






Abstract
Earlier work described a mutation in DEC2 also known as BHLHE41 (basic helix-loophelix family member e41) as causal in a family of short sleepers, who needed just 6 h sleep per night. We evaluated whether there were other variants of this gene in two well-phenotyped cohorts.
Sequencing of the BHLHE41 gene, electroencephalographic data, and delta power analysis and functional studies using cell-based luciferase.
We identified new variants of the BHLHE41 gene in two cohorts who had either acute sleep deprivation (n = 200) or chronic partial sleep deprivation (n = 217). One variant, Y362H, at another location in the same exon occurred in one twin in a dizygotic twin pair and was associated with reduced sleep duration, less recovery sleep following sleep deprivation, and fewer performance lapses during sleep deprivation than the homozygous twin. Both twins had almost identical amounts of non rapid eye movement (NREM) sleep. This variant reduced the ability of BHLHE41 to suppress CLOCK/BMAL1 and NPAS2/BMAL1 transactivation in vitro. Another variant in the same exome had no effect on sleep or response to sleep deprivation and no effect on CLOCK/BMAL1 transactivation. Random mutagenesis identified a number of other variants of BHLHE41 that affect its function.
There are a number of mutations of BHLHE41. Mutations reduce total sleep while maintaining NREM sleep and provide resistance to the effects of sleep loss. Mutations that affect sleep also modify the normal inhibition of BHLHE41 of CLOCK/BMAL1 transactivation. Thus, clock mechanisms are likely involved in setting sleep length and the magnitude of sleep homeostasis.
INTRODUCTION
Sleep is an essential physiological state of reduced activity and alertness in all mammals and several other animal species. In mammals, sleep is identified by characteristic electroencephalogram (EEG) patterns and can be divided into non rapid eye movement (NREM) sleep and rapid eye moment (REM) sleep.1 Loss of sleep, i.e., sleep deprivation, results in performance impairments including performance lapses as determined by the psychomotor vigilance reaction time test (PVT).2–5 This is a biological trait, i.e., there are individuals who are resistant to or very sensitive to the effects of sleep loss as assessed by increases in performance lapses.4 We have recently shown, using a classic twin study, that this is a highly heritable trait.6
Classically, sleep is proposed to be regulated by the interaction between two independent processes: the drive for sleep or sleep homeostasis (process S) and the circadian clock (process C).7–12 In the past, these processes have been considered distinct. Recent data, however, challenge this concept.13 Expression of clock genes such as PER2 (PERIOD 2) in the cerebral cortex is increased with sleep deprivation, i.e., their expression is affected by sleep loss in addition to circadian rhythms.14 Moreover, recent studies in mutant mice have demonstrated that circadian clock genes such as NPAS2 (neuronal PAS domain protein 2; MIM:603347), ARNTL, also known as BMAL1 (aryl hydrocarbon receptor nuclear translocator-like; MIM:602550), and CRY1/CRY2 (cryptochrome 1 [photolyase-like] and 2; MIM:601933; MIM:603732) can affect not only the timing of sleep, but also sleep homeostasis and sleep architecture.13,15–20
Perhaps the most convincing evidence of the role of clock genes in determining sleep duration and the amount of sleep recovery following deprivation (sleep homeostasis) is the demonstration that individuals with a mutation in exon 5 of BHLHE41 (class E basic helix-loophelix protein 41), also known as DEC2, have short sleep duration.21 This mutation is at the amino acid position 384, with a missense mutation, where a proline is replaced by an arginine (c.1151C > G, p.Pro384Arg; MIM:612975). Knocking in this mutation of BHLHE41 into Drosophila or mice results in shorter sleep and less recovery sleep following sleep deprivation.21
BHLHE41 is part of the transcription factor family that is regulated by the mammalian molecular clock.21–24BHLHE41 influences other molecules such as BMAL/CLOCK. The CLOCK and BMAL1 proteins form a heterodimer and through E-box elements in promoter regions activate the transcription of PERIOD genes. Honma et al.23 demonstrated the capacity of the transcriptional repressors BHLHE40 (DEC1) and BHLHE41 (DEC2) to inhibit Clock/Bmal1-induced transactivation of the mouse Per1 gene promoter. The inhibition was either through direct protein–protein interactions with BMAL1 and/or competition for E-box elements. The p.Pro384Arg mutation found by He et al.21 resulted in the inability of the BHLHE41 protein to interact properly with the circadian clock transcription factors CLOCK and BMAL1. The p.Pro384Arg BHLHE41 variant was deficient in its ability to repress CLOCK and BMAL1.
Based on this result, we questioned whether there might be other variants of BHLHE41 in human populations that affect sleep duration and response to sleep deprivation. We found that there are other variants of BHLHE41 in the human population. The effect of these variants on sleep duration in humans is associated with their effect on suppression of CLOCK/BMAL activation. Mutations that alter the suppression of CLOCK/BMAL activation are associated with short sleep and resistance to the effects of sleep deprivation, whereas a mutation that does not affect suppression has no effect on sleep. This has relevance not only for understanding the genetic basis of short sleep in humans, but also adds support for this molecular mechanism setting the duration of sleep that individuals need.
METHODS
Subjects and Phenotypes
Ethics Statement
For all subjects, consent was obtained prior to entry and had the approval of the University of Pennsylvania institutional review board; all subjects received compensation for participation.
Twin Cohort: Total Sleep Deprivation
The twin cohort was collected at the University of Pennsylvania and a consent form approved by the institution was signed. The cohort was studied to assess the heritability of the response to sleep deprivation.6 A total of 59 pairs of monozygotic (MZ) twins (mean age ± standard deviation [SD] 29.2 ± 6.8 y; 15 male and 44 female pairs) and 41 same-sex dizygotic (DZ) twins (mean age ± SD 26.6 ± 7.6 y; 15 male and 26 female pairs) were initially assessed for zygosity by a questionnaire. The details of subject recruitment and assessment as well as the protocol used are described in Kuna et al.6 In brief, subjects were healthy with no chronic conditions such as psychiatric disorder, neurological problems including migraines, or medication use. A greater percentage of the DZ twin pairs reported being African American (n = 15 pairs; 36.6%) compared with MZ twin pairs (n = 7; 11.9%). There were three Asian MZ pairs and no Asian DZ pairs. There was one MZ pair who self-identified as having more than one race. There were 48 Caucasian MZ pairs (81.5%) and 26 Caucasian DZ pairs (63.4%).The subjects underwent a medical history and physical examination, which also included a battery of standardized questionnaires including the Epworth Sleepiness Scale (ESS)43 and Pittsburgh Sleep Quality Index (PSQI).44 Illicit drug use was excluded by a urine drug screen and sleep apnea excluded by an in-laboratory sleep study (1 night of EEG recording of sleep with respiratory variables). Subjects had assessment of sleep duration at home by actigraphy and twice daily sleep diary. They had 2 nights of in-laboratory sleep studies with EEG recording of sleep followed by 38 h of sleep deprivation and then recovery sleep with assessment of the EEG. During the deprivation period, subjects performed a 10-min psychomotor vigilance reaction time test (PVT)6 every 2 h (19 times). A sensitive measure of effect of sleep loss is the number of performance lapses, i.e., responses with reaction times > 500 msec. The number during each trial was recorded. For EEG recording, four leads were placed according to the International 10/20 system including C3A2 and C4A1 electrodes. We used the C3A2 derivation to score sleep stages and wakefulness in 20-sec epochs using our Sandman system (Mallinckrodt, Elite Sleep System Version 6.1). Scoring was based on the rules of Rechtschaffen and Kales.45 The EEG during sleep was sampled at 128 Hz. Power spectra during each episode of NREM sleep (N2 and N3) were estimated using a fast Fourier transform routine with 4-sec epochs, linear detrending, and Hanning window.46 This results in a frequency resolution of 0.25 Hz. We assessed delta power (0.75 Hz to 4.5 Hz). The study was approved by the institutional review boards at the University of Pennsylvania. Written informed consent was obtained from each participant. All testing was performed at the University of Pennsylvania.
Nonrelated Cohort (Partial Chronic Sleep Deprivation)
The second cohort was subjects involved in a study of the effects of chronic partial sleep deprivation (PSD) at the Sleep and Chronobiology Laboratory at the Hospital of the University of Pennsylvania. The subject recruitment for this study and protocol details have been described previously.27 The habitual sleep duration for these subjects ranged between 6.5–8.5 h daily with regular bedtimes and wake up times between 06:00–09:00 (verified by sleep logs and wrist actigraphy for at least 1 w before study entry). For the first 2 nights of the study (baseline), subjects received 10 h time in bed from 22:00–08:00 to reduce any preexisting sleep debt; for the subsequent 5 nights, subjects received 4 h time in bed per night (04:00–08:00) for sleep. The subjects underwent a number of neurobehavioral assessments including the PVT (see previous text).
Identification of BHLHE41 Variants
The blood sample collection and DNA preparation were performed using Qiagen DNAeasy mini kit (Qiagen, CA) and the FX (Beckman Coulter – Indianapolis, IN) Agencourt automation following the manufactures recommendations. We designed five set of primers covering the five exons of BHLHE41 gene originally for the first screening. Sanger sequencing was performed at Center for Applied Genomics and the runs were performed at the Core Facility of University of Pennsylvania according to their procedures and standard protocol. Primers are available upon request.
Cell Culture, Transfections, and Luciferase Assay and Western Blot Analysis
HEK 293T and Neuro2A cells were used for all transfections and maintained in Dulbecco modified medium (DMEM, Gibco, Life Technologies, CA, USA) with 10% fetal bovine serum (FBS, Gibco, L-glu and Pen/Strep). Cells were transiently transfected with expression vectors for CLOCK, NPAS2, BMAL1, wild-type BHLHE41, and BHLHE41 variants (p.Tyr362His, p.Pro384Arg, p.Pro384Gln, and p.Ala380Ser) by reverse transfection in 96-well plates. Then transfection reactions were conducted in each well with 8 × 105 cells in DMEM containing 20% FBS. Approximately 18 h after transfection, the luciferase assays were preformed according to the manufacturer's instructions (Promega, CA, USA). Transfection efficiencies were normalized by cotransfecting 5 ng of the HSV-TK generating Renilla luciferase activity. All transfections were repeated at least three times.
Total protein was extracted from the HEK 293T transiently transfected with expression plasmids of BMAL1, CLOCK, wild-type, or BHLHE41 variants. Cells were lysed with RIPA buffer (20 mM Tris-HCl (pH 7.5) 150 mM NaCl, 1 mM EDTA, 1% NP-40, and a protease inhibitor tablet from Roche, Indianapolis, USA). After the clarification by centrifuge 21.000 g, a total of 30 μg of protein was separated by 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred onto a polyvinyldifluoride (PVDF) membrane. The membrane was blocked with 5% BSA and incubated with mouse anti-FLAG antibody (Sigma-Aldrich, St. Louis, MO, USA) 1 h at room temperature. The blots were incubated with the appropriate secondary antibody, HRP-conjugated ant-imouse, and the blots were assayed with the ECL (Pierce, Waltham, MA, USA) detection system.
Mutagenesis Assays
Random mutagenesis of the BHLHE41 region (362-385aa) was performed with degenerate primers (see Table S1, supplemental material) using polymerase chain reaction (PCR). Briefly, PCR was performed in a total volume of 50 μL containing approximately 50 ng of plasmid samples, 20 pmol of each primer, 0.2 mM deoxyribonucleotide triphosphates, and 2.5 unit pfx DNA polymerase (Life Technologies, CA, USA) for the 12 cycles (95°C for 30 sec, 50°C for 30 sec, and 68°C for 14 min). Samples were then treated with DpnI restriction enzyme to remove the template DNA and transformed into Escherichia coli DH5α. Transformed cells were plated and selected on kanamycin contacting an LB agar plate. We randomly picked 50 colonies and isolated plasmid DNA. The presence of variants was verified by DNA sequencing. Site-directed mutagenesis was carried out using the Stratagene QuickII-change Mutagenesis kit (La Jolla, CA, USA). The PCR products were digested with DpnI to remove template plasmid DNA and transformed into E. coli DH5α. The presence of the variants in BHLHE41 was confirmed by DNA sequencing.
RESULTS
Sleep Phenotypes: Twin Cohort Undergoing Total Sleep Deprivation
Based on the study of He et al.,21 we investigated BHLHE41 variants in a cohort of 59 MZ (mean age ± SD 29.2 ± 6.8 y; 15 male and 44 female pairs) and 41 DZ (mean age ± SD 26.6 ± 7.6 y; 15 male and 26 female pairs) twin pairs evaluated for response to 38 h of sleep deprivation.6 All twin pairs were the same sex. In the week prior to the sleep deprivation protocol, the mean estimated daily sleep time on actigraphy ± SD was 528 ± 103 min in the MZ twins and 502 ± 101 min in the DZ twins. We split the twins into two reference groups, Odds and Evens. Converting to hours, the mean (SD) were 7.04 (0.673) and 6.9 (0.818), respectively. The mean of the three unrelated subjects reported in Table 1 is very similar to these values (7.3 h). Pooling the reference distributions, the mean (SD) is 7.0 (0.75). Based on this reference, the z-scores for the mutation carrier and nonmutation carrier are (4.9-7.0)/0.75 = -2.80, which is smaller than one-third of the first percentile (0.00255). This would suggest that this is an extremely short mean sleep. In comparison, the nonmutation carrier z-score is (6.07-7.0)/0.75 = -1.24, which corresponds to approximately the 11th percentile (0.1075). Although on the short side, the non mutation carrier had mean sleep that was not an outlier. The specific phenotypic characteristics of subjects studied are shown in Table 1 and detailed information can be found in Kuna et al.6
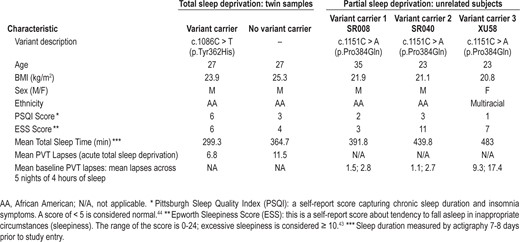
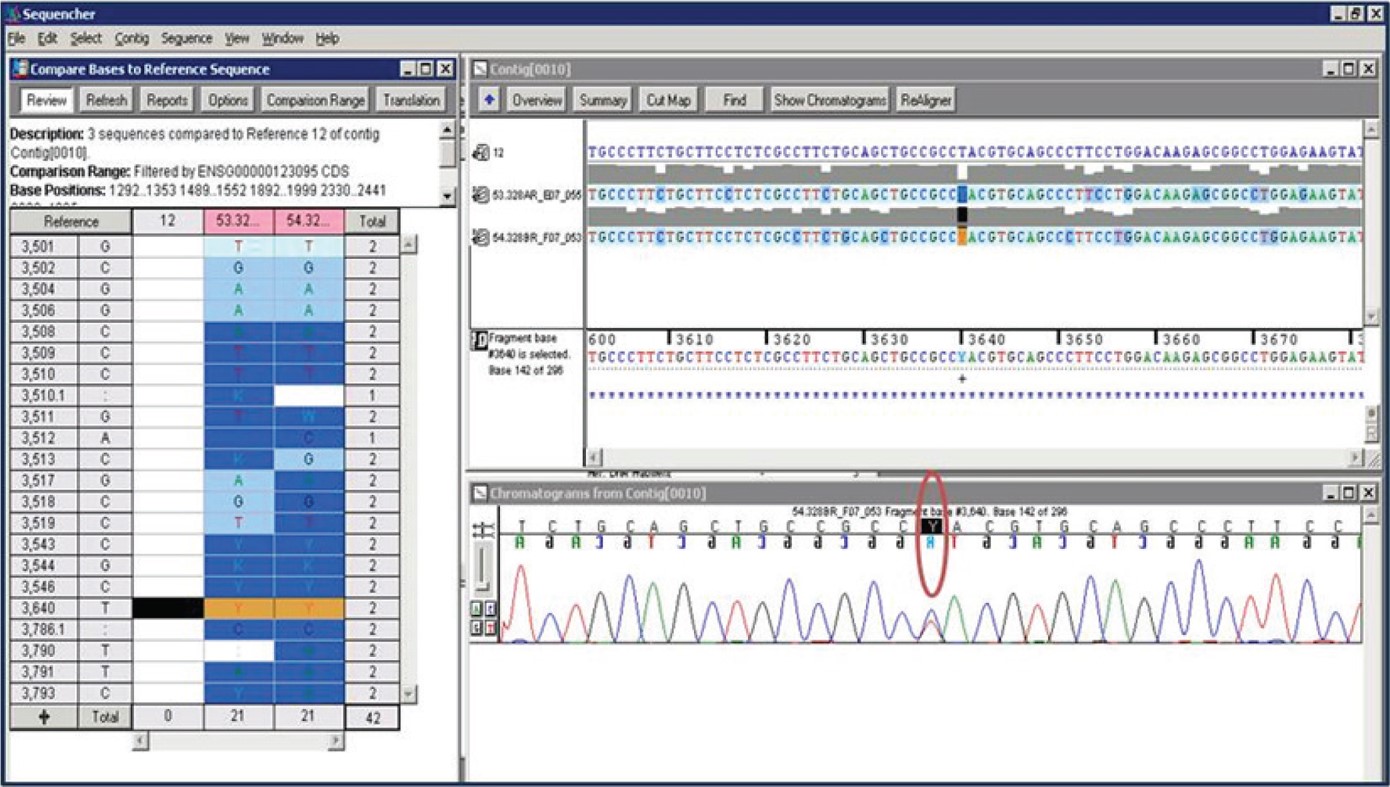
Using Sanger sequencing approach on the five exons of the gene, we identified a new variant of the BHLHE41 at position c.1086C > T (p.Tyr362His) at Exon 5 in one member of one DZ pair (Figure S1, supplemental material). This variant is a non synonymous mutation that alters the amino acid sequence of BHLHE41 protein at position 362, shifting a tyrosine to histidine. Based on previous findings,2 we hypothesized that the mutation carrier might have shorter mean sleep duration than the non carrier and tested this hypothesis using a one-sided t-test using data obtained from multiple nights of actigraphy. In addition, we questioned the possible role of the variants not only in sleep length but also in the response to sleep deprivation. The average sleep duration ± SD by actigraphy over 7 to 8 nights for the mutation carrier was more than 1 h shorter (299.3 ± 62.7 min) compared to 364.7 ± 61.8 min for the non carrier twin (one-sided t-test; P = 0.03) (Table 1).
Subsequently, in the laboratory we recorded sleep by EEG on 3 nights and compared the differences between the mutation carrier (c.1086C > T p.Tyr362His) and non carrier for total sleep time (TST), NREM sleep, and REM sleep amounts (Table 2). The differences for TST between the twins (mutation carrier minus non carrier) in min over the 3 nights were as follows: -41.0, -51.5, and +21.5. The amounts of NREM, however, were very similar. The NREM amount after recovery sleep was 388 for the carrier and 371 for the noncarrier twin. NREM differences (min) were: -8.5, +5, and +13. The differences (min) in REM sleep were larger: -36.5, -58.5, and +6.5. During unrestricted recovery sleep for 1 night following 38 h without sleep, the carrier had much shorter TST (482.0 min) than the non carrier (572.5 min). Spectral analyses of the EEG during recovery sleep showed that the twin with the novel variant (p.Tyr362His) had higher delta power during NREM sleep, a putative measure of sleep drive26 (Figure 1). It has been demonstrated previously that short sleepers have increased delta power in comparison with long sleepers.27 To measure cognitive vulnerability to sleep deprivation, we used the PVT and assessed subjects at baseline and in multiple test bouts during sleep deprivation. Despite evidence of higher sleep drive, the twin with the new variant had significantly fewer average lapses of performance on each 2-h (n = 19) administration of the 10-min PVT (6.8 compared to 11.5, signed-rank one-sided P = 0.0004) during sleep deprivation as shown in Table 1. Thus, the mutation was associated with resistance to the neurobehavioral effects of sleep deprivation (see further details in Tables 1 and 2).
Polysomnography data of total sleep, nonrapid eye movement, and rapid eye movement sleep in twins
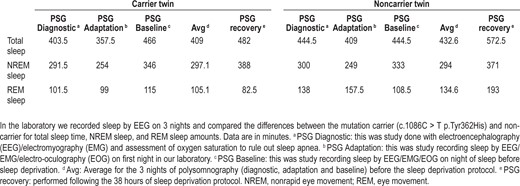
Polysomnography data of total sleep, nonrapid eye movement, and rapid eye movement sleep in twins
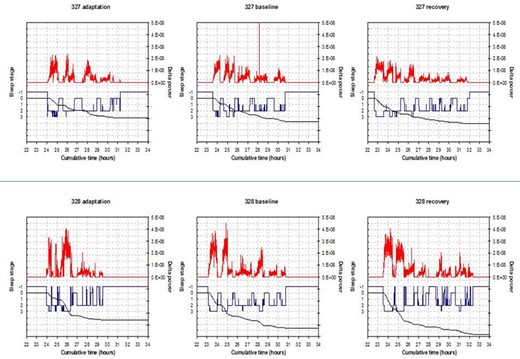
Delta power spectra data. Each of the three panels for each subject represents 1 night of sleep recording and the corresponding values of delta power. The top graphs are for the noncarrier variant (327) and the bottom graphs (328) are for the carrier of BHLHE41 mutation (c.1086C > T p.Tyr362His). In each graph the hypnogram is in blue (-1 = wake, 0 = rapid eye movement, 1-3 = nonrapid eye movement (NREM)1-NREM3) and the delta power aligned with it is in red (for NREM sleep only). The black line shows the cumulatively dissipated delta power.
Sleep Phenotypes: Nonrelated Cohort Undergoing Chronic Partial Sleep Deprivation
We also searched for additional variants in BHLHE41 in 213 other nonrelated healthy adults participating in experiments on the effects of chronic partial sleep deprivation.27 We found a common non synonymous variant c.1151C > A (p.Pro384Gln) in three non-related individuals. This variant was in the same codon as that originally described by He et al.,21 but resulted in a glutamine rather than arginine substitution. The sleep patterns for those individuals were, however, heterogeneous with unclear phenotypic differences (e.g., their average sleep durations based on 7 nights of recording by actigraphy were 6.5, 7.3, and 8.1 h), suggesting that this variant had distinct effect on BMAL/CLOCK transactivation (see details in Table 1). In a reference population of 162 subjects with actigraphy information, the mean ± SD pre study actigraphic sleep duration was 8.03 ± 0.66 h. Based on the nonrelated sample as reference, the z-scores for the nonrelated mutation carriers are -2.27, i.e., 1.2 percentile of normal distribution, -1.06, i.e., 14th percentile, 0.03, i.e., 51st percentile. However, the molecular data suggest an enhanced suppression of BMAL/CLOCK transactivation when 10 ng of plasmid is used and compared to the wild-type and the other mutants. In addition, the PVT for those individuals revealed distinct and heterogeneous results of the mean lapses across 5 nights of chronic partial sleep deprivation (details available in Table 1).
Molecular Functional Studies of Identified Variants
Because one variant had an effect on sleep duration while the other did not; we used functional cell-based assays to address how the new variants we found affect the BMAL-CLOCK-NPAS2 machinery.
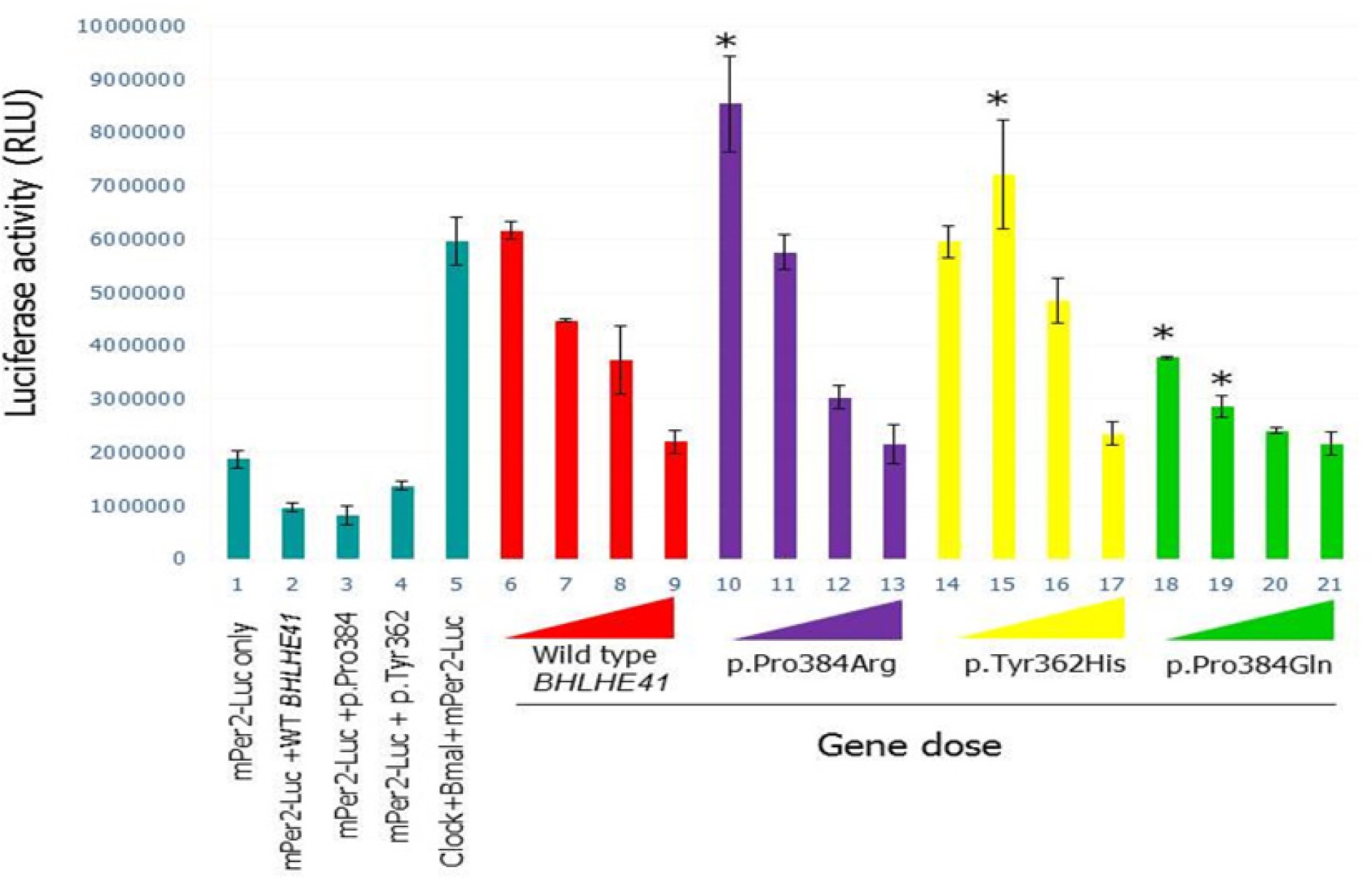
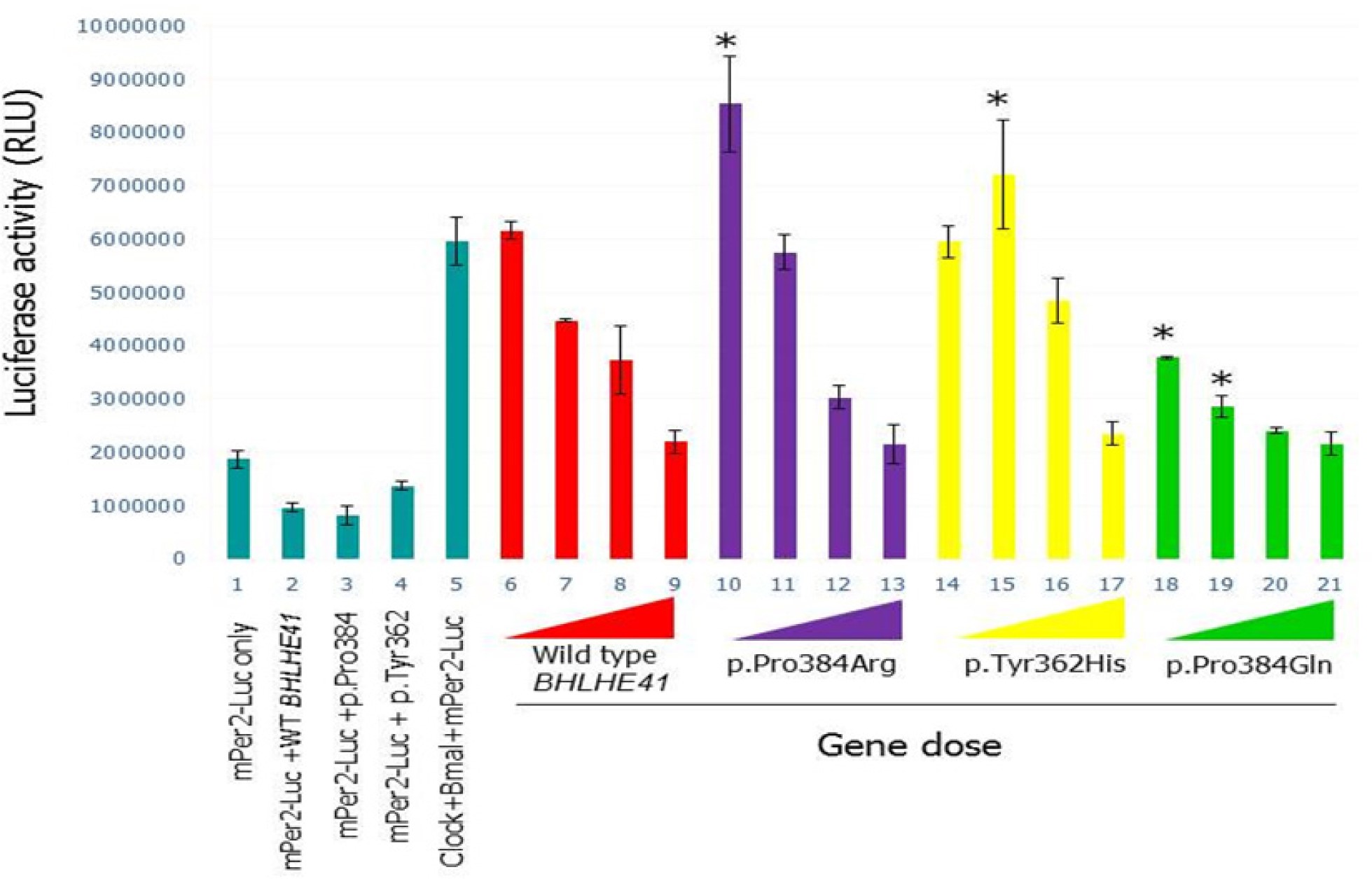
Therefore, at the molecular level we investigated the functional effects of both of these new variants, c.1086C > T (p.Tyr362His) and c.1151C > A (p.Pro384Gln) of BHLHE41, on CLOCK/BMAL1and NPAS2/BMAL1 transactivation using an in vitro PER2:luciferase reporter assay in HEK 293T cell line (Figure 2). We also reproduced the previous experiments using constructs based on the mutation described by He et al.,21 which also involves position 384 but a different amino acid change (p.Pro384Arg).
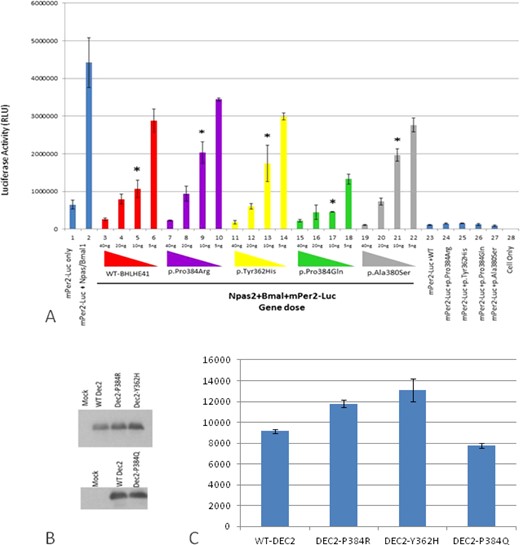
In vitro analyses of wild-type and variants mBHLHE41-NPAS2-Bmal1 transactivation (using HEK293T functional assays). (A) Luciferase assays were carried out with identical methodology to that described in Figure 2. Column 1 indicates background activity of the reporter plasmid (mPer2 promoter-Luc) in HEK 293T cell line in the absence of the Npas2/Bmal1 genes. Column 2 shows transactivation of the luciferase gene on the reporter plasmid by Npas2/Bmal1. Columns from 3 through 22 indicate the effect of the variant and wild-type BHLHE41 on the transactivation of Npas2/Bmal1 in gene doses (40 ng, 20 ng, 10 ng and 5 ng) dependent manner. Red, purple, yellow, green, and gray colors represent BHLHE41-wild-type, BHLHE41 (p.Pro384Arg), BHLHE41 (p.Tyr362His), BHLHE41 (p.Pro384Gln), and BHLHE41 (p.Ala380Ser), respectively. Cells expressing reporter plasmid (mPer2-Luc) along with wild-type BHLHE41 (column 23) and variants of BHLHE41 (columns 24-27) show comparable suppressor activity in the absence of Npas2/Bmal1. Column 28 indicates the background luciferase reading from HEK 293T. Experiments were carried out with triplicates in each point. Luciferase activities (means ± standard error of the mean; n = 9) were measured after a 20-h cell incubation and statistically compared with repression activity of the wild-type BHLHE41 (* P < 0.05). The effect of the variants can be seen when 10 ng of plasmid were used in the assay. (B) Western Blot (WB) analysis of wild-type and variant's mBHLHE41. WB was carried out using anti-Flag with 30 μg of total protein. Enhanced chemiluminescence (ECL) method was used detect the presence of the proteins. The analysis of WB indicates that wild-type and variants were expressed in a comparable level. Arrows indicate the expressed BHLHE41 proteins. (C) Quantification of the protein levels by Western blot results was performed using the ImageJ software. No statistical difference was found in the protein levels.
The new p.Tyr362His variant resulted in an inability of BHLHE41 to suppress CLOCK/BMAL1 transactivation on the Per2:luciferase assay in HEK 293T cells (Figure S2, column 14-16, supplemental material) when a low amount of plasmid is used in the assay. Thus, this novel variant, even in a different amino acid residue, has an effect similar to the variant p.Pro384Arg described previously.21 In contrast, the other new variant, p.Pro384Gln, resulted in strong inhibition of CLOCK/ BMAL1 transactivation in a luciferase assay system. This is an opposite effect compared to the new p.Tyr362His variant and the previously defined p.Pro384Arg variant (Figure S2, column 18-21). Thus, this variant p.Pro384Gln apparently had functional consequences at a molecular level similar to the wild-type without totally affecting the sleep phenotype, but showing a trend in sleep increase. As complementary data we assessed the effect of wild-type and BHLHE41 variant alleles on transactivation driven by NPAS2/BMAL1, also using the PER2:luciferase reporter system (Figure 2A). The PER2:luciferase reporter assay results indicate that NPAS/BMAL1 transactivation is affected in a manner similar to CLOCK/BMAL1 according to each variant. The new p.Tyr362His variant resulted in an inability of BHLHE41 to suppress NPAS2/BMAL1 transactivation when compared to the wild-type (Figure 2A, column 13). We also constructed a PER2:luciferase assay reporter for the mutation p.Pro384Arg,20 which demonstrated a similar inability to inhibit NPAS2/BMAL1 transactivation at a low gene dose (Figure 2A, column 9). In contrast, p.Pro384Gln demonstrated an opposite inhibition of NPAS2/BMAL1 transactivation as shown in our CLOCK/BMAL1 transactivation assay, where p.Pro384Gln exhibits a strong inhibition on transactivation (Figure 2A, column 17). To see the effect of the mutations on the stability and expression levels of the BHLHE41, we carried out Western blot analysis followed by quantification analysis. Analysis of the blots indicated that all alleles had comparable levels of expression as wild-type BHLHE41 (Figure 2). These results indicate that the charge of amino acid R groups at the position of 384 is important on the suppression activity of the BHLHE41. Both p.Tyr362His and p.Pro384Arg mutants have positively charged R groups whereas p.Pro384Gln has a negatively charged group. Although p.Pro384Gln has a strong affinity toward the E-box, it is possible that duration BHLHE41 p.Pro384Arg variant is the same as the wild-type BHLHE41 on E-box, and therefore we do not see the phenotype at a physiological level. However, this observation needs to be shown in the future.
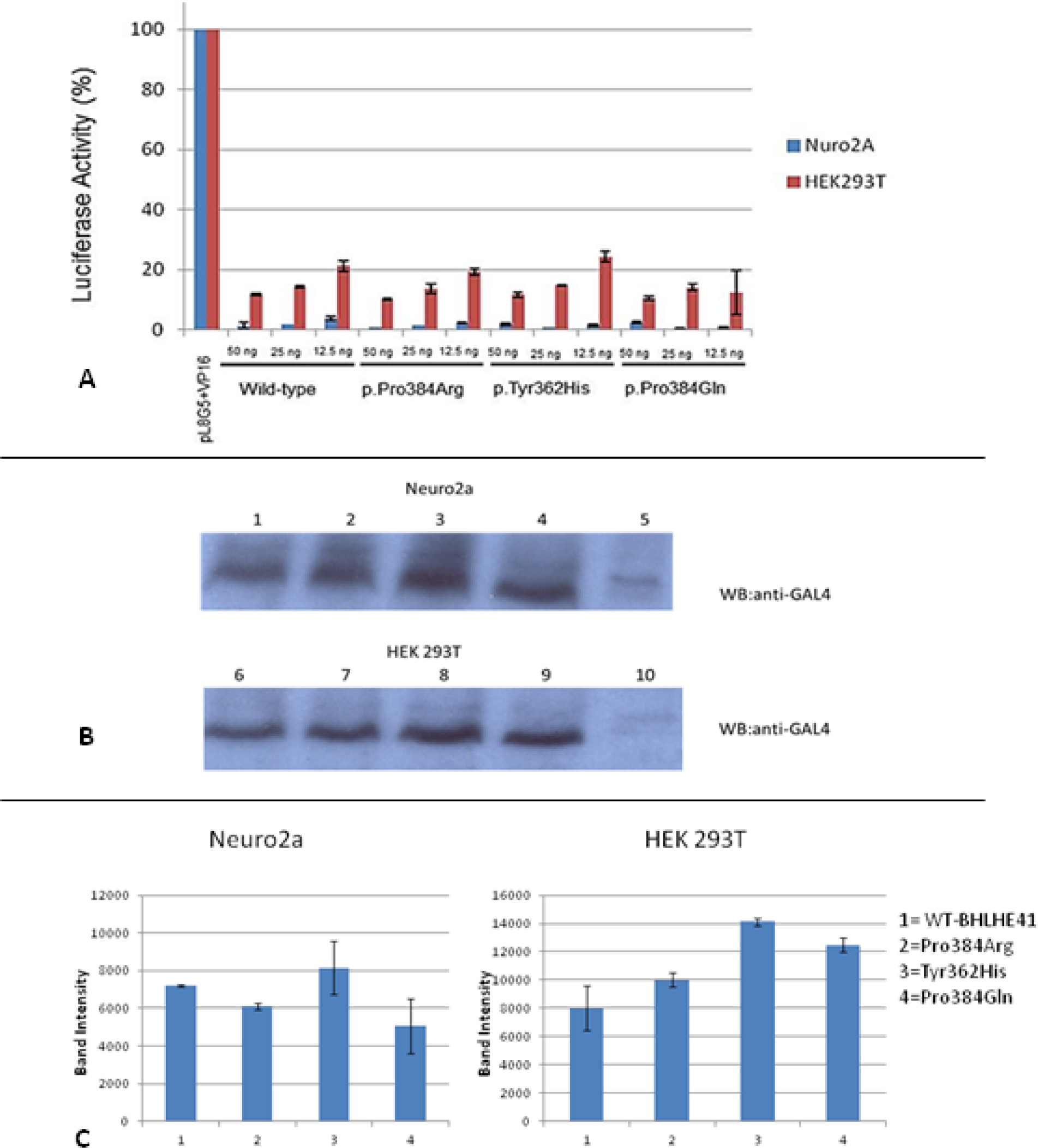
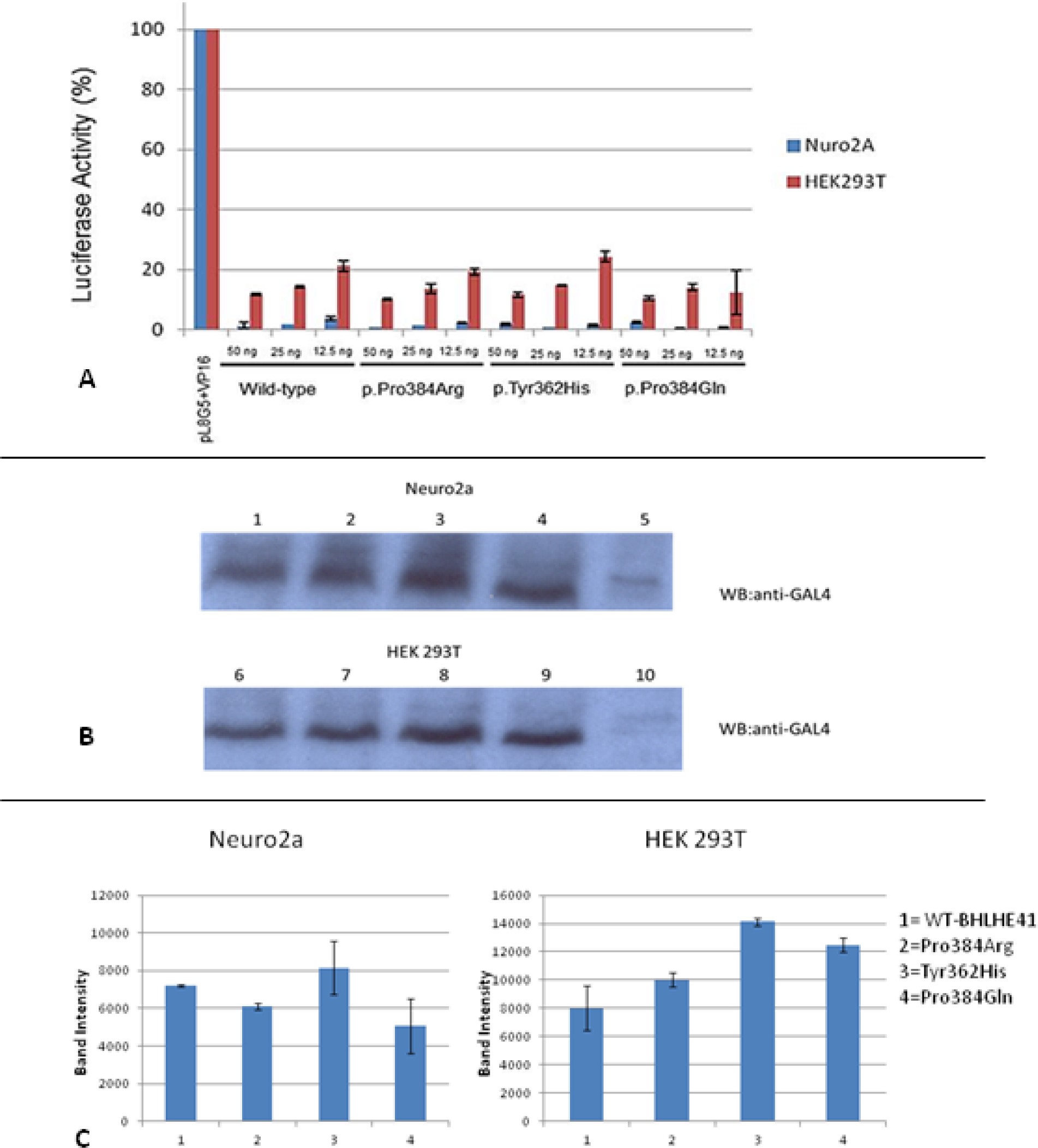
To evaluate whether the described variants changed the transcriptional repressor activity of BHLHE41 independently of E-box binding, we carried out GAL4-based transcription regulatory assays (Figure S3, supplemental material). HEK293T and Neuro2A cells were transiently transfected with pL8G5 (a reporter plasmid with Upstream Activation Sequence (UAS) and LexA operator sites) along with pBind-BHLHE41 and pLexA-VP16 plasmid. In this assay, transcriptional activity is normally high as the LexA-VP16 chimera is strongly active. Repressors can then be evaluated independently from their DNA binding activity as they are tethered to the UAS, an enhancer to which Gal4 specifically binds to activate gene transcription site in the reporter by GAL4. Cells expressing either the wild-type or variant constructs all had comparable activity, indicating the repressor activity of the BHLHE41 gene is intact. This indicates variants have no effect on the other transcriptional activity of BHLHE41 independently of E-box binding. To see if this result is not a consequence of the different expression level and protein stability, cell extracts obtained from both cell lines were subjected to Western blot analysis and then expression levels of both mutant and wild-type were quantified (Figure S3, supplemental material). The results indicated that both mutant and wild-type were expressed at comparable level. Neither WT nor BHLHE41 variants were active in this assay, suggesting they do not inhibit E-box transcription by physically interacting with CLOCK or BMAL1, which is consistent with previous work showing that BHLHE41 directly binds to the E-box and either activates or suppresses gene activation.28 Thus, functional variants of BHLHE41 can reduce affinity toward E-box binding.
Results From Random Mutagenesis
To determine if other possible mutations in the exon 5 region of BHLHE41 also alter repression of CLOCK/BMAL1 transactivation, we did random mutagenesis of amino acids between positions 362 to 384 in exon 5 by PCR using site-specific degenerate primers (Table S1, supplemental material). We randomly selected 50 mutagenized plasmids and tested them in the CLOCK/BMAL/Per2:luciferase transactivation assay (Figure 3). We found that several mutagenized plasmids altered transcriptional repression, highlighting the importance of exon 5 in suppression of CLOCK/BMAL1 activity and indicating that this area is a potential “hot spot” for functional variants. Other variants in this region result in the loss of repression activity of the BHLHE41 on CLOCK/BMAL1 activation (Figure 3). In particular, we named the possible mutants in our constructions with the letter M followed by a number representing the in vitro mutations. The M1, M5, and M35 mutants lost its repression activity on CLOCK/BMAL1 transactivation. Interestingly, one of these constructs, M33, harbored an Ala to Ser amino acid change at position 380 (close to the 384 candidate area). Analysis of the 1000 Genome Project data and National Center for Biotechnology Information shows that this allele (p.Ala380Ser) (rs1057206) occurs naturally in the CEPH population (European from Centre d'Etude du Polymorphisme Humain), although it is rare and sleep data are not available. This in silico mutation has a similar effect on CLOCK/BMAL1 transactivation as BHLHE41-p.Tyr362His and BHLHE41- p.Pro384Arg variants found in our subjects. Overall, these results indicate that variants in the region between p.Tyr362His and p.Pro384Arg result in altered repression activity of BHLHE41 on CLOCK/BMAL1 activation and likely will affect sleep duration and response to sleep deprivation.
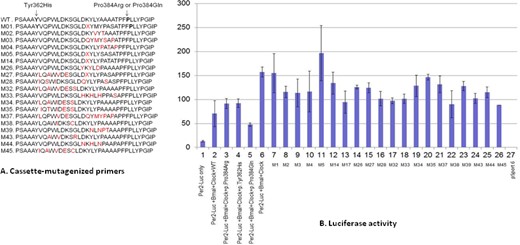
Analyses of the cassette-mutagenized mBHLHE41 by luciferase assay. (A) The region of mBHLHE41 between the 361 and 384 (codons) was mutagenized using appropriate primers by polymerase chain reaction. The effect of the variants was tested with the luciferase assay, which were carried out using reporter construct containing the Per2 E-box in HEK 293T. M stands for the variant and X indicates the presence of the stop codon. (B) The amounts of the vectors are 50ng along with plasmids containing BMAL1 and Clock cDNAs. Column 1 indicates self-activation of the reporter plasmids in HEK 293T. Columns 2 through 5 show repression activity of wild-type BHLHE41, BHLHE41-c.1151C > A (p.Pro384Arg), BHLHE41-c.1086C > T (p.Tyr362His), and BHLHE41-c.1151C > A (p.Pro384Gln) on the transactivation mediated by Clock/Bmal1, respectively. Column 6 shows transactivation of Clock/Bmal1 on reporter plasmids. Columns 7 through 26 indicate the effect of the BHLHE41 variants (indicated by M) listed in Figure 3A on Clock/Bmal1 trans-activation. Column 27 indicates that cells transfected with empty mammalian expression vector (pCMV-Sport 6). The figure shows that variants BHLHE41 (c.1086C > T (p.Tyr362His) and C.1151C > A (p.Pro384Arg) variants have less suppression activity on Clock/Bmal1 trans-activation compared with wild-type BHLHE41 and c.1151C > A (p.Pro384Gln) variant activity indicated by lines. Values of luciferase activity (y-axis) are the averages of three independent transfections (means ± standard error of the mean; n = 9), normalized to cotransfected CMV-Renilla, and expressed as a percentage of the values obtained with Gal4 alone for each cell line. The following abbreviations were used in x-axis: BM, Bmal1; M, variant of BHLHE41.
DISCUSSION
Our studies reveal that there are other variants of BHLHE41 in the human population that affect sleep duration, i.e., p.Tyr362His. The variant that led to an altered sleep phenotype affected the ability of BHLHE41 to inhibit both CLOCK/ BMAL1 and NPAS2/BMAL1 transactivation. Another variant found in the current study had no obvious effect on sleep duration (p.Pro384Gln) and no effect on CLOCK/BMAL1 or NPAS2/BMAL1 transactivation. Thus, it seems reasonable to propose that this aspect of the molecular machinery of the circadian clock is involved in setting sleep duration and also the level of sleep drive. Thus, BHLHE41 likely plays a role in both the clock machinery and sleep homeostatic mechanisms.
The mammalian molecular clock system includes a number of genes and their protein product complexes29–30 involved in interconnected feedback loops of transcriptional and translational regulation through enhancer elements such as CACGTG E-box, D-box, and ROR/REV-ERB binding elements (RORE).31 The E-box is believed to be an important element in the molecular oscillatory system, because it is the binding site for the CLOCK/BMAL1 heterodimer, which upregulates expression of various clock genes, including BHLHE41, BHLHE40, PER1, Dbp, and Rev-erb. In this regulatory model, PER, CRY, and BHLHE41 work as negative factors for transcription from E-box determined promoters, and the E-box-like element E-box (CACGTT) has been shown to be involved in the direct regulation of PER2 and CRY1 genes by CLOCK/BMAL1.22
Our study extends the results of He et al.,21 who found a point mutation in the BHLHE41 gene in two members of a family of short sleepers. The individuals in their study had habitual self-reported total sleep time per 24-h day that was much shorter in mutation carriers (average 6.25 h) compared with the noncarriers (average 8.06 h). We have identified two new variants of BHLHE41. One of these variants had an effect on sleep duration and inability to suppress CLOCK/BMAL1 activation of gene transcription, and the other did not. Given that the sample sizes used are not particularly large (four variants in a total of 589 samples sequenced) it seems likely that variants of BHLEH41 will occur fairly commonly. The phenotypic information we obtained from the twin with the novel variant extends previous descriptions of the effect of BHLEH41 variants on sleep. The carrier twin demonstrated significantly fewer average lapses of performance alertness compared with his brother. We can hypothesize that this gene plays a role in sleep length and affects resistance to sleep loss. We observed, however, in addition, that although sleep duration was shorter, there was a relative preservation of the amount of NREM sleep. There was minimal difference in the total amounts of NREM sleep between the twin with the mutation and the sibling. The twin with the mutation also had higher delta power, a putative measure of sleep drive. That behavioral impairment with sleep loss is lower in the twin with the mutation but delta power is higher, adding further questions about what delta power is actually assessing. Previous studies of short sleepers, albeit not evaluating genetic associations, have shown that short sleepers have higher delta power throughout the day than long sleepers.26 The rate of increase in delta power during wakefulness and decline during sleep are, however, the same.8,26 Thus, short sleepers terminate sleep at higher delta power than long sleepers. Data from our twin with the mutation are compatible with this concept.
Even in the absence of sleep, i.e., when kept awake, short sleepers show an earlier termination of the rise of the nocturnal melatonin in the blood and an earlier increase in morning cortisol than long sleepers.32 Investigators who observed these differences conjectured that the different timing of these neurohormonal changes, even in the absence of sleep, must result from variations of clock genes. Our data, and that of He et al.,21 support this assertion, and it will be of value in the future to investigate neuro-hormonal profiles in the absence of sleep in individuals with functional BHLEH41 variants. It will also be of value in future studies to evaluate other family members.
Chronic partial sleep deprivation in healthy humans leads not only to performance impairment but also to metabolic consequences, i.e., insulin resistance,33–36 alteration in phosphorylation of AKT in fat cells in response to insulin,36 decreased leptin, increased ghrelin, increased appetite37 and caloric intake,38 weight gain,38 and preference for high calorie foods,37–40 particularly during late-night hours.38 This has led to the concept that sleep deprivation is a risk factor for obesity.9 Our data do not suggest that there are metabolic consequences for the BHLHE41 variations because the BMI of the twins is almost identical (23.9 kg/m2 for the carrier and 25.3 kg/m2 for the noncarrier). However, further study of the consequences of BHLHE41 variants on metabolism is warranted.
In addition to Clock, NPAS2 is also a binding partner of BMAL1 and has been proposed as a specific energy sensor for the control of rest/activity.17,19,20 NPAS2 alters sleep/wake amounts and responses to stimuli other than light, in particular restricted feeding, as revealed by studies of NPAS2 knockout mice.13,17,19 We found variants in BHLHE41 have identical effects on both CLOCK/BMAL1 and NPAS2/BMAL1 transactivation. Thus, whether the functional effect of BHLHE41 variants is through NPAS2 or CLOCK remains an open question. We show that like p.Tyr362His, this new variant (in the twin subject) results in a loss of repression activity of NPAS2/BMAL1 whereas the p.Pro384Gln has a strong E-box binding activity. Our results indicate that the charge of amino acid R groups at the position of 384 is important in the suppression activity of the BHLHE41. Both p.Tyr362His and p.Pro384Arg mutants have positively charged R groups, whereas p.Pro384Gln has a negatively charged group. The presence of the negatively charged amino acids enhances the BHLHE41 activity on E-box. We speculate p.Pro384Gln has a strong and fast ability to bind to the E-box region, but has an even faster release from the E-box, which makes it difficult to detect and characterize the behavior phenotype. Cell culture studies have shown that BHLHE41 does not always act as a transcriptional repressor of E-box-controlled genes because this varies by cell type depending in part on whether or not BMAL1 is present.22,23,42
Overall, the new variant (p.Tyr362His) also alters the architecture of sleep, allowing the same amount of NREM sleep in a shorter total sleep time and having a protective effect on neurobehavioral performance in response to sleep deprivation. We explored the biochemical alteration on BMAL/ CLOCK/NPAS transactivation caused by different variants of BHLHE41 in the circadian machinery. We found that structural changes in the protein can affect transient transactivation. It was demonstrated previously that BHLHE41 binds to class B E-box elements (CACGTG) as a homodimer and to repress the transcription of target genes in an HDAC-dependent manner.47 Thus, although the role of clock genes such as BHLHE41 is well established in controlling the diurnal rhythm of gene expression in many organs,41–43 they may also play a fundamental role in determining the duration of sleep that an individual needs. We further show; based on random mutagenesis, that there are likely other variants of BHLHE41 that can alter sleep duration and sleep loss response. In summary, our study provides complete phenotype information including the polysomnography, delta power, and performance evaluation in humans, not simply measuring less rebound sleep. We implemented additional approaches: prima facie estimate of the frequency of BHLHE41 alleles in humans, and evidence that more alleles with this phenotype are likely to emerge. Also, we suggest the role of canonical clock factors in sleep homeostasis.
Important Considerations
Our results suggest that variants of BHLHE41 affecting sleep duration and response to sleep loss may be more common than previously appreciated. Moreover, there are a number of other variants of this gene that have similar functional effects on the molecular machinery of the clock. But, whether these variants occur in human populations is unknown. Larger studies of variants of this and other relevant clock genes in human populations with short sleep are needed. Our data also suggest that altering the repression of CLOCK/BMAL1 and/or NPAS2/ BMAL1 has a direct effect on setting the duration of sleep and sleep architecture with a relative preservation of amounts of NREM sleep in the face of shorter sleep. The neuronal basis for this effect is unknown and needs to be studied. This adds additional evidence that at a molecular level the circadian clock and sleep homeostasis are not independent processes. However, these findings about a clock-related gene could be independent of its function in the circadian clock. This work provides an important second allele of BHLHE41 associated with sleep deprivation and for the first time shows the role of BHLHE41 in resistance to sleep deprivation in humans, confirming previous work on the resistance to sleep deprivation in rodents.21 We also highlighted the possible role of canonical clock factors in sleep homeostasis. Future research may elucidate possible mechanisms of the gene variants' role in sleep length as well as reveal target molecules that enhance sleep homeostasis and improve neurobehavioral responses to sleep deprivation.
ACKNOWLEDGMENTS
The authors thank Dr. Fu for providing the plasmids constructions. We thank Saarene Panossian for help with sequence analysis. We thank Cuiping Hou, Micah Romer, and Nada Abdel-Magid for their collaboration in sample organization and DNA plate preparation. We thank Ron Anafi-and Diego Mazzotti for the critical reading of this manuscript. Author contributions: Dr. Pelligrino performed and analyzed the data of the genetic analyses including the DNA extraction, Sanger sequencing and data analysis. She contributed to writing the manuscript. Dr. Kavakli performed all functional assays and cloning experiments and contributed to writing the manuscript. Both contributed equally to the manuscript. Dr. Goel provided the cohort of non-related individuals and the respective sleep phenotypes. Dr. Cardinale helped with the Sanger primer selection and data interpretation. Dr. Dinges evaluated and studied the cohort of nonrelated individuals who underwent chronic partial sleep deprivation. Dr. Kuna was primarily responsible for the studies of twins involving assessment of response to sleep deprivation. Dr. Maislin was responsible for statistical analysis and data interpretation (EEG phenotypes). Dr. Van Dongen contributed to study design of the twin study and EEG data analyses. Dr. Tufik added fundamental comments on sleep deprivation and data interpretation. Dr. Hogenesch designed and provided the resources to conduct the functional studies and co-authored the manuscript. Dr. Hakonarson provided laboratory structure and supervision for the Sanger sequencing and genetic analysis and edited and co-authored the manuscript. Dr. Pack designed the project, coordinated the main cohort sample collection (twin cohort), provided the phenotypic information and co-authored the manuscript.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments