





Abstract
Endocrine disrupting chemicals (EDCs) interfere with the biosynthesis, metabolism, and functions of steroid hormones, including estrogens and androgens. Aromatase enzyme converts androgen to estrogen. Thus, EDCs against aromatase significantly impact estrogen- and/or androgen-dependent functions, including the development of breast cancer. The current study aimed to develop a biologically relevant cell-based high-throughput screening assay to identify EDCs that act as aromatase inhibitors (AIs), estrogen receptor (ER) agonists, and/or ER antagonists. The AroER tri-screen assay was developed by stable transfection of ER-positive, aromatase-expressing MCF-7 breast cancer cells with an estrogen responsive element (ERE) driven luciferase reporter plasmid. The AroER tri-screen can identify: estrogenic EDCs, which increase luciferase signal without 17β-estradiol (E2); anti-estrogenic EDCs, which inhibit the E2-induced luciferase signal; and AI-like EDCs, which suppress a testosterone-induced luciferase signal. The assay was first optimized in a 96-well plate format and then miniaturized into a 1536-well plate format. The AroER tri-screen was demonstrated to be suitable for high-throughput screening in the 1536-well plate format, with a 6.9-fold signal-to-background ratio, a 5.4% coefficient of variation, and a screening window coefficient (Z-factor) of 0.78. The assay suggested that bisphenol A (BPA) functions mainly as an ER agonist. Results from screening the 446 drugs in the National Institutes of Health Clinical Collection revealed 106 compounds that modulated ER and/or aromatase activities. Among these, two AIs (bifonazole and oxiconazole) and one ER agonist (paroxetine) were confirmed through alternative aromatase and ER activity assays. These findings indicate that AroER tri-screen is a useful high-throughput screening system for identifying ER ligands and aromatase-inhibiting chemicals.
Abbreviations
- AI
aromatase inhibitor
- AR
androgen receptor
- BPA
bisphenol A
- DMSO
dimethyl sulfoxide
- EDCs
endocrine disrupting chemicals
- ER
estrogen receptor
- ERE
estrogen response element
- E2
17β-estradiol
- FDR
false discovery rate
- FRD
Flying Reagent Dispenser
- IGF-1
insulin growth factor 1
- IPA
Ingenuity Pathway Analysis
- PGR
progesterone receptor
- qHTS
quantitative high-throughput screening
- TAM
tamoxifen
- T
testosterone
Endocrine disrupting chemicals (EDCs), which include both natural and synthetic compounds, have the ability to interfere with the biosynthesis, metabolism, and normal functions of hormones such as estrogens and androgens. Exposure to EDCs in the environment during development or in later life may result in disturbances in the physiological actions of hormones through multiple mechanisms (Casals-Casas and Desvergne, 2011; Diamanti-Kandarakis et al., 2009). Aromatase is an enzyme (CYP19) that converts androgen to estrogen. Alterations in aromatase activity and expression can lead to changes in androgen and estrogen levels, thereby altering normal physiological functions that involve these steroid hormones. Aromatase is therefore also an important target of EDCs (Chen, 2002; Kao et al., 1998).
Estrogen-dependent breast cancer is the most common form of breast cancer, accounting for 60% and 75% of breast cancers in premenopausal and postmenopausal women, respectively. Although three estrogen receptor (ER) isoforms (i.e., ERα, ERβ, and membrane-bound ER) have been reported in humans, only the roles of ERα are well defined clinically in breast cancer. In this paper, ER stands for ERα unless it is specified differently. ER and aromatase enzyme play critical roles in the development and recurrence of estrogen-dependent breast cancer. Sometimes, ER is overexpressed and/or overactivated in estrogen-dependent breast cancer, promoting the growth of the tumors. Studies have shown that uncontrolled ER activation plays a critical role in breast cancer resistance to antiestrogens, such as tamoxifen (TAM), and aromatase inhibitors (AIs; e.g., Masri et al., 2008). Abnormal expression of aromatase in breast cancer cells and/or surrounding adipose stromal cells has been shown to significantly influence tumor development and growth (Martin et al., 2003; Masri et al., 2008; Yue et al., 2002).
In female mice, ovariectomy increases body weight through increased food intake (Asarian and Geary, 2006) and reduces energy expenditure (Rogers et al., 2009), suggesting that lowering the level of estrogens may induce obesity. In addition, androgens were found to promote food intake through altered leptin sensitivity and enhanced obesity in ovariectomized mice (Kanaya et al., 2013). Obesity may induce or promote breast cancer through increased production of leptin, insulin, insulin growth factor 1 (IGF-1), and proinflammatory cytokines (Vucenik and Stains, 2012). Elevated androgen levels are also associated with increased fat mass in young women (Sowers et al., 2001). Disruption of the normal androgen/estrogen ratio due to altered aromatase levels or activity may therefore indirectly promote breast cancer development and other metabolic diseases.
Despite extensive efforts, a definitive understanding of the physiological impact of AI-like EDCs has not yet been established, and there are currently no reliable biologically relevant high-throughput screening tools with which to identify EDCs that act as AIs. We previously generated the MCF-7aro cell line, which stably overexpresses aromatase and is a model cell line to study ER- and aromatase-dependent proliferation in vitro and in vivo (Sun et al., 1997; Yue et al., 1994; Zhou et al., 1990). The major ER isoform in MCF-7 cells is ERα (Al-Bader et al., 2011; Zivadinovic et al., 2005). To screen EDCs that act as AIs, ER agonists, or ER antagonists, we subsequently developed an MCF-7aroERE cell line that expresses luciferase under the control of the SV40 promoter regulated by an estrogen response element (ERE; Lui et al., 2008). However, MCF-7aroERE cells had a high background level of luciferase activity, preventing the assay from identifying weak acting EDCs and hampering efforts to adapt the assay to a high-throughput screening format. The objective of the current study was to develop a biologically relevant cell-based high-throughput screening assay to reliably identify EDCs that act as AIs, ER agonists, and/or ER antagonists. This is an important tool to reduce the use of animals in EDC research.
MATERIALS AND METHODS
Materials
Unless they are defined differently, the biochemicals used in this study were purchased from Sigma-Aldrich Corporation. (St. Louis, MO, USA).
Generation of the AroER tri-screen (MCF-7aroERE.2) Cells
Construction of pGL4.26 (ERE)3
We constructed a luciferase reporter vector with the goal of low background and high 17β-estradiol (E2)-induced luciferase activity. Three tandem copies of the ERE consensus sequence insert were synthesized, annealed, and subcloned into the pGL4.26 luciferase reporter vector (Promega Corporation, Madison, WI). The nucleotide sequences of two strands of the ERE insert are: 5′-CCAGGTCAGAGTGACCTGCCAGGTCAGAGTGACCGC CAGGTCAGAGTGACCTG-3′ and 5′-CAGGTCACTCTGA CCTGGCAGGTCACTCTGACCTGGCAGGTCACTCTG ACCTGGGTA-3′. The orientation and sequences of the ERE insert were verified by DNA sequencing. The resulting reporter vector was named pGL4.26 (ERE)3.
Transfection, hygromycin selection, and cell cloning
MCF-7aro cells were seeded in 6-well plates and transfected with pGL4.26 (ERE)3 using Lipofectamine 2000 transfection reagent (Invitrogen, Grand Island, NY). Forty-eight hours after transfection, cells were cultured in media containing hygromycin (200 μg/mL). Hygromycin-resistant mass clones were then cloned by serial dilution in 96-well plates and grown for two weeks. The wells containing single colonies were then identified by microscopy and subcultured into 24-well plates and then into 6-well plates.
Clone selection for maximum E2-induced luciferase activity
Individual MCF-7aroERE cell clones isolated by serial dilution were analyzed for background luciferase activity and E2-induced luciferase activity. Cells from expanded individual MCF-7aroERE clones were maintained in Eagle's Minimal Essential Medium (MEM; Thermo Scientific, Rockford, IL) supplemented with 10% fetal bovine serum (FBS), 2mM L-glutamine, 1mM sodium pyruvate, 1% nonessential amino acids, and 100 U/mL penicillin-streptomycin (maintenance medium). Two days before treatment, the medium was changed to phenol red-free MEM medium with 5% charcoal/dextran-treated FBS (Omega Scientific Inc., Tarzana, CA) (assay medium). The day before treatment, cells were seeded in 24-well plates at 2 × 105 cells per well and continuously cultured in the same medium overnight.
After overnight incubation, cells were treated with either E2 (1nM), E2 plus ICI 182780 (ICI) (100nM) (ICI is an ER antagonist and suppresses ERE-luciferase activity in the presence of E2), or dimethyl sulfoxide (DMSO) (solvent control). Cells were treated for 24 h, washed once with PBS, and lysed in Passive Lysis Buffer (Promega Corporation). Luciferase activity in the cell lysates was measured using the Luciferase Assay Substrate (Promega Corporation) and normalized by protein concentration. Each assay was performed in triplicate. The clone with the highest E2-induced luciferase activity was named MCF-7aroERE.2 (defined as AroER tri-screen) and selected for further analysis.
AroER Tri-screen in 96-Well Format
Evaluation of the assay
AroER tri-screen cells were cultured in maintenance medium. Prior to the assay, cells were incubated for 2 days with phenol red-free MEM containing 10% charcoal-treated-FBS and then seeded in 96-well plates at 2 × 105 cells per well. After overnight incubation, cells were treated with E2 (0.5nM), testosterone (T) (0.5nM), E2 plus ICI (100nM), or DMSO for 24 h.
To further examine the performance of the assay, cells were treated with bisphenol A (BPA), an established EDC with estrogen-like activity. Prior to screening, cells were incubated as described above and then treated with E2 (0.5nM), E2 plus ICI (100nM), T, T plus letrozole (a known AI) (200nM), BPA alone (10μM), BPA plus ICI, BPA plus letrozole, or DMSO for 24 h. Letrozole was purchased from Selleckchem Corporation (Houston, TX).
Luciferase signal was measured using the One-Glo Luciferase Assay System (Promega Corporation) and normalized by protein concentration against the DMSO control. Each assay was performed in triplicate.
Screening for EDCs affecting aromatase and/or ER activity
Prior to screening, AroER tri-screen cells were cultured in phenol red-free MEM containing 10% charcoal-treated FBS for 2 days and then seeded into 96-well plates at 2 × 105 cells per well. The following day, 446 drugs (10μM) from a National Institutes of Health (NIH) Clinical Collection [Evotec (US) Inc., San Francisco, CA] were individually transferred onto the assay plates using the Biomek FX liquid handler (Beckman Coulter, Brea, CA), in the presence of T (0.3nM) for AI screening, DMSO (control) for ER agonist screening, and E2 (0.5nM) for ER antagonist screening in a total volume of 100 μL. Cells were then incubated at 37ºC for 24 h. After incubation, 100 μL of 1X One-Glo reagent (Promega Corporation) was added to each well and incubated for 5 min. Luciferase activity was read using a PHERAstar plate reader (BMG Labtech, Cary, NC). The results were interpreted as follows: EDCs that acted as ER agonists induced ERE-luciferase activity in the absence of E2; EDCs that acted as ER antagonists suppressed ERE-luciferase activity in the presence of E2; and EDCs that acted as AIs suppressed ERE-luciferase activity in the presence of T by blocking the conversion from T to E2 (Fig. 1). Letrozole (200nM) was used as a positive AI control and ICI (100nM) was used as an ER antagonist positive control.
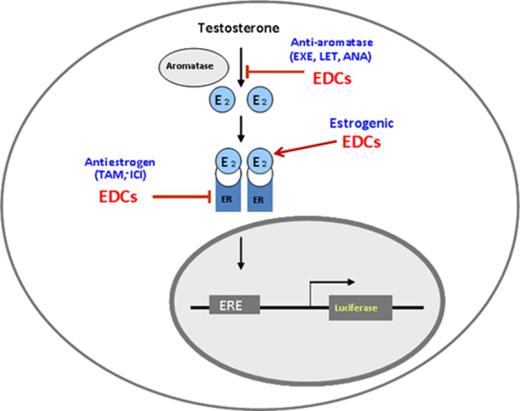
Working principles of MCF-7aro/ERE.2 cells—AroER tri-screen.
Validation of EDCs
Aromatase enzyme kinetic assay
To determine if and how the selected EDCs inhibited aromatase (competitively or noncompetitively), we performed an aromatase kinetic analysis using a tritiated water-release assay (Grube et al., 2001). AroER tri-screen cells were incubated with androst-4-ene-3,17-dione [1-β-3H(N)] (NEN-Dupont, Boston, MA) (the androgen substrate) at varying concentrations (25nM to 400nM) plus bifonazole (10nM or 50nM) or oxiconazole (30nM or 120nM) for 1 h. Tritiated water is produced when tritiated androstenedione is converted to estrone by aromatase. After incubation, the unreacted tritiated androstenedione was first absorbed by dextran-coated charcoal, and the tritiated water product was quantified by a liquid scintillation counter. Aromatase activity was expressed as pmol of tritiated water released per mg protein per hour (pmol/mg/h).
ERE luciferase assay
Two ER-positive breast cancer cell lines, T47D and MCF-7, were transiently transfected with pGL3 (ERE)3 using the Lipofectamine Plus reagent system (Invitrogen). After 24 h, the cells were treated with the test chemicals (100nM to 10μM) alone and/or with 0.5nM of E2 in a total volume of 100 μL, and incubated at 37ºC for an additional 24 h. The treated cells were lysed using passive lysis buffer. Luciferase activity was measured on a TD 20/20 luminometer (Turner Designs, Sunnyvale, CA) and the protein concentration was determined using the bicinchoninic acid (BCA) Assay (Thermo Scientific). The estrogen-like chemicals were defined as those that induced the ERE luciferase activity by themselves. The antiestrogen-like chemicals were identified by their ability to suppress the E2-induced ERE luciferase activity.
AroER Tri-screen in a 1536-Well Format
AroER tri-screen cells were cultured in phenol red-free MEM containing 10% charcoal stripped FBS for 2 days prior to the assay. Cells were then resuspended in the same medium and dispensed at 1500 cells, 4 μL per well, in 1536-well white tissue cultured plates using a Thermo Scientific Multidrop Combi (Thermo Fisher Scientific Inc., Waltham, MA). Cells were incubated at 37ºC for 5 h. For agonist mode, 23 nL of T or E2 at 16 concentrations ranging from 0.3 fM to 4.6nM was added to the assay plates by a pin tool (Kalypsys, San Diego, CA). For antagonist mode, 23 nL of letrozole at 16 concentrations ranging from 1.4nM to 46μM was added to the plates by a pin tool (Kalypsys), followed by addition of 1 μL of the same medium plus or minus T (final concentration of 0.5nM). Plates were incubated at 37°C for 24 h and 4 μL of ONE-Glo Luciferase Assay reagent (Promega) was added using a Flying Reagent Dispenser (FRD) (Aurora Discovery, Carlsbad, CA). Plates were then incubated at room temperature for 30 min, and luminescence intensity was measured by a ViewLux plate reader (Perkin Elmer, Shelton, CT).
RNA-Seq Analysis
Sample preparation
AroER tri-screen cells were treated with DMSO, paroxetine (20μM), or E2 (1nM) for 24 h. For RNA-seq analysis, total RNA was extracted using RNeasy Mini Kit (QIAGEN, Valencia, CA). Preparation of the mRNA samples for RNA-seq analysis was performed using the TruSeq RNA Sample Preparation Kit V2 (Illumina, San Diego, CA). Briefly, sample preparation began with 0.5 μg of total RNA from each sample. Polyadenylated RNA was purified from the sample with oligo dT magnetic beads, and the poly (A) RNA was fragmented with divalent cations under elevated temperature. First-strand cDNA synthesis produced single-stranded DNA copies from the fragmented RNA by reverse transcription. After second-strand cDNA synthesis, the double-stranded DNA underwent end repair, and the 3’ ends were adenylated. Finally, universal adapters were ligated to the cDNA fragments, and polymerase chain reaction was performed to produce the final sequencing library. These template molecules were used for cluster generation and sequencing on the Illumina HiSeq 2000 (Illumina) instrument. Four samples per lane were used to generate 40 base pair single end reads.
Statistical processing of RNA-seq data
Reads were aligned to the human genome (hg19) using TopHat (Trapnell et al., 2009). Partek Genomics Suite (Partek Inc., St. Louis, MO) was used for mRNA quantification and statistical analysis. Reads were attributed to transcripts. Reads Per Kilobase per Million (RPKM; Mortazavi et al., 2008) expression values were calculated using an expectation-maximization algorithm (Xing et al., 2006) using the RefSeq (Pruitt et al., 2012) genomic coordinates for all known transcripts. To better evaluate the data and to focus on the most biologically relevant genes, we added a factor of 0.1 to all RPKM values (effectively censoring transcripts with very low expression values). For statistical analysis, RPKM values were log2 transformed (although fold-change values were calculated on a linear scale). Fold-change values were calculated based upon the least-squares mean, p-values were calculated using 1-way ANOVA with appropriate linear contrast, and false discovery rate (FDR) values were calculated using the method of Benjamini and Hochberg (1995). Differentially expressed genes were defined as those showing a fold-change >1.5 and an FDR < 0.05. Our microarray data are MIAME compliant and the raw data have been deposited in NCBI Gene Expression Omnibus with accession number of SRP035276.
Ingenuity pathway analysis (IPA)
IPA is a Web-based software program that identifies the biological functions, pathways, and mechanisms most relevant to a given set of genes. To obtain a comprehensive view of the differentially expressed genes, IPA provided core analysis, including network generation, functional analysis, and canonical pathway analysis on the filtered genes (fold change >2, p < 0.05).
RESULTS
Generation of AroER Tri-screen
To construct an ERE-luciferase reporter with low luciferase background and high E2 induction, the pGL4.26 vector reporter (Promega Corporation) was used. This vector was chosen because that it has only a minimal promoter sequence instead of the full SV40 promoter in the pGL3 vector and there are far fewer consensus transcriptional factor binding sites on its backbone as well as on its Luc2 gene, compared with the pGL3 vector used for the generation of MCF-7aro-ERE cells. These unique features greatly reduce the background luciferase activity when the transfected cells are cultured in DMSO. In addition, the pGL4.26 vector carries a hygromycin resistance selectable marker that allows selection of stable clones using hygromycin on G418-resistant MCF7aro cells.
A 3X ERE insert was ligated into pGL4.26 to increase ER-binding affinities and thus increase hormone induction sensitivity. After transfection, hygromycin selection, and cell cloning by serial dilution, 28 clones of ERE-luciferase reporter-containing MCF-7aro were generated. Of these, eight individual cell clones had minimal background luciferase activity (nine luciferase units/μg protein) when cultured in the presence of the DMSO control, and high luciferase activity when cultured in the presence of either E2 (1nM) or T (1nM), yielding an excellent signal-to-background ratio. E2 induction ranged from 10 to 24-fold, and E2- and T-induced luciferase activities were blocked by ICI and letrozole, respectively. The cell line with the highest E2-induced luciferase activity (215 luciferase unites/μg protein) was chosen for further analysis and named AroER tri-screen, to differentiate it from the previously generated MCF-7aroERE cell line (Lui et al., 2008)
Assay Development in a 96-Well Screening Format
The high-throughput screening utility of the AroER tri-screen cell line was evaluated in a 96-well format. When seeded at 2 × 105 cells/well, the cells responded to 0.5nM E2 with a signal-to-background ratio of 17.6 and a screening window coefficient (Z-factor) (Zhang et al., 1999) of 0.57.
We then examined the performance of the AroER tri-screen cells (Fig. 1) under three conditions. An example run shown in Figure 2(A) shows that we conducted the screens under the following conditions: test compound-only, E2 + test compound, or T + test compound. These protocols identify chemicals acting as estrogens (examples A and C), antiestrogens (example E), or AIs (example B), respectively. Cells in the example D column showed cytotoxicity because all activities were suppressed under all three conditions. We then examined the activity of BPA under these three conditions and found that BPA functions as an ER agonist in a concentration-dependent manner and cannot block the activity induced by E2 or T (Fig. 2B). ICI and letrozole effectively suppressed E2- and T-induced activities, respectively. We also found that BPA-induced ERE-luciferase reporter activity was inhibited by ICI (Fig. 2C, right panel), but was unchanged by letrozole (Fig. 2B, left panel).
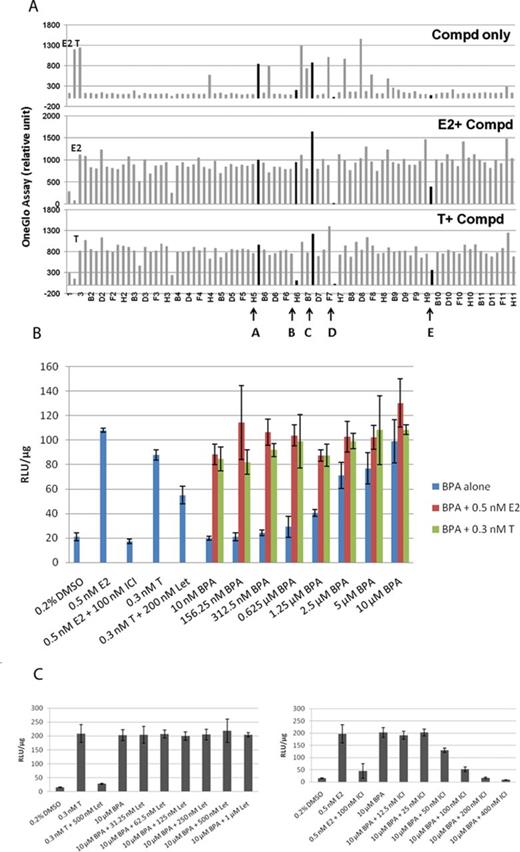
Establishment and evaluation of the AroER tri-screen assay for EDCs using a 96-well plate format. (A) Graphic interpretation of the AroER tri-screen data analyses. Cells were treated for 24 h with E2 (0.5nM), T (0.3nM), E2 plus ICI (100nM), or DMSO (solvent control) as controls. Compounds A–E are examples (highlighted in black) that explain the basis of our analyses. Compounds A and C are estrogenic compounds because they induce the ERE-luciferase reporter activity. Compound C may be a strong ER agonist because it further enhances the reporter activity when examined in the presence of E2 or T. Compound B is an AI because it suppresses the reporter activity only in the presence of T but not E2. In contrast, compound E is an ER antagonist because it suppresses the reporter activity in the presence of both T and E2. Because compound D suppresses the reporter activity in all three screening formats, it is thought to be a cytotoxic agent. (B) Evaluation of BPA by AroER tri-screen. Cells were treated for 24 h with either E2, E2 plus ICI (100nM), T, or T plus Let (200/500nM) as controls. BPA activity was assessed by treating the cells with BPA alone (10nM to 10μM) or in the presence of either 0.5nM E2 or 0.3nM of T. (C) Cells were treated with 10μM of BPA alone or with increasing concentrations of Let (31.25nM to 1μM) or ICI (12.5nM to 400nM). Luciferase reporter activity was measured using the One-Glo Luciferase Assay System and normalized by protein concentration. All experiments were performed in triplicate.
AroER Tri-screen in a 1536-Well Format
To expand the capabilities of the screening system, the AroER tri-screen assay was miniaturized to a 1536-well plate format with a final assay volume of 5 μL per well. T-induced luciferase activity was evaluated in a concentration-dependent manner (Fig. 3A). The 50% effective concentrations (EC50) of T and E2 were 92 pM and 110 pM, respectively. Because T is as effective as E2 in this assay, T is thought to be efficiently converted to E2 by the expressed aromatase in AroER tri-screen cells. The maximum induction of luciferase activity was more than eightfold the basal level. Letrozole effectively blocked T-induced luciferase activity with a 50% inhibitory concentration (IC50) of 11nM (Fig. 3B), confirming that the T-induced luciferase activity in AroER tri-screen cells is a measurement of aromatase activity.
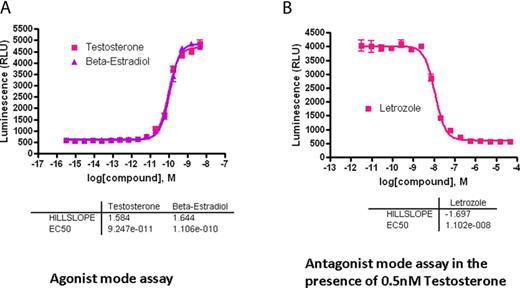
Development of a 1536-well screening format for high-throughput analysis. The AroER tri-screen assay was miniaturized to a 1536-well plate format with a final assay volume of 5 μL per well. After cells were incubated for 24 h, T-induced luciferase activity was evaluated in a concentration-dependent manner. (A) Agonist mode assay: The 50% effective concentration (EC50) of T was 92 pM, and the maximum induction of aromatase activity was more than eightfold the basal level. E2 serves as the positive control. (B) Antagonist mode assay in the presence of 0.5nM T. Letrozole blocked T-induced luciferase activity, with an IC50 value of 11nM.
To evaluate assay performance, the cells were tested in the presence of 0.5nM of T. The signal-to-background ratio was 6.9-fold, the coefficient of variation (Reed et al., 2003) was 5.4%, and the Z-factor was 0.78.
Validation of EDCs That Inhibit Aromatase
To evaluate the utility of the AroER tri-screen assay, we screened the NIH Clinical Collection library of 446 drugs in a 96-well plate format. The screening results revealed 106 drugs that showed ER and/or aromatase activities. Sixty-seven drugs induced luciferase-reporter activity, indicating that they could function as ER agonists. Furthermore, 39 drugs suppressed T-induced reporter activity; among them, 10 drugs only suppressed T-induced activity, indicating that they were AIs, and 29 drugs suppressed both E2- and T-induced activities, indicating that they were ER antagonists. Three drugs were chosen for verification of their effects against aromatase and ER. Bifonazole and oxiconazole (AIs) demonstrated potent AI activity in a dose-dependent manner through the tritiated water-release assay (Fig. 4A), whereas paroxetine (ER agonist) did not show AI-like activity (data not shown). In the presence of 50nM of androst-4-ene-3,17-dione [1-β-3H(N)], bifonazole and oxiconazole inhibited aromatase activity with IC50 values of 4.1nM and 40.4nM, respectively (Fig. 4A), and enzyme kinetic analysis showed that both bifonazole and oxiconazole are noncompetitive inhibitors of aromatase with respect to the androgen substrate (Fig. 4B).
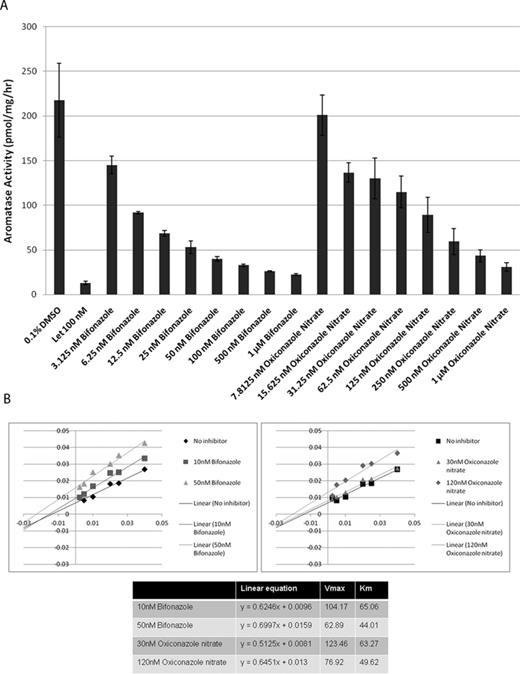
Screening of the NIH Clinical Collection library with AroER tri-screen assay. (A) Dose-dependent inhibition of aromatase by bifonazole and oxiconazole through the tritiated water-release assay. (B) Molecular basis of aromatase inhibition by bifonazole and oxiconazole, examined by enzyme kinetic analysis. The double reciprocal Lineweaver Burk plot reveals that both bifonazole and oxiconazole are noncompetitive inhibitors of aromatase with respect to the androgen substrate.
Validation of EDCs That Affect the ER
To determine if bifonazole, oxiconazole, or paroxetine interacts with the ER, the ER-positive cell lines, MCF-7 and T47D, were treated with each chemical up to 10μM. In both MCF-7 (Fig. 5A) and T47D (Fig. 5B) cells, paroxetine induced a significant, concentration-dependent increase in ERE-luciferase activity compared with the DMSO control whereas bifonazole and oxiconazole are not ER ligands in either cell type (data not shown), consistent with the initial AroER tri-screen. At 10μM of paroxetine, ERE-luciferase activity was 50–80% of that in the E2 positive control, depending on the cell type.
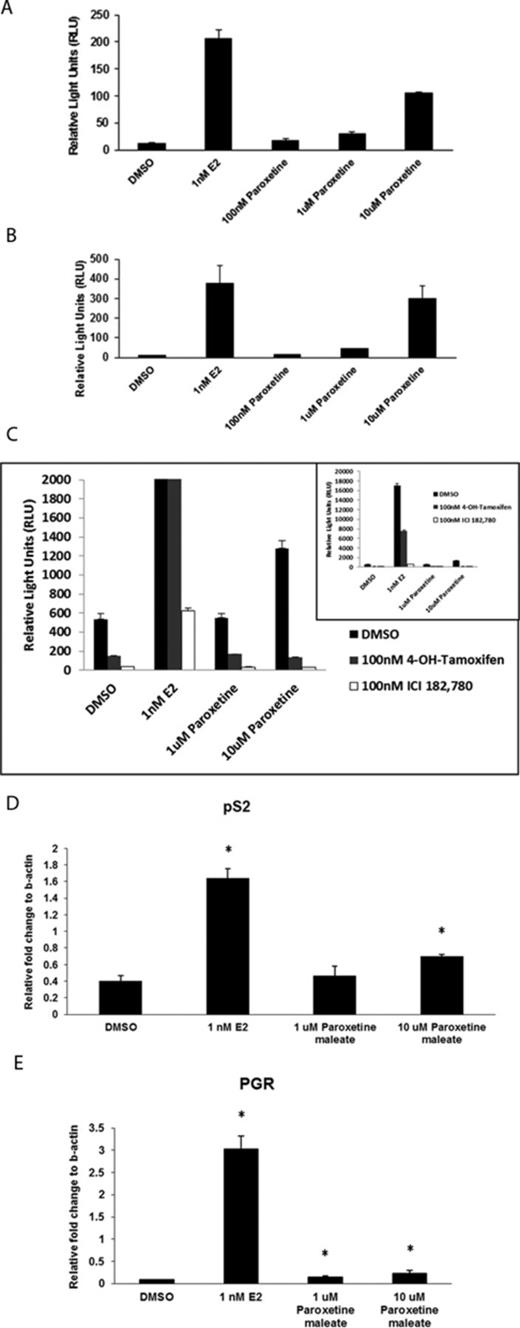
Evaluation of estrogen-like activity of paroxetine in MCF-7 and T47D cell lines. The ER-positive cell lines MCF-7 (A) and T47D (B) were treated with paroxetine alone, at concentrations of 100nM, 1μM, and 10μM. Paroxetine induced a significant dose-related increase in ERE-luciferase activity, compared with the DMSO control. E2 serves as the positive control. (C) Paroxetine-induced ERE-luciferase activity in AroER tri-screen cells in a dose-dependent manner. In the presence of either 4-OH TAM or ICI, this activity was suppressed. The inset shows the results in full scale. The expression of ER-responsive genes pS2 (D) and PGR (E) in MCF-7 cells was significantly induced by 10μM paroxetine. *p < 0.05, Student's t-test when comparing with the DMSO control.
To determine the mechanism by which paroxetine induced ERE-luciferase activity, AroER tri-screen cells were treated with 4-OH TAM or ICI (ER antagonists). Paroxetine alone induced ERE-luciferase activity in AroER tri-screen cells in a concentration-dependent manner, but in the presence of either 4-OH TAM or ICI, this activity was suppressed (Fig. 5C). We also observed a significant induction of the ER-responsive genes pS2 and progesterone receptor (PGR) in MCF-7 cells with 10μM paroxetine treatment (Figs. 5D and E), consistent with direct induction of ER activity by paroxetine.
To examine whether the tested chemicals function as ER antagonists, MCF-7 and T47D cells were treated with E2 (1nM) and each of the three chemicals (bifonazole, oxiconazole, and paroxetine) at three concentrations (100nM, 1μM, and 10μM). None of these compounds inhibited E2-induced ER activity (data not shown).
To provide further evidence of the estrogenic activity of paroxetine, we performed RNA-seq analysis on RNA preparations from E2- and paroxetine-treated AroER tri-screen cells. We found that 1116 of 3166 (35%) of paroxetine upregulated genes were present in the group of 3682 E2 upregulated genes, and 1441 of 3296 (44%) paroxetine downregulated genes were present in the group of 3688 E2 downregulated genes (Fig. 6A). Figure 6(B) shows that the top 10 E2-upregulated and -downregulated genes are also upregulated and downregulated by paroxetine.
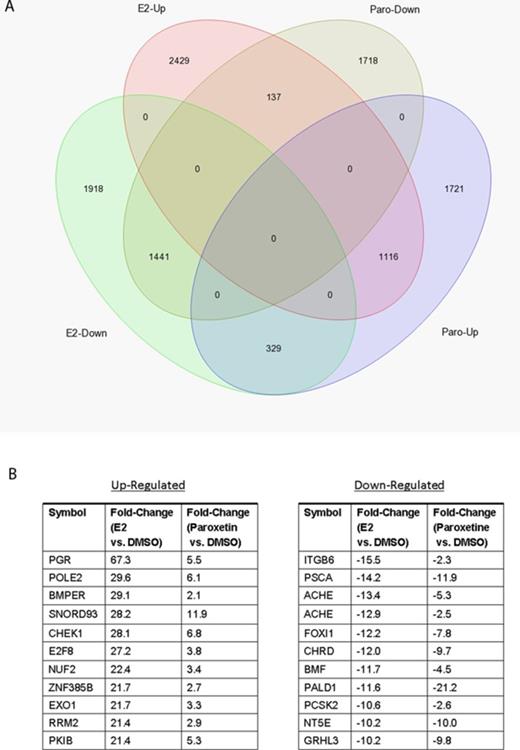
RNA-seq analysis of E2-regulated and paroxetine-regulated genes in AroER tri-screen cells. (A) Using the data from RNA-seq analysis on RNA preparations from DMSO-treated cells as the references, 1116 of 3166 paroxetine upregulated genes (35%) are present in E2 upregulated genes (3682 genes total). Furthermore, 1441 of 3296 paroxetine downregulated genes (44%) are presented in E2 downregulated genes (3688 genes total). (B) The list of top overlapping genes from comparison of E2 and Paroxetine.
To identify unique regulatory pathways associated with paroxetine/E2-up and -downregulated genes, we performed IPA analysis. The top five upregulated gene networks identified by IPA analysis are associated with cell cycle, DNA replication/recombination/repair, and cellular assembly and organization. The top five downregulated gene networks are associated with connective tissue disorders, cellular movement, development and function, and cell death and survival.
DISCUSSION
In the current study, we reported the establishment and validation of the novel AroER tri-screen, a reliable high-throughput screening system to identify antiaromatase EDCs as well as EDCs that act as ER agonists and/or ER antagonists. This is a cell-based assay that will reduce the use of animals in the screening of EDCs that target ER and aromatase.
The Endocrine Disruptor Screening and Testing Advisory Committee, defines an EDC as “an exogenous chemical substance or mixture that alters the structure or function(s) of the endocrine system and causes adverse effects at the level of the organism, its progeny, populations, or subpopulations of organisms, based on scientific principles, data, weight-of-evidence, and the precautionary principle” (Kavlock, 1999). The Tox21 program is a collaborative effort among the U.S. Environmental Protection Agency (EPA), the Food and Drug Administration (FDA), the NIH Chemical Genomics Center (now part of the National Center for Advancing Translational Sciences), and the National Toxicology Program (NTP/NIEHS). The goals of the Tox21 program are to identify and develop target-specific, mechanism-based, and biologically relevant in vitro assays to screen for health-hazard chemicals (Tice et al., 2013). Based on the excellent technical and biological performance characteristics of the AroER tri-screen assay, as reported here, it has been selected for screening the Tox21 10K compound library for identification of AI-like EDCs.
Nuclear receptors are essential components of the endocrine system. They have lipophilic binding sites that bind small molecules, primarily endogenous hormones, but they can also bind small lipophilic exogenous molecules. The ligand-activated nuclear receptors act as transcriptional factors that drive gene expression by binding to specific response elements. Most known EDCs act as ligands of nuclear receptors, specifically the ERs, the androgen receptor (AR), and the thyroid hormone receptor (TR), and they can act either as agonists or antagonists of these nuclear receptors. Nuclear receptor-mediated reporter assays have been well established and several high-throughput screening systems for nuclear receptor signaling have been developed (Huang et al., 2011).
Endocrine function can also be altered by modifications in steroid hormone synthesis pathways. Among the enzymes involved in steroid biosynthesis, aromatase is particularly important because it converts androgens to estrogens. Our laboratory was the first to demonstrate that aromatase is a target of EDCs that can modulate its activity and expression, leading to changes in both androgen and estrogen levels. Although aromatase assays using H295R cells and recombinant aromatase have been included in the EPA's Endocrine Disruptor Screening Program tier 1 battery, these assays are not suitable for a high-throughput screening because the aromatase in these systems is assayed using the traditional tritiated water release method that requires solvent extraction, transfer, and scintillation counting steps. The AroER tri-screen, a homogenous assay, overcomes this difficulty by eliminating the solvent extraction and transfer steps, and it can be adapted to large-scale screening efforts.
We used BPA to validate the utility of the AroER tri-screen that is used to screen EDCs that bind to nuclear ER, and we clearly demonstrated that in this assay BPA functions as an ER agonist at concentrations between 10nM and 10μM, as has been reported previously (Gould et al., 1998). However, BPA is known to be active at significantly lower concentrations in vivo (e.g., Welshons et al., 2006). As summarized in a recent review (Schug et al., 2012), BPA can fit into the ligand binding pocket of nuclear ERs and binds to both ERα and ERβ. It binds to both ER isoforms with EC50 values between 200nM and 700nM. In the review by Schug et al. (2012) and several publications (e.g., Zsarnovszky et al., 2005; Vinas and Watson, 2013), BPA was shown to also bind to membrane-bound ER and GPR30 (a transmembrane ER) with much higher affinities. Furthermore, BPA is known to bind to estrogen-related receptor gamma (ERRγ) with an EC50 value of 5.5nM (Matsushima et al., 2007). Although the differential sensitivities of BPA in vitro versus in vivo are not fully understood, the possibilities that BPA concentrates in target tissues or acts through membrane-bound receptors should be considered. The ability of BPA to bind to different forms of ER with different affinities may also explain its nonmonotonic concentration behavior. At this moment, there is no high-throughput screen system for membrane-associated ER, including GPR30. The structure of the ligand binding pocket of GPR30 is not exactly identical to those of nuclear ERs, e.g., ICI is not an antagonist of GPR30. The design of a screen system for membrane-bound ER will be different from AroER tri-screen, which has been demonstrated to be an effective system to screen EDCs targeting aromatase and nuclear ER.
Several promoters regulate the expression of human aromatase in a tissue-specific manner. The expression of ER and aromatase can be altered through a number of mechanisms during breast cancer development. Thus, all environmental chemicals that modify the expression of ER and aromatase in human breast cancer are biologically relevant EDCs (Diamanti-Kandarakis et al., 2009). Considering the complexity of the regulatory mechanisms involved in aromatase expression (Simpson et al., 1993), it remains technically challenging to develop efficient screening systems to search for chemicals that modulate aromatase expression. As a proof-of-concept analysis, we have generated the pGL4.14-firefly-pI.3/II plasmid that contains human aromatase promoters I.3/II (Chen et al., 2010). Although LBH589, an histone deacetylase inhibitor, has been shown to suppress the function of promoters I.3/II using cells stably expressing this reporter, we have not been able to adapt this screening system into a high-throughput screening format. On the other hand, it is possible to develop a screening system to identify EDCs that inhibit aromatase activity because there is only one aromatase enzyme in humans. AroER tri-screen is the first high-throughput screening system that has been developed to identify antiaromatase chemicals.
Many azole fungicides are known for their ability to inhibit cytochrome P450's, such as those of steroidogenesis including aromatase (Zarn et al., 2003). There are three azole fungicides in the NIH Clinical Collection library: bifonazole, oxiconazole, and clotrimazole. We have confirmed two of these, bifonazole and oxiconazole, as AIs in a noncompetitive manner with respect to the androgen substrate. These results agree with reports suggesting that both bifonazole and oxiconazole are AIs (Ayub and Levell, 1988; Trösken et al., 2006). It is thought that the azole group of these fungicides interacts with the heme prosthetic group of the aromatase enzyme.
Identification of the antidepressant, paroxetine, as a weak estrogenic chemical was an important finding in our study. This activity of paroxetine (Paxil), a commonly prescribed selective serotonin reuptake inhibitor, may be responsible, in part, for the observed reduction in the effectiveness of TAM in some women with breast cancer (Kelly et al., 2010). To confirm its estrogen-like action, we performed RNA-seq analysis and found that 35% of paroxetine upregulated genes and 44% of its downregulated genes are estrogen-regulated genes. As shown in Figure 6(B), paroxetine modulated the expression of the genes well known to be regulated by estrogen, such as PGR, although the effects were weaker. This suggests that paroxetine can act as a weak ER agonist, although it is still unknown whether paroxetine binds to the ER directly or whether it indirectly disrupts ER signaling pathways.
The strength of a high-throughput screen is that one can identify a number of active compounds in a large chemical collection in a relatively rapid manner. However, confirmation of the compound activity using orthogonal methods is essential. Using the AroER tri-screen, we identified a number of active compounds in the NIH Clinical Collection library, which includes FDA-approved drugs. Although our screen strongly suggests that a number of these chemicals have the potential to affect endocrine function, we need to verify their activities in vivo in order to confirm their endocrine disrupting potential.
In conclusion, we have developed the AroER tri-screen assay for use in high-throughput screening of large chemical libraries to identify modulators of ER and aromatase signaling. The advantages of this technology include the ability to assess three readouts in one system and the successful adaptation of the assay to a 1536-well plate robotic platform. We have demonstrated the suitability of this assay for identifying both ER agonists and antagonists as well as AIs, which suggests that the AroER tri-screen assay can be used to screen a larger compound collection and search for novel modulators of ER and aromatase signaling in a single-assay system.
FUNDING
CBCRP (17UB-8701 to S.C.); NTP-NCATS Interagency Agreement (ES-7020-01); National Cancer Institute of the National Institutes of Health (P30CA33572). Research reported in this publication included work performed in the Bioinformatics core, High Throughput Screening core, and Integrative Genomics core supported by the National Cancer Institute of the NIH under award number P30CA33572.
Acknowledgments
The authors would like to thank Nicola Solomon for assistance in writing and editing the manuscript, and Charles Warden for assistance with bioinformatics analysis. We also want to thank Drs B. Alex Merrick, Scott Auerbach, and Raymond Tice for reading the manuscript and providing valuable comments.
Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments