
| Vital signs (Blood pressure, heart rate, weight) | X | X | X | X |
| Drug review including vaccination status | X | X | X | X |
| Bloods | ||||
| Full blood count | X | X | X | X |
| Other tests for anaemia | Xa | Xa | Xa | Xa |
| Renal function | X | X | X | X |
| Bone profile | X | Xa | Xa | X |
| Liver function tests | X | Xa | Xa | X |
| Creatine kinase | X | Xa | Xa | Xa |
| CRP | X | Xa | Xa | Xa |
| Vitamin D3 | X | Xa | Annually | X |
| Thyroid function | X | Xa | Xa | X |
| Immunology | ||||
| ANA | X | – | – | Xa |
| Anti-dsDNA titre, C3/C4 level | X | X | X | X |
| aPL (LA, aCL, anti-beta2-glycoptroteinI) | X | Xa | Xa | Repeat if negative in the past |
| Anti-Ro/La, anti-RNP and anti-Sm antibodies | X |
| – | Repeat if negative in the past |
| Immunoglobulins | X | Xa | Annuallya | Xa |
| Direct Coombs' test | X | Xa | Xa | Xa |
| Urine | ||||
| Urinalysis (screen for proteinuria, haematuria, leucocyturia and nitrites to exclude infection) | X | X | X | X |
| Urine random protein:creatinine ratio Or 24-h urine collection for protein | Xa | Xa | Xa | Xa |
| Urine microscopy (and culture) | Xa | Xa | Xa | Xa |
| Other investigations | ||||
| Microbiology (other) | Xa | Xa | Xa | Xa |
| Biopsy (e.g. skin, kidney) | Xa | Xa | Xa | Xb |
| Lung function tests | Xa | Xa | Xa | Xa |
| Neurophysiology | Xa | Xa | Xa | Xa |
| ECG | X | Xa | Xa | Xa |
| Imaging | ||||
| Chest X-ray | X | Xa | Xa | Xb |
| Other imaging (US, CT, MRI) | Xa | Xa | Xa | Xb |
| Modifiable cardiovascular risk factors | ||||
| Hypertension | X | Xa | Annually | X |
| Dyslipidaemia | X | Xa | Annually | Xa |
| Diabetes mellitus | X | Xa | Annually | X |
| High BMI | X | Xa | Annually | X |
| Smoking | X | Xa | Annually | X |
| Disease activity and damage scores | ||||
| BILAG (BILAG 2004 index) or | X | Xa | Annually | BILAG2004Pc |
| SLEDAI (SLEDAI–2K or SELENA SLEDAI) | X | Xa | Annually | SLEPDAId |
| SLICC/ACR Damage Index | X | Xa | Annually | X |
| Quality of life questionnaires | ||||
| Short-form 36 or LupusQoL | X | Xa | Annually | Xa |
When indicated;
when indicated and benefit > risks;
BILAG2004 pregnancy version;
SLEDAI pregnancy version. Anti-Sm antibodies: anti-Smith antibodies.
Assessment and monitoring of SLE in lupus patients
| Item | Initial assessment | Assessment (active disease) | Monitoring (stable disease) | Pregnancy |
|---|---|---|---|---|
| Patients with active disease should be reviewed at least every 1–3 months | Patients with stable/low disease activity should be reviewed every 6–12 months | Pregnancy counselling and follow-up | ||
| History and examination | ||||
| Detailed history | X | focused history | focused history | obstetric history |
| Clinical examination | X | X | Xa | X |
| Vital signs (Blood pressure, heart rate, weight) | X | X | X | X |
| Drug review including vaccination status | X | X | X | X |
| Bloods | ||||
| Full blood count | X | X | X | X |
| Other tests for anaemia | Xa | Xa | Xa | Xa |
| Renal function | X | X | X | X |
| Bone profile | X | Xa | Xa | X |
| Liver function tests | X | Xa | Xa | X |
| Creatine kinase | X | Xa | Xa | Xa |
| CRP | X | Xa | Xa | Xa |
| Vitamin D3 | X | Xa | Annually | X |
| Thyroid function | X | Xa | Xa | X |
| Immunology | ||||
| ANA | X | – | – | Xa |
| Anti-dsDNA titre, C3/C4 level | X | X | X | X |
| aPL (LA, aCL, anti-beta2-glycoptroteinI) | X | Xa | Xa | Repeat if negative in the past |
| Anti-Ro/La, anti-RNP and anti-Sm antibodies | X |
| – | Repeat if negative in the past |
| Immunoglobulins | X | Xa | Annuallya | Xa |
| Direct Coombs' test | X | Xa | Xa | Xa |
| Urine | ||||
| Urinalysis (screen for proteinuria, haematuria, leucocyturia and nitrites to exclude infection) | X | X | X | X |
| Urine random protein:creatinine ratio Or 24-h urine collection for protein | Xa | Xa | Xa | Xa |
| Urine microscopy (and culture) | Xa | Xa | Xa | Xa |
| Other investigations | ||||
| Microbiology (other) | Xa | Xa | Xa | Xa |
| Biopsy (e.g. skin, kidney) | Xa | Xa | Xa | Xb |
| Lung function tests | Xa | Xa | Xa | Xa |
| Neurophysiology | Xa | Xa | Xa | Xa |
| ECG | X | Xa | Xa | Xa |
| Imaging | ||||
| Chest X-ray | X | Xa | Xa | Xb |
| Other imaging (US, CT, MRI) | Xa | Xa | Xa | Xb |
| Modifiable cardiovascular risk factors | ||||
| Hypertension | X | Xa | Annually | X |
| Dyslipidaemia | X | Xa | Annually | Xa |
| Diabetes mellitus | X | Xa | Annually | X |
| High BMI | X | Xa | Annually | X |
| Smoking | X | Xa | Annually | X |
| Disease activity and damage scores | ||||
| BILAG (BILAG 2004 index) or | X | Xa | Annually | BILAG2004Pc |
| SLEDAI (SLEDAI–2K or SELENA SLEDAI) | X | Xa | Annually | SLEPDAId |
| SLICC/ACR Damage Index | X | Xa | Annually | X |
| Quality of life questionnaires | ||||
| Short-form 36 or LupusQoL | X | Xa | Annually | Xa |
| Item | Initial assessment | Assessment (active disease) | Monitoring (stable disease) | Pregnancy |
|---|---|---|---|---|
| Patients with active disease should be reviewed at least every 1–3 months | Patients with stable/low disease activity should be reviewed every 6–12 months | Pregnancy counselling and follow-up | ||
| History and examination | ||||
| Detailed history | X | focused history | focused history | obstetric history |
| Clinical examination | X | X | Xa | X |
| Vital signs (Blood pressure, heart rate, weight) | X | X | X | X |
| Drug review including vaccination status | X | X | X | X |
| Bloods | ||||
| Full blood count | X | X | X | X |
| Other tests for anaemia | Xa | Xa | Xa | Xa |
| Renal function | X | X | X | X |
| Bone profile | X | Xa | Xa | X |
| Liver function tests | X | Xa | Xa | X |
| Creatine kinase | X | Xa | Xa | Xa |
| CRP | X | Xa | Xa | Xa |
| Vitamin D3 | X | Xa | Annually | X |
| Thyroid function | X | Xa | Xa | X |
| Immunology | ||||
| ANA | X | – | – | Xa |
| Anti-dsDNA titre, C3/C4 level | X | X | X | X |
| aPL (LA, aCL, anti-beta2-glycoptroteinI) | X | Xa | Xa | Repeat if negative in the past |
| Anti-Ro/La, anti-RNP and anti-Sm antibodies | X |
| – | Repeat if negative in the past |
| Immunoglobulins | X | Xa | Annuallya | Xa |
| Direct Coombs' test | X | Xa | Xa | Xa |
| Urine | ||||
| Urinalysis (screen for proteinuria, haematuria, leucocyturia and nitrites to exclude infection) | X | X | X | X |
| Urine random protein:creatinine ratio Or 24-h urine collection for protein | Xa | Xa | Xa | Xa |
| Urine microscopy (and culture) | Xa | Xa | Xa | Xa |
| Other investigations | ||||
| Microbiology (other) | Xa | Xa | Xa | Xa |
| Biopsy (e.g. skin, kidney) | Xa | Xa | Xa | Xb |
| Lung function tests | Xa | Xa | Xa | Xa |
| Neurophysiology | Xa | Xa | Xa | Xa |
| ECG | X | Xa | Xa | Xa |
| Imaging | ||||
| Chest X-ray | X | Xa | Xa | Xb |
| Other imaging (US, CT, MRI) | Xa | Xa | Xa | Xb |
| Modifiable cardiovascular risk factors | ||||
| Hypertension | X | Xa | Annually | X |
| Dyslipidaemia | X | Xa | Annually | Xa |
| Diabetes mellitus | X | Xa | Annually | X |
| High BMI | X | Xa | Annually | X |
| Smoking | X | Xa | Annually | X |
| Disease activity and damage scores | ||||
| BILAG (BILAG 2004 index) or | X | Xa | Annually | BILAG2004Pc |
| SLEDAI (SLEDAI–2K or SELENA SLEDAI) | X | Xa | Annually | SLEPDAId |
| SLICC/ACR Damage Index | X | Xa | Annually | X |
| Quality of life questionnaires | ||||
| Short-form 36 or LupusQoL | X | Xa | Annually | Xa |
When indicated;
when indicated and benefit > risks;
BILAG2004 pregnancy version;
SLEDAI pregnancy version. Anti-Sm antibodies: anti-Smith antibodies.
Validated instruments for the assessment of lupus
The most reliable way of assessing disease activity is to use a defined instrument for this purpose that has been validated and is available with an appropriate glossary and scoring instructions [265, 266]. For example, the NHS England Interim Clinical Commissioning Policy Statement for rituximab in lupus published in 2013 [267] recommended the use of two lupus-specific disease activity indices: the BILAG index and the SLEDAI. For such purposes, the currently recommended revised versions are the BILAG-2004 index [268, 269] (for BILAG-2004 index data collection form, glossary and scoring see supplementary data, available at Rheumatology Online) and SLEDAI-2K [270] or the SELENA-SLEDAI [271, 272] (see supplementary data, available at Rheumatology Online, for SLEDAI-2K and SELENA-SLEDAI index data collection forms). Modifications have been made for use in pregnancy [273, 274]. For optimal performance, training in the use of these instruments is advised. It is essential that only manifestations/items due to SLE disease activity are recorded and that the data collection forms are used in conjunction with the appropriate glossary and scoring rules. There is one validated instrument for assessing damage, the SLICC/ACR Damage Index (SDI) [275]. It is recommended that patients’ assessment of their disease be captured using health status or quality of life questionnaires such as the generic Short-form36 (SF-36), which has been validated for use in lupus patients [276], or a lupus-specific questionnaire such as the Lupus Quality of Life (LupusQoL) [277]. There is agreement that for best practice these instruments should be used [74, 278], although there are no data confirming that their use improves the outcomes for patients. Better outcomes are achieved if lupus in-patients are managed in centres with experience in managing lupus [279–282].
Definitions of mild, moderate and severe lupus
For the purpose of planning appropriate treatment, disease activity has been broadly categorized as mild, moderate or severe [8], and worsening disease activity is termed flare, which can be similarly categorized as mild, moderate or severe [283, 284]. Examples are shown in Table 7. The term mild disease activity reflects clinically stable disease with no life-threatening organ involvement and that is not likely to cause significant scarring or damage. Examples of scores for such patients when using formal assessment tools would include a SLEDAI-2K score of <6 [270] and/or one BILAG B score [269]. Patients with moderate disease have more serious manifestations, which if left untreated would cause significant chronic scarring. Examples of scores for such patients when using formal assessment tools would include a SLEDAI-2K score in the range of 6–12 [270] and/or two or more BILAG B scores [269]. Severe disease is defined as organ or life threatening and reflects the most serious form of systemic disease that requires potent immunosuppression. Examples of scores for such patients when using formal assessment tools would include a SLEDAI-2K score of >12 [270] and/or at least one BILAG A score [269].
SLE treatment strategies for examples of mild, moderate and severe lupus
| Item | Mild activity/flare BILAG C scores or single B score; SLEDAI <6 | Moderate activity/flare BILAG 2 or more systems with B scores, SLEDAI 6–12 | Severe activity/flare (non-renal) BILAG 1 or more A scores; SLEDAI >12 |
|---|---|---|---|
| Typical manifestations attributed to lupus | Fatigue, malar rash, diffuse alopecia, mouth ulcers, arthralgia, myalgia, platelets 50–149 × 109/l | Fever, lupus-related rash up to 2/9 body surface area, cutaneous vasculitis, alopecia with scalp inflammation, arthritis, pleurisy, pericarditis, hepatitis, platelets 25–49 × 109/l | Rash involving >2/9 body surface area, myositis, severe pleurisy and/or pericarditis with effusion, ascites, enteritis, myelopathy, psychosis, acute confusion, optic neuritis, platelets <25 × 109/l |
| Initial typical drugs and target doses if no contra-indications |
|
|
|
| Aiming for typical maintenance drugs/doses providing no contra-indications |
|
|
|
| Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission |
| Item | Mild activity/flare BILAG C scores or single B score; SLEDAI <6 | Moderate activity/flare BILAG 2 or more systems with B scores, SLEDAI 6–12 | Severe activity/flare (non-renal) BILAG 1 or more A scores; SLEDAI >12 |
|---|---|---|---|
| Typical manifestations attributed to lupus | Fatigue, malar rash, diffuse alopecia, mouth ulcers, arthralgia, myalgia, platelets 50–149 × 109/l | Fever, lupus-related rash up to 2/9 body surface area, cutaneous vasculitis, alopecia with scalp inflammation, arthritis, pleurisy, pericarditis, hepatitis, platelets 25–49 × 109/l | Rash involving >2/9 body surface area, myositis, severe pleurisy and/or pericarditis with effusion, ascites, enteritis, myelopathy, psychosis, acute confusion, optic neuritis, platelets <25 × 109/l |
| Initial typical drugs and target doses if no contra-indications |
|
|
|
| Aiming for typical maintenance drugs/doses providing no contra-indications |
|
|
|
| Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission |
The lowest effective dose of prednisolone or other CSs should be used at all times.
SLE treatment strategies for examples of mild, moderate and severe lupus
| Item | Mild activity/flare BILAG C scores or single B score; SLEDAI <6 | Moderate activity/flare BILAG 2 or more systems with B scores, SLEDAI 6–12 | Severe activity/flare (non-renal) BILAG 1 or more A scores; SLEDAI >12 |
|---|---|---|---|
| Typical manifestations attributed to lupus | Fatigue, malar rash, diffuse alopecia, mouth ulcers, arthralgia, myalgia, platelets 50–149 × 109/l | Fever, lupus-related rash up to 2/9 body surface area, cutaneous vasculitis, alopecia with scalp inflammation, arthritis, pleurisy, pericarditis, hepatitis, platelets 25–49 × 109/l | Rash involving >2/9 body surface area, myositis, severe pleurisy and/or pericarditis with effusion, ascites, enteritis, myelopathy, psychosis, acute confusion, optic neuritis, platelets <25 × 109/l |
| Initial typical drugs and target doses if no contra-indications |
|
|
|
| Aiming for typical maintenance drugs/doses providing no contra-indications |
|
|
|
| Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission |
| Item | Mild activity/flare BILAG C scores or single B score; SLEDAI <6 | Moderate activity/flare BILAG 2 or more systems with B scores, SLEDAI 6–12 | Severe activity/flare (non-renal) BILAG 1 or more A scores; SLEDAI >12 |
|---|---|---|---|
| Typical manifestations attributed to lupus | Fatigue, malar rash, diffuse alopecia, mouth ulcers, arthralgia, myalgia, platelets 50–149 × 109/l | Fever, lupus-related rash up to 2/9 body surface area, cutaneous vasculitis, alopecia with scalp inflammation, arthritis, pleurisy, pericarditis, hepatitis, platelets 25–49 × 109/l | Rash involving >2/9 body surface area, myositis, severe pleurisy and/or pericarditis with effusion, ascites, enteritis, myelopathy, psychosis, acute confusion, optic neuritis, platelets <25 × 109/l |
| Initial typical drugs and target doses if no contra-indications |
|
|
|
| Aiming for typical maintenance drugs/doses providing no contra-indications |
|
|
|
| Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission | Aim to reduce and stop drugs except HCQ eventually when in stable remission |
The lowest effective dose of prednisolone or other CSs should be used at all times.
Conclusions
The assessment of a patient with lupus, as with making the initial diagnosis, is dependent on a careful history and examination of the patient, with relevant haematological, biochemical and immunological testing as well as other investigations as necessary (shown in Table 6) to establish the degree of disease activity and accumulation of chronic damage, and to identify other complications or co-morbid conditions that will influence the treatment plan. The LOEs and GORs for the components of the assessment and monitoring of lupus disease are shown in Table 1.
Recommendations for monitoring of SLE
Patients with lupus should be monitored on a regular basis for disease manifestations, drug toxicity and co-morbidities (LOE 2 ++, GOR B, SOA 99%).
Those with active disease should be reviewed at least every 1–3 months (2+, C/D), with blood pressure (1+/A), urinalysis (1+/A), renal function (1+/A), anti-dsDNA antibodies (2 ++/B), complement levels (2+/C), CRP (2+/C), full blood count (3/C), and liver function tests (4/D) forming part of the assessment, and further tests as necessary (4/D). Patients with stable low disease activity or in remission can be reviewed less frequently, for example, 6–12 monthly (4/D) (SOA 99%).
The presence of aPLs is associated with thrombotic events, damage, and adverse outcomes in pregnancy (2 ++/B). If previously negative, they should be re-evaluated prior to pregnancy or surgery, or in the presence of a new severe manifestation or vascular event (4/D) (SOA 96%).
Anti-Ro and anti-La antibodies are associated with neonatal lupus (including CHB) and should be checked prior to pregnancy (1+/A) (SOA 100%).
Patients with lupus are at increased risk of co-morbidities, such as atherosclerotic disease, osteoporosis, avascular necrosis, malignancy and infection (2+/C). Management of modifiable risk factors, including hypertension, dyslipidaemia, diabetes, high BMI and smoking, should be reviewed at baseline and at least annually (4/D) (SOA 98%).
Immunosuppressive therapy may lead to toxicities. Close monitoring of drugs by regular laboratory tests and clinical assessment should be performed in accordance with drug monitoring guidelines (4/D) (SOA 98%).
Rationale
Frequency of monitoring lupus/follow-up visits
There are no randomized controlled trials (RCTs) comparing different monitoring strategies in terms of frequency and details of assessments performed; however, data from various cohort studies have informed our expert opinion and previous guidelines in this respect [22, 71, 74, 278]. Patients should be told to report to clinicians if they develop any new or significant worsening of clinical manifestations. In most patients with active clinical disease, clinic visits should be approximately every 4 weeks initially, reducing gradually down to about 3-monthly reviews as the disease comes under control. There remains a significant risk of flare and the development of damage, even for patients who achieve early remission [72]. For most patients with mild features, including those who are clinically quiet but serologically active, 3-monthly visits are adequate [77]. Review should become more frequent if the disease becomes more active, especially if there is renal involvement, as the patients will require clinical, renal and serological evaluation (see below) [285]. For patients with inactive disease, without previous renal involvement or organ damage (that can predict increased risk of further active disease and damage), review may be less frequent, for example every 6 months providing treatment is stable and suitable drug monitoring is in place [74]. Patients should be seen more regularly, however, if treatment is being withdrawn or has been stopped, due to the risk of disease flare, even if they appear to be in remission [72].
Reasons for clinical monitoring in lupus patients
Regular monitoring of clinical and laboratory features of active disease should take place, with additional investigations as necessary (Table 6), to assess and monitor changes in disease activity, the development of chronic damage, and to detect the presence of (and changes in) co-morbid conditions that may be confused with lupus (such as FM, hypothyroidism, iron deficiency anaemia, infection), and drug-induced conditions [22, 74, 265]. LOEs for the laboratory parameters are shown in Table 1. Proteinuria (and renal function in particular [24]), high DAS [16, 48, 73, 286], new and different types of cutaneous lesions [50], arthritis [72], NP disease [16, 51] and cytopenias [52, 53] have been shown to correlate with disease severity and can predict future flares and the development of damage [11, 32, 49, 54, 55]. Only measurement of proteinuria and renal function have been shown to have strong predictive value for outcome [22, 24, 56]. Chest X-ray, ECG and other specific tests such as lung function, echocardiography and neurophysiology should be repeated during the course of the disease as necessary. When major organs are involved, additional imaging (such as brain MRI) and pathology (renal/skin biopsy) can add significant prognostic information, particularly renal biopsy, and may need to be repeated to assess response to treatment [22–24, 287, 288].
Interpretation of haematological, renal and other biochemical parameters
Lymphopenia is a common manifestation of lupus (supplementary Table S3, available at Rheumatology Online), and some patients will have leucopenia and neutropenia regularly with active disease [53]. This needs to be remembered when monitoring patients on cytotoxic therapy, as a fall in cell counts may signify the need to increase therapy for lupus rather than reduce or discontinue therapy if drug toxicity is suspected. It also means that the usual drug-monitoring limits of tolerance may need to be reviewed and personalized in the context of an individual with SLE. Thrombocytopenia may be acute and indicative of a disease flare, or low grade and chronic as part of of lupus and/or associated with APS [57].
ESR is often raised in active SLE [70], but can also reflect persistent polyclonal hypergammaglobulinaemia, and is not a reliable marker of disease activity. CRP is usually normal [66–68] or slightly elevated in the presence of serositis or arthritis [69]. A significantly raised CRP is more likely to indicate infection, and patients with raised CRP will need therefore to be thoroughly screened for infection, given that infection is the commonest cause of death in lupus patients. In contrast, a raised ESR does not discriminate between active lupus and infection [69]. Immune complexes of CRP and anti-CRP antibodies may form in lupus patients, possibly explaining the low levels of CRP observed with active disease [67].
Proteinuria should be quantified using the urine protein:creatinine ratio or 24-h urine collection. Microscopic examination of the urine to look for red cells and red cell casts is useful for identifying active renal disease and renal flares, but the assessment of casts is now rarely done [24, 289, 290]. When assessing haematuria, it is important to exclude infection, menstrual blood loss and calculi. White cells in the urine are most often due to urine or vaginal infection and can be hard to interpret, but as an otherwise unexplained finding, are associated with active tubulointerstitial inflammation.
Serum immunoglobulins should be measured prior to starting drugs such as MMF, CYC and rituximab which have the most risk of inducing immunoglobulin deficiency that might increase the risk of infection. The initial repeat measurement of the serum immunoglobulins should take place about 3–6 months later and can then be spaced out to annual checks [74, 199, 291, 292, 293]. Specific antibodies, for example, pneumococcal antibodies, may be assessed (if tests are available) to assess the need for and response to immunization. Screening for chronic infections (such as TB, hepatitis B and C, HIV, HPV) is recommended before starting immunosuppressants and repeated if reactivation of infection is suspected.
It is important to measure creatinine kinase at baseline and to continue to follow it in patients with myositis or myalgias that might be due to lupus or statins used to prevent atherosclerosis [75]. Monitoring of cholesterol and of other lipids, and remaining vigilant for and treating the development of diabetes mellitus and features of the metabolic syndrome (which may increase cardiovascular risk, particularly in patients on glucocorticoids), are important and should be as successful as in the general population [71, 74, 76]. Additional monitoring investigations should include Vitamin D3, which is often low as a consequence of sun avoidance and/or chronic kidney disease [294]. Vitamin D is required for optimal bone health, especially in patients on chronic glucocorticoid therapy and/or following the menopause [295]. Clinicians should have a low threshold for assessing thyroid function, as hypothyroidism can present with similar features to lupus; it co-exists with lupus in ∼7% of patients, and thyroid antibodies are found in 14% [296–298].
Monitoring of lupus autoantibodies and complement
Serial anti-dsDNA antibodies and C3 and C4 levels are useful because rising, high anti-dsDNA antibodies and falling, low complement levels are associated with flare [49, 58], particularly in patients with LN [24]. In general, concomitantly rising anti-dsDNA titres [39, 43, 46, 49, 59, 60] and decreasing C3 and/or C4 levels [43–46] are more important predictors of current or impending flares than the absolute levels, and levels of anti-dsDNA antibodies may actually fall at the time of flare [299].
It can be helpful to combine a sensitive but less specific anti-dsDNA antibody assay (e.g. ELISA) with one that only measures more specific, high affinity or high avidity antibodies (such as Farr radioimmunoassay or the Crithidia test), because only tests measuring high affinity and high avidity antibodies are strongly associated with renal disease; however, other ELISAs can be used to monitor disease activity [40]. Stable active serology without clinical features does not necessarily warrant therapy [71], but patients need to be followed closely, with individual care decisions made to prevent over- or undertreatment. Many physicians would avoid reducing therapy in this situation as patients may develop renal disease [300], but the serological tests do not always predict flare [61, 62, 71]. About 40% of lupus patients do not have anti-dsDNA antibodies, so for this group of patients, they are not useful for monitoring disease activity [63]. Some patients are heterozygous for the C4 allele and due to a null allele have a persistently low C4 level (at about 50% of normal), without having active disease, but C4 levels can still fluctuate with disease activity.
ANA, anti-Sm and anti-RNP antibodies tests should be carried out at baseline and do not need to be repeated at each visit, as levels do not fluctuate with disease activity. Anti-Ro and anti-La antibodies should be measured in women planning pregnancy or in early pregnancy, as they may be transferred across the placenta and are associated with CHB in ∼1–2% of babies [64, 65]. Fetal heart-rate monitoring should be instituted from week 16 of pregnancy and continued throughout pregnancy in women with either of these antibodies. Neonatal lupus rash develops in ∼10% of babies born to mothers with these antibodies (especially if exposed to UV light), and laboratory abnormalities (cytopenias and abnormal liver function tests) have also been observed in babies exposed to these antibodies [64].
aPLs should be assessed at baseline and, if previously negative, they should be re-evaluated in the presence of a new vascular event, adverse pregnancy outcome or other new manifestation that might have a thrombotic component, as well as prior to a planned pregnancy [47, 241, 252, 253]. Positive tests for APS include LA, aCL (IgG, IgM) and/or anti-beta-2 glycoprotein 1(IgG, IgM), and these tests should be repeated after 12 weeks to confirm positivity [241, 252], although LA cannot be evaluated if anticoagulation has been started, as this would interfere with the assay.
Monitoring for the development of co-morbidities
Patients with lupus are at increased risk of co-morbidities [71, 74], such as infection, premature cardiovascular and peripheral vascular disease, osteoporosis, avascular necrosis and some malignancies (non-Hodgkin’s lymphoma, cervical, vulval, lung and thyroid cancer [301, 302]). The management of these issues is beyond the scope of this guideline and should follow national/international guidelines for each condition and include appropriate vaccinations [22, 71, 74, 278]. Nevertheless, screening for and managing these conditions is an integral part of the assessment and regular monitoring of lupus patients, as described in the EULAR recommendations for monitoring patients with SLE in clinical practice and in observational studies [74]. A preventative approach should be adopted, since the commonest causes of death in lupus patients in the UK are infection and cardiovascular disease, followed by malignancy [15, 16, 18]. Modifiable risk factors for co-morbidities to address include vaccination status, hypertension, dyslipidaemia, diabetes, high BMI and smoking. These should be reviewed at baseline and at least annually thereafter [22, 24, 71, 74]. These co-morbidities may occur at a younger age than in the normal population, and clinicians should screen regularly for them, even though there are no RCTs to suggest that more intense screening than that applied in the general population improves outcome in lupus patients [22, 24, 71, 74]. Routine cancer screening (particularly for cervical cancer, given the increased risk of HPV infection in lupus patients [303]) should not be forgotten due to emphasis on lupus disease management [304].
Monitoring of drugs
This should be similar to that for drugs used in other rheumatic diseases, but due to the occurrence of cytopenias and abnormal renal and liver function possibly caused by lupus disease itself, monitoring tests may need to be undertaken more frequently, and the interpretation of laboratory results is more difficult. Adherence to drugs may be confirmed by measuring drug levels (e.g. of ciclosporin, tacrolimus, mycophenolate [171] and HCQ [80]), but these tests are not widely available (except that for tacrolimus, which is tested in order to guide optimal dosing and to prevent renal toxicity). There is little lupus-specific data about target drug levels, and detailed discussion is beyond the scope of these recommendations, but this topic has been reviewed for rheumatic diseases in general [78] as well as for lupus [305]. It should be noted that, like other chronic conditions, adherence levels are suboptimal in lupus, and therefore specific consideration of this issue is needed in patients showing poor response to therapy [79].
Conclusions
It is important to monitor lupus patients regularly to assess and monitor changes in disease activity, chronic damage, and in drug-induced and co-morbid conditions that may be confused with lupus and that are associated with an increased risk of death. The LOEs and GORs for the main components of monitoring of lupus patients are shown together in Table 1, and a suggested protocol is shown in Table 6.
Recommendations for the management of mild SLE
Treatments to be considered for the management of mild non–organ-threatening disease include the disease-modifying drugs HCQ (1 ++/A) and MTX (1+/A), and short courses of NSAIDs (3/D) for symptomatic control. These drugs allow for the avoidance of or dose reduction of CSs (SOA 94%).
Prednisolone treatment at a low dose of ⩽7.5 mg/day may be required for maintenance therapy (2+/C). Topical preparations may be used for cutaneous manifestations, and IA injections for arthritis (4/D) (SOA 93%).
High–Sun Protection Factor (SPF) UV-A and UV-B sunscreen are important in the management and prevention of UV radiation–induced skin lesions (2 ++/B). Patients must also be advised about sun avoidance and the use of protective clothing (4/D) (SOA 97%).
Rationale
Overview of treatment of mild lupus
Mild lupus features (Table 7) are distressing for patients and warrant treatment to relieve symptoms and signs. Such treatment may prevent progression to severe manifestations requiring more intense immunosuppression. These manifestations can be managed with CSs, HCQ and other antimalarials, MTX, NSAIDs and sunscreens. The LOEs and GORs for the drugs used to treat lupus disease are summarized in Table 2, and the SOAs with the recommendations are above. There are little data to support the use of topical therapies, dapsone, retinoids, thalidomide or danazol in the treatment of refractory cutaneous lupus rashes and vasculitis, and as these drugs are not used for other systemic features of lupus, they are not discussed here but have been reviewed [287, 288].
CSs for mild lupus
Summary
Topical preparations should be used initially for cutaneous manifestations, and intra-articular (IA) or intramuscular (i.m.) injections of CSs for arthritis. Short courses of oral prednisolone (up to 20 mg/day) are used for short periods of time (up to 14 days and reduced rapidly) to induce remission in some cases of mild lupus where local treatment is not sufficient or practical (evidence discussed below in moderate lupus). Prednisolone can be used in women who are trying to conceive, are pregnant or are breast-feeding [239].
Evidence
There are no RCTs comparing different types of CS administration, such as skin creams and ointments, intralesional, IA and i.m. injections, and oral CS drugs (usually prednisolone in the UK). CSs contribute to the development of chronic damage and co-morbidities such as cataracts, osteoporotic fractures, diabetes, atherosclerosis and infection [12, 14]. It has been shown that a 1 mg/day increase in maintenance prednisone dose is associated with a 2.8% increase in the risk of new organ damage, and that prednisolone dosing of ⩽7.5 mg/day is associated with less risk of cataracts, osteoporotic fractures and cardiovascular damage than higher doses [306].
Conclusions
The lowest possible dose/amount of CSs should be used due to their side effects, including the risk of contributing to chronic damage and infection. Prednisolone treatment at a low dose of ⩽7.5 mg/day may be required for maintenance therapy and has less risk of side effects than higher doses (2+/C).
HCQ and other anti-malarial agents
Summary
There is good evidence (Table 2) for the efficacy and safety of HCQ, the most commonly prescribed anti-malarial agent and one of the few licensed drugs for lupus. Providing that the patient has normal renal and liver function, HCQ can be used at doses of up to 6.5 mg/kg/day and is compatible with pregnancy and breast-feeding. It is used (Table 7) for skin and joint involvement, myalgia, fever, fatigue, pleurisy, to reduce the development of renal disease and chronic damage [14, 121] and for its steroid-sparing properties (even in patients with more severe disease) [71]. Chloroquine is used if HCQ is not available or not tolerated; however, there is less evidence for benefit and it has a greater risk of retinal toxicity than HCQ [121]. Mepacrine (quinacrine) is used predominantly for cutaneous lupus and has the least risk of ocular toxicity [287, 307–309].
Evidence
The benefits of anti-malarials on lupus activity were reported in four RCTs [81–84], five prospective cohort studies [87–91], three retrospective cohort studies [92–94] and an open-label extension of the first RCT [95]. There have been two other double-blind RCTs confirming that lupus rashes significantly improve with HCQ [85] and chloroquine [86]. The cohort studies have shown that response often takes 3–4 months [94], but at 6 months only 60% of patients with discoid rash show some response [94]. Another study showed that 20% of patients with an adequate response lose it within 2 years and need other therapies [310]. Higher drug levels were associated with increased cutaneous response in a prospective study [311]. In a double-blind RCT [80], low drug levels were associated with increased disease activity. Systemic features and smoking are also associated with an increased risk of poor response [94, 96, 122].
Many of the studies showing increased flare rates in patients who discontinued HCQ involved pregnant patients. A RCT in lupus patients [84] and two prospective [87, 90] cohort studies support the use of this drug before conception and in pregnancy to reduce flares in the mother. Although HCQ can cross the placenta, exposure is not associated with significant adverse effects on the fetus [87, 90, 97–100]. HCQ has anti-thrombotic as well as anti-inflammatory properties and by reducing disease activity in the mother may improve the outcome for the child by improving placental function [101, 102]. There is increasing evidence that HCQ reduces the risk of CHB in babies born to mothers with anti-Ro antibodies [103, 312, 313]. Further evidence supporting the use of HCQ in pregnant women as well as in those planning pregnancy and breast-feeding is reviewed in the BSR Guidelines on drugs in pregnancy in the rheumatic diseases [239].
There is further evidence from high-quality prospective and retrospective cohort studies that patients treated with anti-malarials (particularly HCQ) not only have lower levels of overall lupus activity and reduced rates of flare [80, 81, 84, 89, 90, 95], but can be managed with lower doses of CSs [83, 84, 90, 104]. The patients are more likely to stay clinically quiescent if HCQ is continued when the disease goes in to remission [105]. Patients on MMF are more likely to achieve renal remission if treated with HCQ [93]. Patients on HCQ are less likely to develop serious renal disease and have delayed time to renal damage [104], lower frequency of seizures [106] and less NP damage [107], greater delay in integument damage [108], less overall damage [109, 110] and, most importantly, improved survival [111, 112]. Some of the benefits on survival may be mediated by the beneficial effects of anti-malarials on total cholesterol, LDL-cholesterol, triglycerides, glucose [113] and/or by the prevention of thrombosis [101, 102, 121] and atherosclerotic plaque formation [114].
Patients take HCQ on average for about 6 years [115–118]. In general HCQ is well tolerated and better tolerated than chloroquine [86, 115, 116, 121]. The commonest adverse effects of anti-malarials are gastrointestinal, but a few patients stop because of headache, dizziness, itching, rash, non-retinal eye problems, hearing loss, myopathy or other rare neuromuscular side effects [121, 287]. The most serious adverse events are cardiac (which are very rare) [119] and retinopathy (which is more common with chloroquine than HCQ) [121, 314]. Retinopathy is unpredictable but unlikely with <7 years treatment with HCQ. It is more common thereafter [120] and with doses of HCQ above 6.5 mg/kg/day, or renal or liver impairment. It requires active screening to detect it early when it is asymptomatic and is most likely to be reversible [120, 314]. Policies on screening for ocular toxicity vary between countries and local guidelines should be followed [314, 315]. In general in the UK, baseline and yearly optician eye tests are recommended initially, with more detailed ophthalmological screening after 5 years of therapy [316].
Conclusions
There are good data from two systematic reviews and a meta-analysis including 7 RCTs and 36 cohort studies supporting the use of HCQ in lupus patients to reduce disease activity and as a steroid-sparing agent: overall LOE 1 ++, GOR A. HCQ should be given to all patients with mild lupus to prevent flares, the development of damage and to improve survival. It is recommended that HCQ be continued or started, even in those developing disease severe enough to warrant immunosuppressive therapies, including LN [22, 24, 25]. However patients with renal or liver dysfunction should have the dose reduced [314]. It is compatible with conception, pregnancy and breast-feeding. Unfortunately, it has a long half-life and takes at least 2 months to be effective [287, 309]. Patients need to be warned about this or they may discontinue the drug prematurely.
MTX in mild SLE
Summary
Although not licensed for the treatment of lupus, low-dose weekly MTX (⩽25 mg/week) has been used to reduce mild and moderate disease activity in lupus, particularly to control inflammatory arthritis and lupus skin rashes, originally on the basis of a variety of case series and cohort studies [317, 318]. MTX was originally used in patients who had failed HCQ and low-dose CSs, but it can be used with HCQ to avoid CSs or to promote CS dose reduction. Caution has been advised on the use of MTX in patients with LN, particularly as those with renal impairment will be at increased risk of MTX toxicity [317]. It is contra-indicated in women trying to conceive or pregnant as it is teratogenic. For these patients AZA would be more suitable (see section on moderate lupus for evidence).
Evidence
A systematic review by Sakthiswary and Suresh [319] summarizes the data from three controlled trials (two double-blind, placebo-controlled trials [123, 124], and a controlled open-label trial comparing MTX and chloroquine [125]) and five observational studies (two open-label prospective studies [126, 127]; a cross-sectional study [128]; a retrospective case–control cohort study [129]; and an open-label controlled study [130]). Another systematic review [133] includes two additional case series [131, 132]. These studies support the use of MTX to reduce mild and moderate lupus disease activity, and some demonstrated steroid-sparing properties. Some of these studies showed benefit specifically in treating lupus arthritis, rashes, vasculitis, serositis, myositis and constitutional symptoms, but there was little change in ESR, anti-dsDNA antibodies, C3 or C4 levels, except in a study with longer duration than previous studies [130]. The reduction in SLEDAI in the five controlled studies reporting these data included in the systematic review [319] was calculated to have an odds ratio = 0.444 (95% CI: 0.279, 0.707; P = 0.001). The analysis of the four controlled studies reporting steroid-sparing properties for MTX provided an odds ratio = 0.335 (95% CI: 0.202, 0.558; P = 0.001). Side effects led to discontinuation in ∼10% of patients but were not serious. It is teratogenic and should not be used in women within 3 months of planning to conceive, or who are pregnant or breast-feeding [239], nor in patients with renal impairment, because reduced renal function increases the risk of adverse events, particularly bone marrow suppression.
Conclusions
There are good data from two systematic reviews including three RCTs and seven cohort studies supporting the use of MTX in lupus to reduce disease activity and as a steroid-sparing agent: overall LOE 1+, GOR A.
NSAIDs in mild SLE
Summary
There are no RCTs of NSAIDs in SLE. Publications support the cautious use of NSAIDs for short periods of time for symptom control in SLE (inflammatory arthralgia, myalgia, chest pain and fever) where potential benefit outweighs the known risks of NSAIDs and paracetamol has been insufficient or not tolerated. The risk of NSAID-induced acute renal failure is increased in patients with LN, so NSAIDs should be avoided in patients with renal involvement. NSAID-induced allergic reactions, aseptic meningitis, cutaneous reactions and hepatotoxicity are increased in SLE patients. Caution is required in pregnancy [240].
Evidence
A review of the literature on non-selective Cox inhibitors and selective Cox-2 inhibitors [320] highlighted the potential increased risk of renal, hepatic and neurological toxicity in lupus patients. A retrospective case series assessing celecoxib, with a detailed literature review of NSAIDs [321] and a more comprehensive systematic review addressing the risk–benefit ratio of non-selective and selective inhibitors of cyclooxygenases in SLE patients, were published subsequently [134]. More recently it has become clear that NSAIDs (except possibly naproxen) can predispose to acute myocardial infarction in individuals with coronary heart disease [322], which is an additional reason for caution in lupus patients.
Conclusions
Based on one systematic review of the evidence from case series and case reports, the overall LOE for NSAIDs in non-renal mild lupus is three and GOR is D.
High-SPF UV-A and UV-B sunblock in SLE
Summary
There is clear evidence that ultraviolet radiation (UV-A and UV-B) can induce various forms of cutaneous lupus [287]. Patients with systemic lupus without cutaneous features have also been found to have an abnormal reaction to UV irradiation [323].
Evidence
Sunscreens were shown to prevent discoid and subacute cutaneous lupus rashes in a case series [141] and to reduce systemic features such as renal disease, thrombocytopenia and hospitalization in a cohort study [136]. Three open-label controlled trials [137–139], a retrospective case series [140] and a double-blind, controlled trial [135] have shown that sunscreens that block UV-A and UV-B can reduce UV radiation–induced lesions of cutaneous lupus.
Conclusions
Lupus patients should be advised about avoidance of sun and other sources of UV irradiation, and about the use of sunscreens (UV-A protection five stars and UV-B protection from SPF factors 30 to 50 products, which can be prescribed on the NHS) and protective clothing. Overall, the LOE is 2++ for sunscreens (one small RCT and six other studies) in lupus patients to prevent cutaneous lesions, and the GOR is B.
Recommendations for the management of moderate SLE
The management of moderate SLE involves higher doses of prednisolone (up to 0.5 mg/kg/day) (2+/C), or the use of i.m. (4/D) or i.v. doses of methylprednisolone (MP) (2+/C). Immunosuppressive agents are often required to control active disease and are steroid-sparing agents (2+/C). They can also reduce the risk of long-term damage accrual (4/D) (SOA 98%).
MTX (1+/A), AZA (2+/C), MMF (2 ++/B), ciclosporin (2+/C) and other calcineurin inhibitors (3/D) should be considered in cases of arthritis, cutaneous disease, serositis, vasculitis or cytopaenias if HCQ is insufficient (SOA 97%).
For refractory cases, belimumab (1+/B) or rituximab (2+/C) may be considered (SOA 98%).
Rationale
Overview of the management of moderate lupus
Immunosuppressive cytotoxic agents should be used with CSs, while continuing anti-malarials and avoidance of UV radiation, to reduce disease activity in moderate lupus (Table 7), prevent the risk of further flares and lower the risk of damage accrual due to disease and CSs, because they act as steroid-sparing agents. Despite their widespread use in clinical practice and as background standard of care therapy in clinical trials, there are only a few RCTs demonstrating the efficacy of CSs and other immunosuppressive agents for the management of moderate lupus. Additional drugs should be considered if HCQ is insufficient or not tolerated and can be used in addition to HCQ. The evidence supporting the use of MTX has been discussed above, and the evidence supporting the use of CSs, AZA, MMF, calcineurin inhibitors (ciclosporin and tacrolimus) and LEF are discussed in this section. For patients who do not respond to these drugs, the biologic drugs rituximab and belimumab may be considered. It should be noted that there is a specific NHS England 2013 Interim Clinical Commissioning Policy Statement for rituximab in adult SLE patients [267], and NICE guidance for the use of belimumab in active autoantibody-positive SLE in adults has been published in 2016 [324]. Patients being considered for these drugs should be discussed with and/or seen by a specialist lupus centre with experience in using these drugs. The patients should meet specific criteria and be entered in to the BILAG Biologics Register (see below and Fig. 1). For patients not requiring biologics, suggested initial target dosing regimens for active disease (as used in most studies) and lower maintenance dosing regimens to prevent recurrence of disease once patients are stable are shown in Table 7. The actual regimen used for individual patients will depend on the clinical picture and the treatment history. It is important to increase the dose and/or change treatment if patients fail to respond in the expected time frame. The LOEs and GORs for all the drugs used to treat lupus are summarized in Table 2.
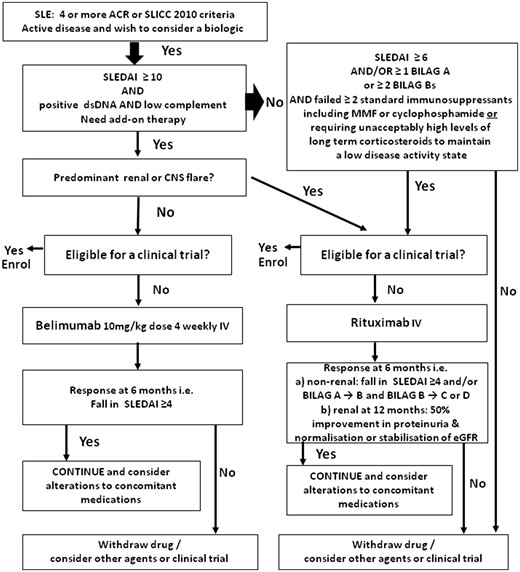
Summary of NICE and NHS England guidance for the use of belimumab and rituximab in patients with SLE
Belimumab is licensed and NICE-approved (Belimumab for active autoantibody-positive systemic lupus erythematosus: TA397, published June 2016) and should be considered first [324]. Rituximab is not licensed and should only be used according to the NHS England Interim Clinical Commissioning Policy Statement: rituximab for the treatment of systemic lupus erythematosus in adults: published September 2013 A13/PS/a [267]. All patients receiving either drug must be enrolled in the BILAG Biologics Register and be managed at or in collaboration with a specialized centre.
CSs for moderate lupus
Summary
Higher doses of oral CSs are required initially than are required for mild lupus, for example prednisolone at up to 0.5 mg/kg/day, and intermittent treatment with i.m. 80–120 mg MP or even i.v. doses of MP (up to 250 mg) are used as well as, or instead of, oral prednisolone to promote a quicker response with less total CS exposure. Prednisolone dosing should be reduced, as disease activity improves, to the lowest possible maintenance dose and stopped, if possible, as other immunosuppressive agents take effect over several weeks or months.
Evidence
There are no data comparing different oral CS regimens for the treatment of moderate lupus. Two controlled studies have shown that treating patients who are clinically stable but showing serological deterioration with a short course of moderate-dose CSs (e.g. 30 mg/day) can prevent more flares than placebo and lead to improvement in serological markers [46, 60]. However, there is a risk of treating patients that will not flare, and this approach is not recommended due to the side effects of CSs.
There are some data supporting the use of 100 mg i.v. MP pulses in non-renal lupus as an alternative to 1000 mg pulses [143], and for 1000 mg pulses on three occasions in patients with moderate or severe lupus, with very little oral prednisolone [146]. The data supporting the use of i.v. pulses of 500 or 1000 mg are discussed further below in the section on the management of severe lupus [148, 326]. There is one open-label RCT [142] comparing triamcinolone 100 mg given as an i.m. injection with a short course of oral MP tapered over 1 week. Overall, there was little difference between the regimens but some improvement was seen more quickly with the triamcinolone injection.
Conclusions
Overall the LOE for CSs by i.m. or i.v. injection in non-renal moderate lupus is 2+ and GOR is C.
AZA for moderate lupus (non-renal disease)
Summary
AZA is not licensed for the treatment of lupus, but has been used for over 40 years, and it is the most frequently used cytotoxic agent [327] in lupus. AZA treatment (1–2.5 mg/kg/day orally) has been associated with prevention of flares and a reduction in CS dosage (see below and Table 2). It is usually started in patients with moderate lupus activity (Table 7) in conjunction with CSs, as it can take up to 3 months to be effective. It is also used for maintenance therapy after remission or significant response has been achieved with other agents used to treat severe lupus (such as CYC) that are less suitable for long-term therapy, particularly in women desiring pregnancy, or who are pregnant or breast-feeding [24, 25, 239, 328]. Most of the evidence (and the only double-blind RCTs) supporting its use relate to the management of LN [24, 25]. Only papers discussing the management of non-renal lupus with AZA are discussed here, although in some cases the studies included renal and non-renal patients. There is no evidence that it prevents atherosclerosis or other forms of damage [12, 329].
Evidence
The first reports of AZA being used for renal and non-renal manifestations of lupus with CSs appeared in the late 1960s and 1970s [149–151, 153, 330, 331]. Reduction in disease activity and flare rate and steroid-sparing effects were demonstrated in most of these open-label, controlled studies and in a case series [158]. AZA 200 mg daily was associated with an increased risk of significant liver dysfunction. There was no increased risk of infection, even starting at 3–4 mg/kg/day, but subsequent studies have used 2–2.5 mg/kg/day.
A prospective longitudinal open-label study [154] involving 17 SLE patients showed that AZA reduced lupus activity and anti-dsDNA antibody levels. Subsequently, in a retrospective study [155] with 61 SLE patients, suppression of anti-dsDNA antibodies by AZA (2 mg/kg/day) and low-dose prednisolone (7–12 mg/day) was associated with efficacy and better long-term outcome. However, the presence of renal disease, persistence of anti-dsDNA antibodies for at least 1 year after the beginning of treatment and reduction in AZA dosage to below 2 mg/kg/day predicted flares and was associated with a higher rate of lupus-related death.
An open-label, multicentre, RCT study of 89 SLE patients requiring 15 mg or more of prednisolone compared AZA (mean dose 2.1 mg/kg/day) with ciclosporin (mean dose 2.2 mg/kg/day) for its steroid-sparing properties [152]. The absolute mean change in prednisolone dose at 12 months, adjusted for baseline prednisolone dose, was not significantly different: 9.0 mg for ciclosporin (95% CI: 7.2, 10.8) and 10.7 mg for AZA (95% CI: 8.8, 12.7). There was no difference between groups in change in disease activity or number of flares, development of new damage, change in quality of life or numbers of patients discontinuing study drugs due to adverse events or lack of efficacy [152]. The conclusion was that both drugs can be used in lupus for their steroid-sparing properties, with appropriate monitoring.
AZA is usually well tolerated [332]. The main adverse events are nausea and vomiting, diarrhoea, flu-like illness with fever, rash, leucopenia and hepatotoxicity [156, 157, 332–334]. Side effects can occur soon after starting AZA and may require drug withdrawal [156, 335]. Hepatic veno-occlusive disease is a rare adverse event, but autoimmune hepatitis can improve on AZA, so this is not a contra-indication to its use [157]. AZA is not excreted by the kidney, and it can be used in patients with renal impairment. Managing patients with lupus-related leucopenia with AZA can be difficult [332, 336]. The enzyme thiopurine S-methyltransferase (TPMT) catalyses the inactivation of AZA. It is worth testing patients for TPMT [334] before starting AZA, as the very low level phenotype (homozygous deficiency that occurs in 0.3% Caucasians) is associated with potentially life-threatening bone marrow toxicity; otherwise, weekly full blood counts are required as the dose is increased over several weeks [337, 338]. Those patients with intermediate TPMT levels due to a heterozygous state have an increased risk of leucopenia as well, and such testing does not remove the need for monitoring the effects of the drug on the full blood count [156, 332] and liver function according to national or local guidelines [337, 338].
AZA does not cause infertility and has not been found to be teratogenic in clinical practice, despite theoretical concerns [339, 340]; thus, it can be used in women planning conception and is compatible with pregnancy and breast-feeding [24, 98, 239]. It may reduce the response to some immunizations [341–344], but this is not a contra-indication to immunization except with live viruses [74, 292]. There is no evidence that AZA increases the risk of malignancy in lupus patients [301, 345], but it may increase the risk of cervical dysplasia [346].
Conclusions
Although the data for AZA in non-renal lupus are much weaker than the data supporting its use in LN (see below), there are four open-label RCTs, three prospective cohort studies, two retrospective cohort studies and one case series supporting the use of AZA for non-renal lupus: overall LOE 2+, GOR C.
MMF for moderate lupus (non-renal disease)
Summary
There are increasing data showing that MMF in combination with CSs reduces moderate and severe lupus disease activity, reduces renal and non-renal flares, is associated with CS-sparing properties and is tolerated well (see Tables 2 and 7 for suggested treatment strategies). However, there are no placebo-controlled double-blind RCTs specifically designed to assess the use of MMF in non-renal lupus. It is teratogenic and is contra-indicated in women trying to conceive, or who are pregnant or breast-feeding.
Evidence
The first systematic review of MMF (2–3 g daily) in non-renal lupus was published by Mok in 2007 [170] and reviewed 20 papers in terms of the response of specific clinical features (up to 2006) and steroid-sparing properties. This systematic review included patients mostly refractory to other therapies who were treated with MMF in uncontrolled studies for arthritis, renal, haematological and cutaneous manifestations, and a few with neuropsychiatric manifestations, and also covered the use of MMF in prevention of flare in a small prospective study of patients with rising anti-dsDNA antibody levels [162–164, 347].
A later systematic review [133] with a literature search up to end of October 2011 provided further evidence that MMF treatment is associated with reductions in disease activity, flare rate and prednisone dose and included data from five cohort studies [162–166] and from the Aspreva Lupus Management Study (ALMS) trial in LN that specifically reported on non-renal lupus manifestations (see below) [159]. Further supporting evidence for MMF comes from a small case series [169] and a study [348] showing that mycophenolic acid (MPA) levels vary between patients and that higher trough levels were associated with less risk of disease flare. MPA levels were more closely associated with efficacy and safety than the dose of MMF. This test is available in some hospitals, but the target trough level of 3.5–4.5 mg/l was recommended to be tested in a controlled trial before being widely applied.
The beneficial effects of MMF on non-renal disease activity [159] were demonstrated in a 6-month open-label RCT (ALMS) that compared oral MMF (target dose 3 g/day, median exposure 2.6 g/day) with pulses of i.v. CYC (0.5–1.0 g/month) as induction treatment for biopsy-proven LN [349]. All patients received prednisone starting at 60 mg/day that was tapered to 10 mg/day. There was induction of remission in >80% of patients treated with MMF for active disease at baseline in mucocutaneous, musculoskeletal, cardiorespiratory and vasculitis systems in addition to renal response in 56% (the primary end point) [349]. There were no flares in the patients on MMF, and complement levels and titres of anti-dsDNA antibodies normalized. Very similar renal and non-renal responses were seen in those given CYC [159]. However, more Black and Hispanic patients responded to MMF than i.v. CYC, and further trials are required to assess the role of race, ethnicity and geographical region on treatment response [350].
In the maintenance phase of ALMS [160], 227 patients from the 6-month induction study who met the renal clinical response criteria were randomized again to MMF (2 g/day) or AZA (2 mg/kg/day) in a 36-month, double-blind, double-dummy, phase III RCT [160]. Prednisolone ⩽10 mg/day or its equivalent was allowed and was taken by 90% of the MMF group (n = 116) and 87% of the AZA group (n = 111). Secondary end points included an analysis of non-renal severe flare. Severe non-renal flare rates did not differ between groups: 6.9% for the MMF group and 6.3% for the AZA group. There were no significant differences in the changes in anti-dsDNA antibodies or complement levels between groups. However, MMF was superior to AZA in various renal parameters related to maintaining a renal response and in preventing renal relapse in these LN patients, irrespective of which induction treatment had led to their initial response, race and geographical region [160]. Adverse events were common in both groups (>95%) (mostly minor infections and gastrointestinal disorders). Serious adverse events occurred in 24% of the MMF group and 33% of the AZA group (P = 0.11). The rate of withdrawal due to adverse events was lower with MMF than AZA (25% vs 40%, P = 0.02).
Another randomized open-label controlled trial [161], in Caucasians predominantly, compared MMF (mean 2 g/day) and AZA (mean 124 mg/day) for maintenance therapy over 36 months, starting at week 12 after induction with a short course of i.v. CYC (6 × 500 mg over 10 weeks) for the management of biopsy-proven proliferative LN. All patients initially received three i.v. pulses of MP and were tapered from 0.5 mg/kg/day prednisone down to 5 mg/day at week 52 and then tapered further and stopped if possible. Both regimens were well tolerated, and there was comparable improvement in renal end points and non-renal parameters, including disease activity indices and C3 levels in both groups. There were less renal flares and less haematological adverse events with MMF than AZA (though this was not statistically significant in this study).
Since the systematic review [133], further studies reporting reduction in disease activity included a retrospective review of patients treated with MMF that found a significant reduction in mean weekly steroid dosage (from about 12.5 to 3 mg/day prednisone) [167]. A single-centre retrospective cohort study [168] involving 135 patients with SLE (50% with renal disease) and 43 patients with systemic vasculitis treated with MMF reported good responses in 46% of patients, and the mean prednisolone dosage was significantly reduced from 22 to 8 mg/day at 12 months. These and other studies have shown that adverse events occur in up to 44% of patients over 5 years: mostly mild gastrointestinal intolerance and infections, with leucopenia and hospitalization rare. In one study most patients tolerated the drug well, with 73% of patients on the drug at 12 months, and there was no relationship between adverse events and dose (250 mg to 3 g daily) [351]. However, there have been increasing reports of teratogenicity, and it should be stopped at least 6 weeks before a planned pregnancy, and MMF should not be taken by women who are pregnant or breast-feeding [239].
Yahya et al. [172] reported on a small open-label prospective study of 14 non-renal lupus patients randomized to mycophenolate sodium (MS) or standard care and showed that MS treatment was safe and was associated with reduced disease activity. A randomized open-label trial [171] of 40 patients with primary systemic vasculitis or SLE compared MMF (2000 mg/day) and enteric-coated MS (1440 mg/day). The composite primary end point was treatment failure and/or drug intolerance over 12 months. MS was anticipated to be tolerated better, but no difference in tolerance was observed. Although MS was associated with slightly better efficacy, this may have been due to imbalance in factors affecting remission and relapse, despite randomization with minimization. This study did not support the use of MS as a better tolerated and efficacious alternative to MMF for routine use, but MS could be considered in patients with gastrointestinal side effects from MMF.
Conclusions
The evidence that MMF reduces disease activity, lupus flare and has steroid-sparing properties in non-renal lupus comes from two systematic reviews, three open-label RCTs in LN and seven cohort studies: LOE 2 ++, GOR B. MPA/sodium (MS) may be considered in patients intolerant of MMF based on two studies (LOE three, GOR D).
Ciclosporin and tacrolimus for moderate lupus (non-renal disease)
Summary
Ciclosporin and tacrolimus do not cause myelosuppression and have the ability to reduce moderate disease activity (Tables 2 and 7). There is more evidence for ciclosporin in non-renal lupus, and it has been particularly helpful in the treatment of cytopenias, where there is likely to be difficulty distinguishing cytopenias due to lupus from cytopenias due to drugs such as AZA, MTX and MMF. Both ciclosporin and tacrolimus can be used (at the lowest possible dose) in women planning pregnancy, and in those who are pregnant or breast-feeding [239].
Evidence
There are two open-label RCTs [152, 173] and eight non-renal cohort studies supporting the use of ciclosporin at doses of ⩽2.5 mg/kg/day in patients with normal renal function, although a systematic review [133] that included details of two open-label RCTs and a brief summary of six of the cohort studies reported that there was not much evidence supporting the use of ciclosporin in lupus because there were no double-blind, placebo-controlled RCTs.
Nevertheless, the open-label RCTs suggested that ciclosporin reduced disease activity as well as AZA did [152] and better than CSs alone [173], and that ciclosporin treatment was associated with significant CS-sparing properties in both RCTs, equivalent to that of AZA in one trial [152] as reported previously by the cohort studies. These included two prospective cohort studies [174, 175] that showed significant reduction in disease activity at 6 months, with most benefit in patients with renal and/or haematological manifestations, and response maintained to 24 months in one study [175]. Three retrospective studies [176–178] reported a reduction in disease activity and/or flares (particularly haematological manifestations such as thrombocytopenia), and significant steroid-sparing properties were reported in two of these studies [175, 177].
In the first of two additional studies not mentioned in the systematic review, ciclosporin was shown to treat thrombocytopenia in six patients [179], three of whom were able to stop CSs. In the second study [180], a retrospective cohort study, ciclosporin was used to manage 40 refractory lupus patients, including 11 patients with neurological conditions and 7 with overlap syndromes, as well as 18 with LN. The study showed reduction in disease activity and only mild transient adverse events not requiring discontinuation.
Adverse events were the focus of another study [181] with doses up to 5 mg/kg/day, so it was not surprising that adverse events were reported in 63%, but these led to discontinuation in only 16% and were reversible within 3 months of stopping the drug, consistent with many other reports. Ciclosporin treatment can cause hypertrichosis, gum hypertrophy, hypertension, paresthesiae, tremor, gastrointestinal symptoms and impaired renal function, especially at higher doses (>3 mg/kg/day). It is best used at lower doses (⩽2.5 mg/kg/day) as that is more tolerable and rarely causes permanent nephrotoxicity if carefully monitored. In the open-label RCT [152], there were no unexpected adverse events, and with appropriate monitoring of renal function and blood pressure, it was not discontinued due to adverse events or inefficacy more often than AZA.
There are two reports of tacrolimus in non-renal lupus and they were included in the systematic review [133]. The first was a small retrospective cohort study [182] with 10 non-renal patients showing significant reductions in SLEDAI and prednisolone over 1 year on 1–3 mg daily. The second was an open-label prospective study [183] with 21 mostly non-renal patients showing reduction in SLEDAI score over 6 months and no serious side effects, but 29% withdrew due to inefficacy and 10% due to adverse events.
Conclusions
Overall, the LOE for ciclosporin in non-renal lupus from two open-label RCTs, eight non-renal cohort studies and one systematic review is 2+ and GOR is C.
The LOE for tacrolimus from two studies in non-renal lupus and one systematic review is three and GOR is D.
LEF in moderate lupus
Summary
The systematic review [133] and our search found little evidence for efficacy and safety of LEF in lupus patients, with only two small studies in the literature. This drug can be considered in patients refractory to, not suitable for or intolerant of MTX, AZA, MMF and calcineurin inhibitors, for whom CYC, rituximab and belimumab are not suitable or not available. It is not suitable for women considering pregnancy, and a cholestyramine washout is required if pregnancy is desired or occurs while it is being taken [239].
Evidence
There was a randomized, double-blind, placebo-controlled trial in moderate SLE patients, with only six patients in each group [184]. A significant reduction in SLEDAI and prednisone occurred in both groups over 24 weeks. The LEF group showed significantly greater mean reduction in SLEDAI score, but there was no difference in steroid reduction between the groups. Side effects included transiently abnormal alanine aminotransferase (ALT), leucopenia and hypertension. There was a retrospective analysis of 18 patients who received LEF [185], but 4 patients withdrew (3 due to adverse events, including 1 with rash), and only 9/14 achieved lower SLEDAI scores after 2–3 months of therapy.
Conclusions
Overall the LOE for LEF for reducing non-renal lupus disease activity from two studies is three and the GOR is D. Caution is advised about its use in those with pre-existing subacute cutaneous lupus, as this may worsen as observed in other non-lupus studies.
Rituximab for refractory moderate lupus
Summary
Rituximab can be prescribed and reimbursed in the UK currently according to the NHS England 2013 Interim Clinical Commissioning Policy Statement for rituximab in adult SLE patients [267] who have two or more systems with BILAG B scores; or have severe BILAG A level disease activity, using the BILAG-2004 index [268, 269]; or have a SLEDAI-2 K score [270] >6 if they have failed two or more immunosuppressive agents (due to inefficacy or intolerance), at least one of which must be MMF or CYC; or need unacceptably high doses of steroids to achieve lower level of disease activity.
The patients must be managed in conjunction with a specialist centre for lupus and be entered in to the BILAG Biologics Register for standardized reporting of outcome (see Fig. 1 flowchart for eligibility and response criteria). This is essential for providing more open-label data in a prospective study with control patients treated with other immunosuppressive therapies, given the failure of the international double-blind, placebo-controlled lupus trials to meet their primary end points, as discussed below (EXPLORER for active non-renal disease [190, 191] and LUNAR for LN [352]). This policy was agreed as a result of the increasing published evidence supporting the efficacy of rituximab in refractory lupus patients, who are likely to differ from those recruited to trials where there was no requirement to have failed conventional therapy. Pregnancy should be avoided for at least 6 months after exposure to rituximab [239].
Evidence
The current evidence supporting the efficacy and safety of rituximab in non-renal lupus was most recently reported in a systematic review [200] in 2014 by Cobo-Ibanez with a literature search up to June 2013. This included the non-renal RCT EXPLORER [190] and its exploratory analysis [191], 2 open-label phase I/II trials [192, 193] and 22 cohort studies which analysed 1231 patients in total [200]. The 2 open-label trials [192, 193] and 5 of the cohort studies had been discussed in a previous systematic review summarizing off-label use in 188 cases (including non-renal and renal patients in 9 cohort studies and 26 case series/reports published up to December 2007) [202].
The non-renal patients discussed in the systematic review by Cobo-Ibáñez et al. [200] were heterogeneous, but in general had active lupus disease unresponsive to steroids and/or immunosuppressants prior to treatment with rituximab. Treatment with rituximab was associated with a reduction in global disease activity over 3–9 months, with 64–91% achieving response, including patients with a reduction in complement and anti-dsDNA antibody levels, arthritis and thrombocytopenia. Evidence for a steroid-sparing effect was based on the 2 open-label trials and 10 of the cohort studies [200]. There were few significant adverse events in the RCT, 2 open-label studies and 20 cohort studies [200]. Relapses/flares did occur at variable times (3.7–18 months), although in the RCT there were numerically fewer severe BILAG A flares and longer time to these flares in the rituximab group compared with the placebo group, and this almost achieved statistical significance (hazard ratio = 0.61, P = 0.052) [191]. Better clinical response after a second course was observed in 2 of the cohorts that studied retreatment [200], and a further report supported this observation and that steroid reduction occurred after each of two courses of rituximab [199]. The evidence for rituximab treating mucocutaneous involvement was deemed weak [200], and this may be explained by a recent report [353] specifically addressing 26 SLE patients with various subtypes of lupus rash, which observed that acute lupus rash responded whereas chronic cutaneous lupus (such as discoid rash) did not respond to rituximab and that new lesions with typical histology may appear despite confirmed B cell depletion.
Rituximab treatment early in the course of lupus disease, followed by AZA, was tried by Ezeonyeji et al. [194] specifically for its steroid-sparing effect in a pilot study with 8 SLE patients whose results were compared with 23 matched historical control patients treated conventionally [194]. Reduction in disease activity, a fall in anti-dsDNA antibodies and complement, and significant lower cumulative prednisolone at 6 months compared with controls was observed. There is also an open-label LN study suggesting that early rituximab with i.v. MP followed by MMF may avoid the use of oral CSs, and this regimen is currently being tested in a controlled randomized RCT called RITUXILUP [354].
The Duxbury systematic review and meta-analysis [201] reported response rates for various disease activity measures for patients in the open-label studies of refractory lupus treated with rituximab also reviewed by Cobo-Ibáñez et al. [200]. The Duxbury review and meta-analysis did include a section on LN (not discussed here) and included a few non-renal studies not in the Cobo-Ibáñez review, although the latter also included a few not in the Duxbury review. The BILAG index was used in 188 patients treated with rituximab in 8 open-label studies (3 prospective, 4 retrospective and 1 small case–control) [201]. The pooled global response in seven of these studies was 83%. The complete response rate was 47% and the partial response rate was 38% in six studies. A significant reduction in anti-dsDNA antibodies was observed in 6 of the 8 studies and a significant rise in complement was observed in 5 of 6 studies. Various versions of the SLEDAI were used in 513 patients treated with rituximab in 12 open-label studies: 5 prospective, 6 retrospective and 1 open-label randomized trial, only 1 of which also analysed BILAG response. With SLEDAI the global response was 77% in 11 studies. In 6 studies the complete response rate was 57% and the partial response rate was 31%. Anti-dsDNA levels fell in 3 of 3 studies and complement rose in 2 of 3 studies [201].
Publications from cohorts in Germany [195], Italy [196] and Japan [197] have confirmed similar levels of efficacy with various disease activity measures and provided further safety data in another 264 patients. Long-term follow-up of 98 SLE patients treated with rituximab over a 12-year period has shown in a retrospective analysis that the group with longer duration of depletion (⩾12 months) was associated with a better response (greater decrease in BILAG score at 6 and 12 months) than those with shorter period of B cell depletion [198].
The results of these open-label studies are much better than the response rates observed in the EXPLORER RCT (for rituximab vs placebo: complete 12% vs 16%, partial 17% vs 13%) [190]. However, EXPLORER used more stringent BILAG response criteria than used in any other study [201], but did observe a reduced rate and time to severe BILAG A flare [191]. High-dose CSs and background immunosuppression were used in both arms of the EXPLORER trial and may have reduced the ability to discriminate benefit from rituximab [201]. Patients on MTX as the background immunosuppressant derived more benefit from rituximab in a post hoc analysis than those in the placebo group [190], and in contrast to those on background AZA or MMF [190]. Patients of Afro-American or Hispanic origin were also shown to benefit from rituximab in the RCT, in contrast to Caucasians [190].
However, two case series reports have suggested that repeat courses of rituximab may increase the risk of hypogammaglobulinaemia and infection [199, 293]. Progressive multifocal leukoencephalopathy (PML) has been reported in 17 SLE patients, of whom 5 had been treated with rituximab. It seems likely that immunosuppression, however it is achieved, is the key factor in the development of PML. Lupus patients may be at increased risk of developing PML compared with other rheumatic diseases [355]. The risk of rituximab causing PML in rheumatic diseases, including RA and SLE, has been estimated at 5/100 000, which is less than the risk observed with some other immunosuppressants in other diseases [356].
Conclusions
There is now considerable evidence for the ability of rituximab to reduce disease activity in refractory non-renal SLE of moderate and severe severity, albeit mostly from cohort studies. There have been relatively few concerns in the individual reports and systematic reviews about adverse events, including infections, in lupus patients on rituximab. There is increasing evidence that rituximab has steroid-sparing properties, but further evidence for its use early in the disease course is needed. Overall, the LOE for rituximab from 3 systematic reviews (including a meta-analysis and 30 studies, including 1 RCT and 3 open-label trials for reducing disease activity and for steroid-sparing properties) is 2+ and the GOR is C.
Belimumab for refractory moderate lupus
Summary
There have been two large phase III RCTs [203, 204] investigating the use of belimumab in moderate–severe seropositive lupus (mostly musculoskeletal and cutaneous disease; as severe active renal and NPSLE disease were exclusions). All patients received steroids, HCQ and/or immunosuppressive drugs, with specific criteria for dosing changes allowed or contra-indicated in the protocol. Both trials showed a significantly increased proportion of responders to belimumab at a 10 mg/kg dose in addition to standard care. A variety of secondary end points were met, and there were no significant differences in adverse events, leading to the drug being approved and licensed by the US Food and Drug Administration and the European Medicines Agency. NICE guidance for use of belimumab in active autoantibody-positive SLE in adults has been published [324] and is summarized in Fig. 1. Patients must have positive anti-dsDNA antibodies, low complement and a SELENA-SLEDAI score ⩾10 despite standard therapy. Patients should be recruited to the BILAG Biologics Register so that outcomes can be recorded, and treatment with belimumab should not be continued for >24 weeks unless the SELENA-SLEDAI score has improved by 4 points or more. Pregnancy should not occur while on belimumab, but first trimester exposure is unlikely to be harmful [239].
Evidence
In the BLISS52 trial [203], at week 52 the response rate with placebo was 44%, with belimumab 1 mg/kg it was 51% (P = 0.013) and with 10 mg/kg it was 58% (P = 0.001). In the BLISS76 trial [204], the placebo response rate at week 52 was 34%, with belimumab 1 mg/kg it was 41% (P = 0.089) and with 10 mg/kg it was 43% (P = 0.017). The response rates at week 76 were a little lower in all groups. A meta-analysis of the response at 52 weeks in the phase II trial of belimumab [205] as well as BLISS 52 and BLISS 76 trials showed benefit for belimumab, with an odds ratio of 1.63 (95% CI: 1.27, 2.09) [209]. Safety data from the phase II trial and its open-label extension have not shown any significant concerns and continued benefit for up to 7 years [207, 208]. The most common side effects have been upper respiratory tract and urinary tract infections, arthralgia, headaches, fatigue and nausea. Serious infusion reactions and infections have been rare [207, 208]. There have been two case reports of progressive multifocal leukoencephalopathy [357, 358], but there is no evidence that belimumab increases the risk more than other immunosuppressive regimens in SLE patients [356].
Further post hoc analyses [359, 360] on the pooled datasets from BLISS 52 and BLISS 76 trials have demonstrated that belimumab therapy was associated with significantly more patients showing improvements than with placebo in the most commonly affected musculoskeletal and mucocutaneous systems, and more immunological abnormalities normalized than with placebo [359]. Improvement was reported less consistently in other systems that were less often affected [359]. There was less worsening in haematological, immunological and renal parameters in those patients on belimumab than in those on placebo [359], but as with improvement, effects were not always dose related. Serological improvements (reduction in anti-dsDNA antibodies and increase in C3/C4 levels, without reduction in memory T or B cell numbers or levels of anti-pneumococcal or anti-tetanus toxoid antibodies) have been reported [361]. This is consistent with the low rate of serious infections in the long-term open-label study of belimumab [207, 208].
Another pooled analysis of BLISS 52 and BLISS 76 trials identified that belimumab had most therapeutic benefit compared with standard therapy alone in patients with higher disease activity (SELENA-SLEDAI ⩾10), positive anti-dsDNA antibodies, low complement, or CS treatment at baseline [206]. Week 52 response rates in the low complement/anti-dsDNA–positive subgroup were 32% for placebo, 42% for belimumab 1 mg/kg (P = 0.002) and 52% for belimumab 10 mg/kg groups (P < 0.001). For the SELENA-SLEDAI ⩾10 subgroup, the response rates were 44%, 58% (P < 0.001) and 63% (P < 0.001), respectively. Belimumab was also shown to reduce severe flares and CS use and to improve health-related quality of life most in these more severe subgroups [206]. These analyses contributed to the decision by the European Medicines Agency to limit the market authorization for belimumab (Benlysta) to add-on therapy in adult patients with active autoantibody-positive SLE with a high degree of disease activity (e.g. positive anti-dsDNA and low complement) despite standard therapy [362].
Conclusions
Treatment with belimumab in addition to standard therapy in autoantibody-positive SLE patients was associated with some improvements in clinical, laboratory and patient-reported outcome measures (compared with placebo in addition to standard therapy) and had a low risk of serious side effects. Based on the results of the two RCTs and the post hoc analyses, belimumab is considered by NICE to be cost-effective in the UK only for patients who meet the specific criteria [324] (see summary above and Fig. 1), so availability is limited. The drug is being used in other countries, particularly in the USA, where the licence covers patients with moderate disease activity and only specifies that patients must have active, autoantibody-positive lupus and be receiving standard therapy (such as CSs, antimalarials, immunosuppressives and NSAIDs) [363]. Overall, the LOE for belimumab in non-renal lupus from a meta-analysis, one phase II study, two phase III RCTs, their open-label extension study and post hoc analyses combining the data from the two RCTs is 1+ and the GOR is B.
Recommendations for the management of severe SLE
Patients who present with severe SLE, including renal and NP manifestations, need thorough investigation to exclude other aetiologies, including infection (4/D). Treatment is dependent on the underlying aetiology (inflammatory and/or thrombotic), and patients should be treated accordingly with immunosuppression and/or anticoagulation, respectively (4/D) (SOA 98%).
Immunosuppressive regimens for severe active SLE involve i.v. MP (2+/C) or high-dose oral prednisolone (up to 1 mg/kg/day) (4/D) to induce remission, either on their own or more often as part of a treatment protocol with another immunosuppressive drug (4/D) (SOA 98%).
MMF or CYC are used for most cases of LN and for refractory, severe non-renal disease (2 ++/B) (SOA 98%).
Biologic therapies belimumab (1+/B) or rituximab (2+/C) may be considered, on a case-by-case basis, where patients have failed to respond to other immunosuppressive drugs, due to inefficacy or intolerance (SOA 98%).
IVIG (2−/D) and plasmapheresis (3/D) may be considered in patients with refractory cytopaenias, thrombotic thrombocytopaenic purpura (TTP) (1+/B), rapidly deteriorating acute confusional state and the catastrophic variant of APS (SOA 93%).
Rationale
Overview of the management of severe lupus
Patients who have serious manifestations with organ- or life-threatening disease require treatment with intensive immunosuppression followed by a prolonged period of less aggressive maintenance therapy to prevent relapse (summarized with suggested dosing regimens in Table 7). In some cases there may be a thrombotic component to the clinical features that requires anticoagulation, for example in patients with APS as well as lupus. There is most evidence for the management of LN, less for neuropsychiatric disease and very little for other organ-specific manifestations.
The authors of this guideline have not reviewed the evidence for the management of LN as they suggest that the EULAR/ERA-EDTA recommendations for the management of adult and paediatric LN [24] are followed. The main recommendations and SOAs with them are shown in Table 3. Further details about these recommendations and the evidence for them have been published [24].
For the management of severe non-renal SLE, the evidence for treatment with high-dose CSs, AZA, CYC, MMF, rituximab, IVIG and plasma exchange (plasmapheresis) is discussed below. The evidence for use of belimumab and of the calcineurin inhibitors ciclosporin and tacrolimus, particularly for cytopenias due to lupus, has already been reviewed above. Suggested initial target dosing regimens and lower maintenance regimens to prevent flares once patients are stable are shown in Table 7. The actual regimen used for individual patients will depend on the clinical picture and the treatment history. Patients with refractory disease, especially those being considered for belimumab and rituximab, should be discussed with and/or seen by a specialist lupus centre (see Fig. 1 flowchart for eligibility and response criteria). It is important to review the response regularly and to increase the dose and/or change the treatment if patients fail to respond.
CSs for severe SLE
Summary
The emphasis in the last 10 years has been on finding steroid-sparing regimens to treat severe lupus, using other immunosuppressants in conjunction with CSs (either orally, intravenously or both), to induce and maintain response with the least risk of adverse events, particularly infection. In general, there is an increasing tendency to use oral prednisolone at a dose of 0.5 mg/kg/day with i.v. MP pulses (3 × 500–750 mg) rather than higher doses of i.v. MP pulses and/or higher dose of oral prednisolone (e.g. 0.75–1 mg/kg/day) as done in the past for all severe manifestations of lupus.
Evidence
I.v. MP pulses as an alternative to, or in addition to, high-dose oral prednisolone was first reported as a treatment for LN [24, 325, 326]. I.v. MP pulses were introduced for the management of non-renal lupus in the early 1980s [147]. An open-label cohort study [146] and an open-label trial [145] using i.v. MP pulses followed by alternate day oral CSs found that pulse therapy led to rapid improvement in clinical symptoms and anti-dsDNA and C3 levels, but that an alternate day oral regimen was associated with relapses. A small double-blind, placebo-controlled RCT with mostly non-renal SLE patients [144] found that 3 i.v. MP pulses resulted in faster and more complete improvement in the first 2 weeks in 12 patients with SLE, but there was no significant difference in efficacy or safety parameters at 4 weeks or 6 months compared with the placebo group; however, all patients received 40–60 mg of oral prednisolone daily [144].
A double-blind RCT [143] comparing three daily i.v. MP pulses of either 1000 or 100 mg in 21 patients with SLE causing fever, cardiorespiratory, renal or NP manifestations (with individualized outcomes based on entry manifestations) suggested no difference in efficacy between the regimens. A retrospective study compared low-dose i.v. MP pulses (⩽1500 mg over 3 days) with high-dose pulses (3–5 g over 3–5 days) for the treatment of severe flares [148]. This study suggested that the lower dose was sufficient and safer for controlling SLE flares than the high-dose regimen, which was associated with an increased number of infections [148].
Conclusions
There is limited evidence for any particular CS regimen for specific manifestations of severe non-renal lupus. Overall the LOE for i.v. MP pulses and oral prednisolone in non-renal severe lupus is 2+ and the GOR is C.
AZA in severe SLE
Summary
AZA (2–3 mg/kg/day) is sometimes used as first-line therapy with CSs in severe non-renal lupus (see Table 7), based on the evidence discussed in the section on the use of AZA for the management of moderate lupus. It is most often used in women planning pregnancy or pregnant, as it is much safer in pregnancy than CYC or MMF, which are contra-indicated in such situations [239].
Evidence
There was only one open-label controlled trial, with 24 patients with severe (life-threatening) multisystem manifestations of lupus [151], which showed no definite benefit from the addition of AZA compared with 40–60 mg prednisone alone for 6 months, before tapering over the next 18 months, although there was some steroid-sparing benefits seen at 12 months. It has been used as primary treatment at a dose of 2 mg/kg/day as an alternative to MMF or CYC in low-risk renal patients without adverse prognostic factors and when these drugs are contra-indicated, not tolerated or unavailable [24].
AZA has been used more often as maintenance therapy after a course of CYC for severe lupus, based on the evidence from studies undertaken in patients with LN [24, 25]. The rate of major extra-renal flares in the maintenance phase of the Aspreva Lupus Management Study (ALMS) study was low in the AZA group at 6.3% (7/111) and similar to the frequency of 6.9% (8/116) in the MMF group [160]. There is some evidence that AZA may be less effective at preventing renal flare in patients in this LN study than MMF, as discussed in the section on MMF [160]. However in a predominantly Caucasian LN population, in the MAINTAIN study, no difference in number or time of severe systemic flares in the AZA group (4/43) compared with the MMF group (3/53) was observed [161]. There are no trials or controlled studies addressing AZA as a primary treatment for neuropsychiatric lupus or any other specific serious non-renal manifestations of lupus, but it has been used after CYC for the treatment and prevention of recurrence of lupus psychosis in 13 patients [328].
The systematic review of non-biologic immunosuppressants in non-renal SLE by Pego-Reigosa et al. [133] only considered the unblinded RCT (showing no benefit) from 1975 [151] and a cohort study (showing a reduced rate of flare [155] in patients on AZA) and concluded that there was little evidence to support the use of AZA in non-renal lupus.
Conclusions
Overall, the LOE for AZA in non-renal severe lupus is 2+ and the GOR is C.
CYC in severe SLE including LN and neuropsychiatric lupus
Summary
CYC, although not licensed for lupus, has been used for the treatment of severe lupus, particularly LN and organ- or life-threatening non-renal disease, since the late 1960s, with the first open-label trial in LN reported in 1971 [364]. Oral CYC is associated with an increased risk of bladder cancer and has been replaced by i.v. CYC pulses in the management of severe lupus. There is most experience with i.v. CYC pulses in LN and NPSLE (Tables 3 and 7). CYC is teratogenic and is contra-indicated in women trying to conceive, or who are pregnant or breast-feeding. It is gonadotoxic and can cause infertility, and men should not father children while on CYC [239].
Evidence
The first controlled trial comparing prednisone with CYC in LN, non-renal lupus and PM was reported in 1973 [365], and a similar design was used to compare oral CYC and AZA in lupus not responsive to 15 mg prednisolone [366], but numbers were small and the aim of matching individual patients and comparing their outcomes was unsuccessful. Since then, studies have used different trial designs and evidence supporting the use of various doses of oral and later i.v. pulse CYC regimens to reduce disease activity and prednisolone dosage and to improve outcomes in patients with LN and non-renal lupus have been reported. The best-known regimens are based on the National Institutes for Health i.v. CYC protocol (monthly i.v. CYC at 500–1000 mg/m2 body surface area for 6 months, followed by 3 monthly i.v. CYC for 2 years) [367] and the Euro-Lupus protocol, which uses lower doses (500 mg fixed dose i.v. CYC 2-weekly for a total of 6 doses, followed by oral AZA) [368] and appears to be as effective and safer for LN in Europe than high-dose regimens [369]. In recent years, the 3-monthly i.v. CYC maintenance pulses for 2 years in the National Institutes for Health protocol have been replaced by oral MMF or AZA [25, 370].
I.v. CYC pulses were the most widely used regimes for all but the mildest cases of acute proliferative glomerulonephritis until MMF was found to be comparable in efficacy and safer [24, 25]. It should be noted that neither of these drugs is licensed for the treatment of LN, but both are supported as appropriate treatment for the management of LN in the EULAR/ERA-EDTA recommendations for the management of adult and paediatric LN [24] (Table 3) and the ACR guidelines for screening, treatment and management of LN [25].
Treatment regimens tested in LN have often been applied to severe non-renal lupus disease as there are fewer non-renal studies and they include heterogeneous patient populations. A systematic review [133] evaluated 29 studies, including 4 unblinded RCTs in which 3742 patients with non-renal lupus were treated with a variety of CYC regimens. There are more data on the efficacy and safety of using CYC to treat non-renal lupus than of any other drug treatment; however, there are fewer high-quality studies than for LN, and diverse end points have been used, making it hard to compare the studies.
Data from the ALMS RCT comparing i.v. CYC (0.5–1.0 g/m2 monthly × 6) and MMF (target 3.0 g/day) as induction therapy for LN [159] showed that i.v. CYC therapy was associated with almost 95% response in all of the non-renal systems, apart from the haematology, which was confounded by drug-induced cytopenias and anaemia of uncertain cause. There was no difference in response between i.v. CYC or MMF in any of the systems studied, including renal.
Some of the best evidence supports the use of pulse i.v. CYC in NP lupus, with one small RCT favouring an i.v. CYC regimen over i.v. MP alone [186]. That trial used more CSs than we would recommend now and was based on a previous retrospective cohort study that suggested that i.v. CYC was useful in the management of NPSLE [371]. The RCT [186] recruited 32 SLE patients with active severe NP manifestations without thrombosis (such as seizures, optic neuritis, peripheral or cranial neuropathy, coma, brainstem disease or transverse myelitis) that had developed within the previous 15 days. All of the patients received oral prednisolone 1mg/kg/day for up to 3 months and then tapered depending on response and 1 g of i.v. MP daily for 3 days. One group received further 1 g of i.v. MP daily for 3 days repeated monthly for 4 months then bimonthly for 6 months and finally 3 monthly for one year. The other group received i.v. CYC 0.75g/m2 body surface monthly for 12 months then this dose was repeated every 3 months for another year. The primary end point was at least 20% improvement from baseline using clinical, laboratory or specific neurological criteria and was met in 18/19 (95%) receiving CYC and 6/13 (46%) receiving MP [186]. A Cochrane systematic review of the treatment of NPSLE [372] calculated a relative risk of 2.05 (95% CI: 1.13, 3.73) for 20% response at 24 months with CYC therapy, but most patients responded by 5 months. CYC treatment was also associated with greater improvement in other lupus manifestations, a significant reduction in SLEDAI score at 6 and 12 months, greater reduction in prednisolone dosage and more patients completing the protocol compared with the MP group. There was no difference in adverse events, including infections and deaths. Recruitment to the study was stopped early due to the higher failure rate of the MP arm. Although the RCT is not of high quality [372] due to the small number of patients studied, the heterogeneity of the NP events, the variable outcome measures used for their assessment, and potential confounding by variable oral CS dosing, it is clear that the i.v. MP regimen was not sufficient and that CYC was better at controlling active NPSLE and preventing relapse.
Further evidence for the use of CYC in NPSLE comes from a previous open-label, controlled pilot study on the use of low-dose i.v. CYC, with a mean dose of 21 mg/day oral prednisone in 37 NPSLE patients, compared with oral prednisone alone in 23 patients (mean dose 21 mg/day) [187], and a cohort study [373] in which a low-dose regimen of i.v. CYC was used in 25 patients with NPSLE with benefit and a low risk of adverse events. A case series [328] found that treating 13 patients with lupus psychosis with oral prednisolone starting at 1 mg/kg/day for 8 weeks and oral CYC (1–2 mg/kg/day) for 6 months followed by oral AZA (1–2 mg/kg/day) led to improvement within a mean of 44 days and only one relapse with psychosis after 2 years; however, 23% developed other NP features and 38% had non-NP flares over the mean follow-up of 7 years. Anti-psychotic agents were used in nine patients for a mean of 6 months. Evidence for CYC and other treatments in neuro-ophthalmic manifestations of lupus have been reviewed in a systematic review [374], but the data on treatment is mostly based on case reports and small case series, for example cases with neuromyelitis optica treated with or without CYC [374].
In contrast to the studies assessing low-dose regimens, high-dose CYC has been studied as well in the hope of achieving better responses in severe lupus. An open-label, uncontrolled study [375] reported the initial safety and efficacy of high-dose CYC (50 mg/kg × 4 days) without stem cell transplantation in 14 patients with refractory moderate to severe SLE despite CSs and at least one immunosuppressant. A prospective RCT [188] was designed to compare the efficacy and safety of a widely used standard i.v. CYC regimen (monthly i.v. CYC at 750 mg/m2 body surface area for 6 months, followed by 3 monthly i.v. CYC for 2 years) with this high-dose i.v. CYC regimen. Entry criteria included moderate-to-severe lupus with renal (22 patients), neurologic (14 patients) or other organ system involvement (11 patients). There was no evidence that response differed between the regimens, but non-responders to monthly i.v. CYC could be rescued with high-dose i.v. CYC. There was no difference in serious adverse events, infections, premature ovarian failure or deaths between the two groups. Leuprolide (a gonadotropin-releasing hormone analogue) was not used to protect against ovarian failure [376]. This should be considered with i.v. CYC moderate- and high-dose regimens [188], as amenorrhoea and ovarian failure are dose- and age-related adverse events of CYC [370, 377], but are rare with the European low-dose i.v. CYC regimen (500 mg 2-weekly for 3 months only) recommended for LN [24].
The remaining data [133] supporting the use of CYC for other serious non-renal manifestations of lupus are obtained predominantly from a variety of cohort studies, small case series and case reports, including 5 patients with systemic lupus vasculitis [378], 11 patients with myocarditis [379] and 5 patients with heart failure due to myocarditis [380]. There is one open-label RCT comparing i.v. CYC with enalapril for 6 months in the treatment of pulmonary hypertension, which showed greater benefit from CYC but an increased risk of infection and gastrointestinal side effects [189].
Conclusions
There is considerable evidence supporting the use of i.v. CYC to reduce disease activity and CS usage in severe lupus, for both renal and non-renal disease, including NPSLE. There is no evidence that CYC prevents chronic damage, and all regimens are teratogenic, but there is less risk with the Euro-Lupus regimen of adverse events (such gastrointestinal side effects, alopecia, infection, amenorrhoea and infertility due to ovarian failure) than with higher dose regimens [12, 16, 24, 25, 133, 372]. Overall, the LOE for the use of CYC in non-renal severe lupus, including NPSLE, from 1 systematic review including 29 studies and 1 systematic Cochrane review of NPSLE is 2 ++, and the GOR is B.
MMF in severe SLE
Summary
There is considerable evidence supporting the use of MMF in the management of LN, and this has been discussed in the Joint EULAR/ERA-EDTA recommendations for the management of adult and paediatric LN [24] (Table 3) and the ACR guidelines for screening, treatment and management of LN [25]. The mean SOA of all of the authors of this guideline with each of the main EULAR/ERA-EDTA recommendations for the management of LN is shown in Table 3. There is very little evidence for the use of MMF in NPSLE, but it is being used to reduce other types of moderate and severe non-renal lupus disease activity (Table 7), to prevent flare and for its steroid-sparing properties, as an alternative to CYC or AZA, especially in cases where inefficacy, drug intolerance and concerns about toxicity arose. It is not compatible with conception, pregnancy or breast-feeding [239].
Evidence
As mentioned in the section on moderate lupus, there is a systematic review of non-biologic immunosuppressants in non-renal SLE [133] that summarizes the data from 8 papers (covering 768 patients with moderate/severe lupus), which assessed the efficacy and safety of MMF in the treatment of non-renal SLE, including the ALMS RCT comparing the use of MMF with that of CYC as induction therapy for LN [159], and 7 cohort studies including 6 discussed above [162–166, 351] and an abstract that does not meet the criteria for this guideline.
Conclusions
Overall, the LOE for MMF in non-renal lupus from 2 systematic reviews, 2 open-label RCTs in LN and 7 cohort studies is 2 ++, and the GOR is B.
Rituximab in severe SLE
Summary
According to the NHS England Interim Commissioning Policy Statement for rituximab in SLE [267], rituximab may be considered in patients with severe or moderate SLE (BILAG system category A or ⩾2B system scores, or SLEDAI >6) who fail treatment with MMF or CYC, either because of lack of effect or due to adverse events, providing they have already failed another immunosuppressant or it would be contra-indicated, or who require unacceptably high long-term CS dosing to control their lupus activity (see Fig. 1 flowchart for eligibility and response criteria).
Evidence
Clinical examples of severe lupus are shown in Table 7, and the evidence for rituximab is summarized in Table 2. The systematic reviews by Duxbury et al. [201] and Cobo-Ibáñez et al. [200] provide evidence supporting the use of rituximab for non-renal severe manifestations of lupus, such as NP involvement (5 cohort studies [381–385]), haematological manifestations (6 cohort studies [383, 385–389]) and at least 10 other cohort studies [382, 383, 385, 387, 390–395]). The data for improvement in NPSLE are still limited and uncontrolled, but showed 73–100% response in small numbers of patients. There is some evidence for improvement (50–100%) in mostly refractory lupus patients and idiopathic autoimmune thrombocytopenia and haemolytic anaemia. There are some specific reports on the use of rituximab in neuro-ophthalmological cases in a systematic review of these conditions [374], and pooled data from European cohorts [396] on the effects of rituximab in LN, as mentioned in the EULAR/ERA-EDTA recommendations for the management of adult and paediatric LN [24]. There are insufficient data to comment on other specific severe lupus manifestations at present, but rituximab is accepted as having steroid-sparing properties (three open-label studies [192, 193, 199]).
Conclusions
Overall, the LOE for rituximab from 3 systematic reviews and 30 studies, including 1RCT and 3 open-label trials for reducing lupus disease activity and for steroid-sparing properties, is 2+, and the GOR is C.
IVIG in severe SLE
Summary
IVIG has been used most in patients with refractory cytopaenias, thrombotic TTP and the catastrophic variant of APS. It can be used in pregnancy (but does not prevent heart block or fetal loss) and in patients with infection. It is rarely indicated as there is not much evidence for its use (Table 2).
Evidence
Much of the initial data are from case reports or small case series reporting treatment of acute events in small numbers of patients [223–226]. A systematic review and meta-analysis covering 3 controlled and 10 observational studies in SLE concluded that IVIG led to a reduction in SLE disease activity scores and a rise in complement levels in 31% of patients (P = 0.001, 95% CI: 22.1, 41.3) . There were insufficient data to assess response using other outcome measures, although serious adverse events were rare and mild [227]. The observational studies often did not report concomitant medication and used a variety of outcome measures and treatment regimens, as discussed below.
IVIG at a dose of 400 mg/kg/day for 5 consecutive days was used monthly for 6–24 months with some benefit in an open-label, uncontrolled trial with 12 refractory SLE patients [210]. Another open-label study [213] assessed 13 female SLE patients with a flare who received 0.4 g/kg body weight IVIG daily for 5 days. Short-term benefit was seen irrespective of concomitant therapy. IVIG‐related adverse effects were mild and rare, and there was no worsening of renal function [213].
Low-dose IVIG was used to treat histologically confirmed cutaneous lupus in 12 patients starting with doses of 1 g/kg × 2, followed by 400 mg/kg monthly until disease remission or for 6 months [214]. Five patients showed complete or almost complete (>75%) clearing of their skin lesions, two had partial improvement (>50%) and three had poor responses (<50%). There were few side effects in this study, but renal patients were avoided because nephrotoxicity has been reported in other studies [397].
A retrospective chart review of 62 patients treated with low-dose IVIG (∼0.5 g/kg) on average every 5 weeks for a mean of 6 courses showed a steady reduction in SLEDAI score over 8 months [215]. Patients with fever, rash, mucosal ulcers, pleurisy, pericarditis, urinary casts and urinary red cells responded in over 50% of cases, but only 30% of arthritis cases responded. Patients with thrombocytopenia, vasculitis and alopecia did not respond. Another group also found a disappointing response to IVIG in thrombocytopenia [216] in a retrospective analysis of 59 patients with immune-mediated severe thrombocytopenia, 44 of whom had definite lupus. A transient response to IVIG was reported in three patients with haemolytic anaemia in another study [217].
The effect of high-dose IVIG (30 g of sulfonated IVIG on days 1–4 and 21–24) in 12 mild to moderate active lupus patients [218] was only temporary in most patients. High-dose IVIG treatment in 17/20 (85%) SLE patients given 1–8 treatment courses consisting of 2 g/kg monthly given over 5 days [219] led to some improvement in arthritis, fever, thrombocytopenia and NP lupus [219]. A retrospective chart review of 17 patients (including 11 with SLE), with a mean follow-up of 30 months and long-term high-dose IVIG treatment monthly for 6 months then every 2–3 months [220], found that there was a significant reduction in the SLEDAI score with significant steroid-sparing effects, and remission was achieved in 12 patients [220].
A case–control study [221] compared 12 pregnant SLE patients with a history of recurrent spontaneous abortions who were on high-dose IVIG (0.5 g/kg every 3–33 weeks) with 12 similar patients treated with prednisolone and NSAIDs. Patients in the IVIG group stopped prednisolone (n = 4) and NSAIDs (n = 9). Disease activity decreased by the end of pregnancy (P < 0.0001) and there was a reduction in autoantibodies and normalization of complement levels in the IVIG group. Such improvements were not seen in the control group, and there were three fetal losses due to spontaneous abortion in this group compared with none in the IVIG group. However, other studies have not confirmed that IVIG can prevent fetal loss [239], and it is possible that NSAIDs contributed to fetal loss in the control group [240].
A multicentre, prospective, open-label study of pregnant women with anti-SSA/Ro antibodies in the mother and birth of a previous child with CHB/neonatal lupus rash was undertaken to determine whether IVIG (400 mg/kg) given every 3 weeks from weeks 12 to 24 of gestation could prevent the development of CHB [211]. CHB was detected at 19, 20 and 25 weeks in 3 babies at a stage when 20 mothers had completed the IVIG protocol before the trial was stopped. An additional child without CHB developed a transient rash consistent with neonatal lupus [211]. Another European prospective study showed similar results [212].
A large retrospective, single-centre cohort study was published by Camara in 2014 [222], which included 52 SLE patients with predominantly cutaneous, haematological, NP and cardiac manifestations who received at least one cycle of IVIG (400 mg/kg/day for 5 days). IVIG was given to 27 patients with infection and active lupus disease, and 17 (63%) patients showed some response. In 18 (69%) of 26 patients with refractory active disease without infection, some response was seen also. This study was too recent to be included in the comprehensive review on the use of IVIG in rheumatic diseases [228] that covered the case–control study in pregnancy by Perricone et al. [221], 4 prospective open-label studies [210, 213, 215, 218, 219], a retrospective cohort study [220] in lupus and a small RCT in LN not discussed here [228].
Conclusions
IVIG, particularly the high-dose regimen, can have some beneficial effects in the short term on disease activity, but has to be continued with intermittent courses for sustained benefit to be seen and only then has steroid-sparing properties. It has a low rate of adverse events in non-renal patients, but can cause nephrotoxicity, especially with pre-existing renal disease. The evidence supporting its use is weak compared with that of other treatments that are cheaper and easier to administer, so it should be reserved for patients in whom other treatments are contra-indicated or have failed. Overall, the LOE for IVIG in non-renal severe lupus from 2 systematic reviews (including a meta-analysis, 3 open-label trials, 10 cohort studies and 4 case series) is 2–, and the GOR is D.
Plasma exchange (plasmapharesis) for severe SLE
Summary
Plasma exchange in SLE has been used in small numbers of patients with conflicting results since the late 1970s. A systematic review was published while this paper was in preparation [238]. It is rarely indicated, because there is inadequate data to support its use except in thrombotic TTP (Table 2).
Evidence
The evidence supporting treatment with plasma exchange, which is expensive and often difficult to organize, remains poor except for thrombotic TTP [229, 398], the catastrophic variant of APS [238] and refractory neuropsychiatric, haematological and renal lupus [238]. Even for rapidly progressive glomerulonephritis, the evidence is limited [399].
Studies have shown that plasmapheresis can reduce immune complexes and anti-dsDNA antibodies, but there is a rapid rebound of complexes and antibodies to pre-treatment levels, as shown originally in 5/8 patients [230]. Marked improvement after plasma exchange was seen in 7/11 (64%) SLE patients in another study [231] lasting up to 3 years, but one (9%) patient with a severe relapse died, and plasma exchange was ineffective in 3 (27%) patients. In another small study of nine patients, 5 (56%) improved, 2 (22%) progressed to end-stage renal failure, and 2 (22%) died due to complications of severe SLE [232].
There was less support for the use of plasma exchange in SLE after a trial comparing plasma exchange in combination with CYC and CSs with standard therapy revealed no benefit from the plasma exchange for 40 patients with severe LN [400]. However, to avoid the rebound increase in autoantibodies after plasma exchange, a synchronized protocol was developed by the Lupus Plasmapharesis Study Group, consisting of plasmapheresis (3 × 60 ml/kg) followed by high-dose pulse CYC (36 mg/kg) then 6 months of oral immunosuppression. This treatment led to rapid improvement in disease activity in the initial 14 patients with various severe SLE manifestations, sufficient for immunosuppressants including CSs to be withdrawn in 12 (86%) patients at 6 months. Treatment-free clinical remission was sustained in 8 (57%) patients for a mean of 5.6 years [233]. However, there has been concern that improvements seen in this and 2 other uncontrolled studies [234, 235] with 23 patients may have been due to the concomitant immunosuppressants. It is notable that the Lupus Plasmapharesis Study Group never reported on the final disappointing results of a randomized international multicentre trial comparing their synchronized protocol [233] with the administration of pulse CYC alone.
The evidence for treating patients who have diffuse alveolar haemorrhage, thrombotic TTP or catastrophic APS with lupus is predominantly from case reports and small case series [229, 236, 237]. Given the high mortality in TTP in general, but especially with lupus [229, 398], it is essential that patients with TTP are referred early for plasma exchange and specialist care [398, 401]. Further details about the experience with and potential use of plasma exchange and immunoadsorption in lupus and APS, including LN, are covered by the systematic review [238].
Conclusions
There remains a need for further research to better define the patients who are most likely to benefit from plasma exchange, but in general they are considered to be those who have TTP, severe refractory disease or contra-indications to conventional treatment (such as pregnancy). Overall, the LOE for plasma exchange for the treatment of non-renal severe lupus from one systematic review and nine studies is weak [3], and the GOR is D, but for TTP it is strongly recommended (grade B), as for non-lupus patients with TTP.
Applicability and utility
Implementation
Diagnosis and assessment of lupus can be difficult due to multisystem involvement and variable laboratory and serological test results. These guidelines will increase knowledge and raise the standard of care for patients with lupus. Only HCQ, CSs and belimumab are licensed treatments for lupus. The evidence for the treatment options discussed in this guideline, which reflect current best practice, has increased considerably in the last 10 years, although there is still relatively little evidence from high-quality RCTs. There should be no barriers to implementation, apart from limitations on the funding for rituximab and belimumab discussed in the relevant sections. The guidelines will be widely presented at local, regional and national meetings for health professionals and patients, carers and supporters of relevant charities.
Key standards of care
Lupus patients should be referred to a physician with experience in managing lupus who can confirm the diagnosis, assess the level of disease activity and provide advice on treatment and monitoring of the disease, its complications and side effects of therapy. Managing immunosuppressive therapies and their potential toxicities in patients with lupus can be a considerable challenge due to the risk of infection, difficulties with attribution of cytopenias to lupus or cytotoxic drugs, and difficulties in distinguishing manifestations of lupus disease activity from damage and co-morbid conditions. Input from a multidisciplinary team including nurse specialists and physiotherapists is usually required, and management may involve a variety of specialists, including rheumatologists, nephrologists, dermatologists, haematologists, cardiologists, chest physicians, neurologists, obstetricians, podiatrists and occupational therapists working as part of collaborative clinical networks involving regional specialist centres, local hospitals and GPs.
It is important to get patients to a low level of disease activity, if not remission, using HCQ, immunosuppressants and the least amount of CSs possible, in order to reduce cumulative damage from the disease and its treatment with CSs [71]. If drug treatment is not working within the expected time frame, it is important to consider adherence to treatment and adjusting the therapy to reduce the accumulation of chronic damage.
Patients need personalized advice, written information and education about the disease and its drug treatment from members of the multidisciplinary team, including specialist nurses and an individual to contact in the event of new symptoms. Additional topics covered should include sun avoidance, adequate vitamin D intake, weight control, exercise, not smoking and other measures to reduce atherosclerotic risk factors, as well as cancer screening, contraception and pregnancy planning when the disease is under good control on appropriate treatment for conception.
Future research agenda
There is a need for more evidence to support decision-making in the management of lupus patients. The guideline development group identified certain priorities for research into lupus to help address this issue, and these are shown in Table 8.
Research priorities to improve the management of lupus patients
| Analysis of the BILAG Biologics Register data is needed to assess the efficacy and safety of using rituximab for treating refractory lupus disease, administered according to the NHS England Interim Clinical Commissioning Policy Statement. |
| Analysis of the BILAG Biologics Register should also provide some data on the use of MMF in non-renal lupus patients; this is needed to support data from previous renal trials. |
| More research into stratified and personalised medicine and the cost-effectiveness of immunosuppressive drugs in lupus patients is warranted to help identify which drug will be most suitable for an individual. |
| Trials of immunosuppressive regimens and biologic therapies that will significantly reduce the need for CSs are needed in renal and non-renal lupus patients. |
| The cost-effectiveness and value of monitoring drug levels in order to improve adherence/compliance with drug therapy and improve the outcome in terms of reduced disease activity, damage and steroid usage should be investigated (e.g. for HCQ, MMF). |
| The role of IVIG and plasma exchange in the management of lupus patients requires further evaluation. |
| More data are required on the long-term outcome for children born to mothers with lupus who were exposed to drugs used pre-conception, while pregnant and/or while breast-feeding. |
| Analysis of the BILAG Biologics Register data is needed to assess the efficacy and safety of using rituximab for treating refractory lupus disease, administered according to the NHS England Interim Clinical Commissioning Policy Statement. |
| Analysis of the BILAG Biologics Register should also provide some data on the use of MMF in non-renal lupus patients; this is needed to support data from previous renal trials. |
| More research into stratified and personalised medicine and the cost-effectiveness of immunosuppressive drugs in lupus patients is warranted to help identify which drug will be most suitable for an individual. |
| Trials of immunosuppressive regimens and biologic therapies that will significantly reduce the need for CSs are needed in renal and non-renal lupus patients. |
| The cost-effectiveness and value of monitoring drug levels in order to improve adherence/compliance with drug therapy and improve the outcome in terms of reduced disease activity, damage and steroid usage should be investigated (e.g. for HCQ, MMF). |
| The role of IVIG and plasma exchange in the management of lupus patients requires further evaluation. |
| More data are required on the long-term outcome for children born to mothers with lupus who were exposed to drugs used pre-conception, while pregnant and/or while breast-feeding. |
NHS: National Health Service.
Research priorities to improve the management of lupus patients
| Analysis of the BILAG Biologics Register data is needed to assess the efficacy and safety of using rituximab for treating refractory lupus disease, administered according to the NHS England Interim Clinical Commissioning Policy Statement. |
| Analysis of the BILAG Biologics Register should also provide some data on the use of MMF in non-renal lupus patients; this is needed to support data from previous renal trials. |
| More research into stratified and personalised medicine and the cost-effectiveness of immunosuppressive drugs in lupus patients is warranted to help identify which drug will be most suitable for an individual. |
| Trials of immunosuppressive regimens and biologic therapies that will significantly reduce the need for CSs are needed in renal and non-renal lupus patients. |
| The cost-effectiveness and value of monitoring drug levels in order to improve adherence/compliance with drug therapy and improve the outcome in terms of reduced disease activity, damage and steroid usage should be investigated (e.g. for HCQ, MMF). |
| The role of IVIG and plasma exchange in the management of lupus patients requires further evaluation. |
| More data are required on the long-term outcome for children born to mothers with lupus who were exposed to drugs used pre-conception, while pregnant and/or while breast-feeding. |
| Analysis of the BILAG Biologics Register data is needed to assess the efficacy and safety of using rituximab for treating refractory lupus disease, administered according to the NHS England Interim Clinical Commissioning Policy Statement. |
| Analysis of the BILAG Biologics Register should also provide some data on the use of MMF in non-renal lupus patients; this is needed to support data from previous renal trials. |
| More research into stratified and personalised medicine and the cost-effectiveness of immunosuppressive drugs in lupus patients is warranted to help identify which drug will be most suitable for an individual. |
| Trials of immunosuppressive regimens and biologic therapies that will significantly reduce the need for CSs are needed in renal and non-renal lupus patients. |
| The cost-effectiveness and value of monitoring drug levels in order to improve adherence/compliance with drug therapy and improve the outcome in terms of reduced disease activity, damage and steroid usage should be investigated (e.g. for HCQ, MMF). |
| The role of IVIG and plasma exchange in the management of lupus patients requires further evaluation. |
| More data are required on the long-term outcome for children born to mothers with lupus who were exposed to drugs used pre-conception, while pregnant and/or while breast-feeding. |
NHS: National Health Service.
Mechanism for audit of the guideline
To assess compliance with these guidelines, an audit proforma is available on the British Society for Rheumatology website.
Funding: No specific funding was received from any funding bodies in the public, commercial or not-for-profit sectors to carry out the work described in these guidelines.
Disclosure statement: D.D.’C. has undertaken consultancies and received honoraria from GlaxoSmithKline/Human Genome Sciences and Roche, has been a member of the speakers’ bureau for GlaxoSmithKline/Human Genome Sciences, Union Chimique Belge (UCB) and Eli Lilly and has received research grant support from Aspreva/Vifor Pharma. C.G. has undertaken consultancies and received honoraria from Bristol-Myers Squibb, Eli-Lilly, GlaxoSmithKline, MedImmune, Merck Serono, Parexel, Roche and UCB, has been a member of the speakers’ bureau for GlaxoSmithKline, UCB and Lilly and has received research grant support from Aspreva/Vifor Pharma in the past and UCB currently. Y.N. has received funding to attend scientific meetings and received honoraria from UCB and GlaxoSmithKline. P.N. has received funding to attend scientific meetings and received honoraria from UCB. I.N.B. has undertaken consultancies and received honoraria from AstraZeneca, GlaxoSmithKline, MedImmune, Merck Serono, Pfizer, Roche and UCB, has been a member of the speakers’ bureau for GlaxoSmithKline, UCB and Pfizer and has received research grant income from Genzyme Sanofi, GlaxoSmithKline, UCB and Roche. B.G. has received honoraria from Pfizer. M.K. has received funding to attend scientific meetings and honoraria from AstraZeneca, MedImmune, GlaxoSmithKline, INOVA Diagnostics and UCB. S.B. has received honoraria from Actelion INB to attend scientific meetings, has undertaken consultancies and received honoraria from AstraZeneca, GlaxoSmithKline, MedImmune, Merck Serono, Pfizer, Roche and UCB and has been a member of the speakers’ bureau for GlaxoSmithKline, UCB and Pfizer. M.G. has received funding to support scientific meetings from Roche, Abbvie and Bristol-Myers Squibb. D.J. has received research grants, honoraria and consulting fees from Roche/Genentech, consulting fees from Boehringer Ingelheim, Chemocentryx, GlaxoSmithKline and Medimmune and is a Board member of Aurinia Pharmaceuticals. D.I. has consulted for Merck Serono, Eli Lilly, Celegene, UCB, XTLBio, Anthera and Baxalta; the honoraria received have been passed on to a local arthritis charity. L.L. has received research funding in grants/in kind from Roche and Genentech, has acted as an advisor to Genentech, Medimmune and Rigel and has received honoraria/travel grants from Genentech, Roche and UCB. K.S. received funding to attend a scientific meeting from Daiichi Sankyo. All other authors have declared no conflicts of interest.
Supplementary data
Supplementary data are available at Rheumatology Online.

{kind=link}
Comments