





Abstract
Low-grade endotoxaemia is detectable in human circulation but its role in thrombosis is still unclear.
We measured serum lipopolysaccharide (LPS) concentration, soluble P-selectin (sP-selectin), a marker of platelet activation, and zonulin, a marker of gut permeability, in peripheral circulation, coronary thrombi, and intracoronary blood of patients with ST-elevation myocardial infarction (STEMI, n = 50) and stable angina (SA) (n = 50), respectively, and in controls (n = 50). Experimental study was carried out in mice to assess if Escherichia coli-LPS (E. coli-LPS) possess thrombotic property. Coronary thrombi from STEMI showed higher concentrations of LPS, sP-selectin vs. intracoronary blood of SA and peripheral blood of controls (P < 0.001). Zonulin was higher in STEMI compared to the other two groups [4.57 (3.34–5.22); 2.56 (0.41–4.36); 1.95 (1.22–2.65) ng/mL; P < 0.001] and correlated with LPS (Rs = 0.585; P < 0.001). Escherichia coli DNA was positive in 34% of STEMI vs. 12% of SA and 4% of controls (P < 0.001). In a subgroup of 12 STEMI, immunohistochemical analysis of coronary thrombi showed positivity for leucocyte Toll-like receptor 4 (TLR4), cathepsin G, and LPS from E. coli in 100%, 80%, and 25% of samples, respectively. E. coli-LPS injected in mice to reach LPS concentrations like those detected in coronary thrombi was associated with enhanced artery thrombosis and platelet activation, an effect blunted by TLR4 inhibitor co-administration. In vitro study demonstrated that LPS from E. coli enhanced platelet aggregation via TLR4-mediated leucocyte cathepsin G activation.
ST-elevation myocardial infarction patients disclose an enhanced gut permeability that results in LPS translocation in human circulation and eventually thrombus growth at site of artery lesion via leucocyte–platelet interaction.
See page 3166 for the editorial comment on this article (doi: )
Escherichia coli lipopolysaccharides (E. coli-LPS), a component of gut microbiota, is detectable in human circulation but its role in thrombosis is not fully understood. In patients with myocardial infarction (MI) coronary thrombi showed positivity for E. coli-LPS and leucocyte Toll-like receptor 4 (TLR4) with a significant association between low-grade endotoxaemia, soluble P-selectin, a marker of platelet activation, and zonulin, a marker of gut permeability. Escherichia coli-LPS infusion in mice to mimic low-grade endotoxaemia of MI patients enhanced experimental thrombosis, an effect blunted by TLR4 inhibitor. Low-grade endotoxaemia may represent a novel pathway amplifying thrombus growth at site of artery lesion.
Introduction
Platelets play a key role in the atherothrombotic process as documented by a significant decrease of myocardial infarction (MI) and stroke in patients with stable or unstable vascular disease treated with antiplatelet drugs such as aspirin or clopidogrel.1 In fact, plaque rupture or erosion with ensuing platelet activation is believed to be the ‘primum movens’ of a sequence of events, which, along with local activation of the coagulation system, ultimately lead to coronary thrombosis formation and growth.2 However, the in vivo mechanism accounting for thrombus growth at the site of coronary atherosclerotic lesion has not been fully elucidated.2 In particular, while platelet adhesion and aggregation on the thrombogenic core of atherosclerotic plaque is an established mechanism for initiation of coronary thrombosis, the role of systemic factors, which may contribute to thrombus growth via amplification and propagation of platelet aggregation, is still unclear.
There is a growing body of evidence that lipopolysaccharides (LPS) are implicated in atherothrombosis3 via several mechanisms such as binding to macrophages and release of inflammatory cytokines like tumour necrosis factor alpha (TNFα) and interleukins 1, 6, 8, 12, or induction of reactive oxidant species, which may deleteriously affect the artery wall.4 Circulating levels of endotoxins have been associated with human atherosclerosis progression, particularly in smokers or in patients with infections.5 Furthermore, endotoxins are implicated in the thrombotic process through several mechanisms including up-regulation of macrophage tissue factor (TF) expression6 and amplification of platelet response to common agonists upon interaction with Toll-like receptor 4 (TLR4).7 Of note, platelet activation by endotoxins occurs with concentrations, which may be detected in human circulation even in absence of septic situations8 and could putatively trigger platelet activation at site of atherosclerotic plaque rupture. Hence, the relationship between endotoxins and platelets may be relevant in the context of acute coronary syndromes as endotoxins could locally amplify platelet-dependent thrombus formation and growth but this issue is still unexplored.
Previous studies from our group demonstrated that Escherichia coli-LPS (E. coli-LPS)-dependent low-grade endotoxaemia is detectable in human circulation, likely as consequence of enhanced gut permeability, and may be responsible for leucocyte–platelet aggregation and eventually thrombosis.8 We hypothesized that low-grade endotoxaemia in patients with coronary heart disease may favour thrombus growth at site of coronary unstable plaques. To explore this issue, we measured LPS concentration and biomarkers of platelet activation in coronary thrombi and intracoronary blood of patients with ST-elevation myocardial infarction (STEMI) and stable angina (SA), respectively, and in peripheral circulation of both patients and controls. Furthermore, to substantiate that LPS could be biologically active as prothrombotic molecule we performed (i) immunohistochemical analysis of thrombi, (ii) in vitro studies to assess the interplay between LPS and platelet activation, and (iii) developed an experimental animal model to assess if low-grade endotoxaemia from E. coli, which mimicked LPS concentration detected in human coronary thrombi, enhanced thrombus growth at the site of endothelial lesion. Here, we provide the first evidence that LPS concentration is higher in the coronary thrombi of patients with STEMI compared to intracoronary blood of SA patients and provide experimental evidence that low-grade endotoxaemia by E. coli amplifies thrombus growth at site of endothelial lesion in an experimental model of thrombosis.
Methods
See Supplementary material online for extended experimental procedures.
Results
In vivo study
Lipopolysaccharide, markers of gut permeability, platelet activation, oxidative stress, tissue factor, and inflammatory cytokines in peripheral blood
Clinical, demographic, and laboratory characteristics for the entire population are summarized in Table 1. Age and gender as well as the prevalence of hypertension and diabetes were not different among patients with STEMI (n = 50), SA (n = 50), and controls (n = 50). On the contrary, dyslipidaemia and smoking habit were more prevalent in STEMI and in SA patients than in controls. Regarding therapy on admission, β-blockers, and antiplatelets use was more common in SA and STEMI patients, compared to controls, while no differences in anti-hypertensive drugs and statins were found among groups. White blood cell (WBC) count and high-sensitivity C-reactive protein (hs-CRP) were higher in STEMI patients compared to SA and controls (Table 1). Serum LPS, zonulin, and soluble P-selectin (sP-selectin) were higher in STEMI patients compared to SA and control patients (Table 1, Figure 1 A–C).
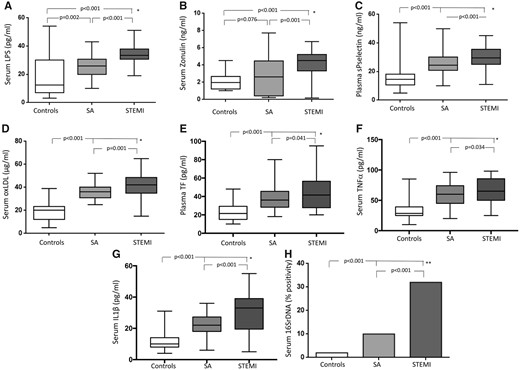
(A) LPS, (B) zonulin, (C) soluble P-selectin (sP-selectin), (D) oxLDL, (E) tissue factor (TF), (F) TNFα, (G) IL1-β, and (H) E. coli DNA positivity in control subjects and in patients with stable coronary disease or ST-elevation myocardial infarction. Data are expressed as median and interquartile range (boxes) and non-outlier range (whiskers) (A–G) or percentage (H) *Mann–Whitney U test (A–G); **Fisher’s exact test.
Clinical and laboratory characteristics of STEMI, stable angina patients, and control subjects
| Controls (n = 50) | Stable angina (n = 50) | STEMI (n = 50) | P-value | |
|---|---|---|---|---|
| Age (years)a | 68 ± 12.5 | 66 ± 15.2 | 67 ± 4.9 | 0.324 |
| Male gender (male, %) | 76 | 76 | 78 | 0.960 |
| Hypertension (%) | 84 | 82 | 68 | 0.121 |
| Dyslipidaemia (%) | 26 | 72 | 54 | <0.001 |
| Diabetes (%) | 22 | 28 | 22 | 0.685 |
| Smoking (%) | 8 | 22 | 50 | <0.001 |
| LVEF (%) | 60 (54–65) | 50 (40–53) | 45 (37–50) | <0.001 |
| WBC (×103)a | 6.73 ± 1.20 | 7.88 ± 2.07 | 11.40 ± 3.64 | <0.001 |
| PLT (×103)a | 215 ± 45 | 218 ± 55 | 212 ± 59 | 0.869 |
| hs-CRP (mg/dL)b | 0.065 (0.050–0.122) | 0.070 (0.050–0.257) | 1.26 (0.677–3.117) | <0.001 |
| ACE/ARBs (%) | 66 | 55 | 66 | 0.454 |
| β-blockers (%) | 6 | 22 | 24 | 0.031 |
| Antiplatelet drugs (%) | 40 | 100 | 100 | <0.001 |
| Statins (%) | 40 | 49 | 35 | 0.357 |
| Plasma sP-selectin (ng/mL)b | 14.5 (10.7–18.0) | 24.0 (20.7–29.2) | 29.5 (25.0–35.5) | <0.001 |
| Serum LPS (pg/mL)b | 12.5 (7.0–30.0) | 25.1 (20.0–30.2) | 33.9 (30.9–38.2) | <0.001 |
| Serum zonulin (ng/mL)b | 1.95 (1.22–2.65) | 2.56 (0.41–4.36) | 4.57 (3.34–5.22) | <0.001 |
| Serum oxLDL (μg/mL)b | 20.0 (12.0–23.2) | 36 (31.0–40.3) | 42.0 (35.0–48.5) | <0.001 |
| Plasma TF (pg/mL)b | 21.5 (15.0–29.2) | 36.0 (28.2–45.5) | 41.5 (27.7–58.0) | <0.001 |
| Serum TNFα (pg/mL)b | 28.5 (25.0–39.2) | 59.0 (45.0–70.2) | 65.0 (50.0–87.2) | <0.001 |
| Serum IL1-β (pg/mL)b | 10 (8–14) | 24 (18–28) | 31 (18–39) | <0.001 |
| Controls (n = 50) | Stable angina (n = 50) | STEMI (n = 50) | P-value | |
|---|---|---|---|---|
| Age (years)a | 68 ± 12.5 | 66 ± 15.2 | 67 ± 4.9 | 0.324 |
| Male gender (male, %) | 76 | 76 | 78 | 0.960 |
| Hypertension (%) | 84 | 82 | 68 | 0.121 |
| Dyslipidaemia (%) | 26 | 72 | 54 | <0.001 |
| Diabetes (%) | 22 | 28 | 22 | 0.685 |
| Smoking (%) | 8 | 22 | 50 | <0.001 |
| LVEF (%) | 60 (54–65) | 50 (40–53) | 45 (37–50) | <0.001 |
| WBC (×103)a | 6.73 ± 1.20 | 7.88 ± 2.07 | 11.40 ± 3.64 | <0.001 |
| PLT (×103)a | 215 ± 45 | 218 ± 55 | 212 ± 59 | 0.869 |
| hs-CRP (mg/dL)b | 0.065 (0.050–0.122) | 0.070 (0.050–0.257) | 1.26 (0.677–3.117) | <0.001 |
| ACE/ARBs (%) | 66 | 55 | 66 | 0.454 |
| β-blockers (%) | 6 | 22 | 24 | 0.031 |
| Antiplatelet drugs (%) | 40 | 100 | 100 | <0.001 |
| Statins (%) | 40 | 49 | 35 | 0.357 |
| Plasma sP-selectin (ng/mL)b | 14.5 (10.7–18.0) | 24.0 (20.7–29.2) | 29.5 (25.0–35.5) | <0.001 |
| Serum LPS (pg/mL)b | 12.5 (7.0–30.0) | 25.1 (20.0–30.2) | 33.9 (30.9–38.2) | <0.001 |
| Serum zonulin (ng/mL)b | 1.95 (1.22–2.65) | 2.56 (0.41–4.36) | 4.57 (3.34–5.22) | <0.001 |
| Serum oxLDL (μg/mL)b | 20.0 (12.0–23.2) | 36 (31.0–40.3) | 42.0 (35.0–48.5) | <0.001 |
| Plasma TF (pg/mL)b | 21.5 (15.0–29.2) | 36.0 (28.2–45.5) | 41.5 (27.7–58.0) | <0.001 |
| Serum TNFα (pg/mL)b | 28.5 (25.0–39.2) | 59.0 (45.0–70.2) | 65.0 (50.0–87.2) | <0.001 |
| Serum IL1-β (pg/mL)b | 10 (8–14) | 24 (18–28) | 31 (18–39) | <0.001 |
hs-CRP, high-sensitivity C-reactive protein; IL1, interleukin 1; LPS, lipopolysaccharide; LVEF, left ventricular ejection fraction; PLT, platelets; TF, tissue factor; TNF, tumour necrosis factor; WBC, white blood cells.
Data are represented as media ± standard deviation.
Data are represented as median and interquartile range.
Clinical and laboratory characteristics of STEMI, stable angina patients, and control subjects
| Controls (n = 50) | Stable angina (n = 50) | STEMI (n = 50) | P-value | |
|---|---|---|---|---|
| Age (years)a | 68 ± 12.5 | 66 ± 15.2 | 67 ± 4.9 | 0.324 |
| Male gender (male, %) | 76 | 76 | 78 | 0.960 |
| Hypertension (%) | 84 | 82 | 68 | 0.121 |
| Dyslipidaemia (%) | 26 | 72 | 54 | <0.001 |
| Diabetes (%) | 22 | 28 | 22 | 0.685 |
| Smoking (%) | 8 | 22 | 50 | <0.001 |
| LVEF (%) | 60 (54–65) | 50 (40–53) | 45 (37–50) | <0.001 |
| WBC (×103)a | 6.73 ± 1.20 | 7.88 ± 2.07 | 11.40 ± 3.64 | <0.001 |
| PLT (×103)a | 215 ± 45 | 218 ± 55 | 212 ± 59 | 0.869 |
| hs-CRP (mg/dL)b | 0.065 (0.050–0.122) | 0.070 (0.050–0.257) | 1.26 (0.677–3.117) | <0.001 |
| ACE/ARBs (%) | 66 | 55 | 66 | 0.454 |
| β-blockers (%) | 6 | 22 | 24 | 0.031 |
| Antiplatelet drugs (%) | 40 | 100 | 100 | <0.001 |
| Statins (%) | 40 | 49 | 35 | 0.357 |
| Plasma sP-selectin (ng/mL)b | 14.5 (10.7–18.0) | 24.0 (20.7–29.2) | 29.5 (25.0–35.5) | <0.001 |
| Serum LPS (pg/mL)b | 12.5 (7.0–30.0) | 25.1 (20.0–30.2) | 33.9 (30.9–38.2) | <0.001 |
| Serum zonulin (ng/mL)b | 1.95 (1.22–2.65) | 2.56 (0.41–4.36) | 4.57 (3.34–5.22) | <0.001 |
| Serum oxLDL (μg/mL)b | 20.0 (12.0–23.2) | 36 (31.0–40.3) | 42.0 (35.0–48.5) | <0.001 |
| Plasma TF (pg/mL)b | 21.5 (15.0–29.2) | 36.0 (28.2–45.5) | 41.5 (27.7–58.0) | <0.001 |
| Serum TNFα (pg/mL)b | 28.5 (25.0–39.2) | 59.0 (45.0–70.2) | 65.0 (50.0–87.2) | <0.001 |
| Serum IL1-β (pg/mL)b | 10 (8–14) | 24 (18–28) | 31 (18–39) | <0.001 |
| Controls (n = 50) | Stable angina (n = 50) | STEMI (n = 50) | P-value | |
|---|---|---|---|---|
| Age (years)a | 68 ± 12.5 | 66 ± 15.2 | 67 ± 4.9 | 0.324 |
| Male gender (male, %) | 76 | 76 | 78 | 0.960 |
| Hypertension (%) | 84 | 82 | 68 | 0.121 |
| Dyslipidaemia (%) | 26 | 72 | 54 | <0.001 |
| Diabetes (%) | 22 | 28 | 22 | 0.685 |
| Smoking (%) | 8 | 22 | 50 | <0.001 |
| LVEF (%) | 60 (54–65) | 50 (40–53) | 45 (37–50) | <0.001 |
| WBC (×103)a | 6.73 ± 1.20 | 7.88 ± 2.07 | 11.40 ± 3.64 | <0.001 |
| PLT (×103)a | 215 ± 45 | 218 ± 55 | 212 ± 59 | 0.869 |
| hs-CRP (mg/dL)b | 0.065 (0.050–0.122) | 0.070 (0.050–0.257) | 1.26 (0.677–3.117) | <0.001 |
| ACE/ARBs (%) | 66 | 55 | 66 | 0.454 |
| β-blockers (%) | 6 | 22 | 24 | 0.031 |
| Antiplatelet drugs (%) | 40 | 100 | 100 | <0.001 |
| Statins (%) | 40 | 49 | 35 | 0.357 |
| Plasma sP-selectin (ng/mL)b | 14.5 (10.7–18.0) | 24.0 (20.7–29.2) | 29.5 (25.0–35.5) | <0.001 |
| Serum LPS (pg/mL)b | 12.5 (7.0–30.0) | 25.1 (20.0–30.2) | 33.9 (30.9–38.2) | <0.001 |
| Serum zonulin (ng/mL)b | 1.95 (1.22–2.65) | 2.56 (0.41–4.36) | 4.57 (3.34–5.22) | <0.001 |
| Serum oxLDL (μg/mL)b | 20.0 (12.0–23.2) | 36 (31.0–40.3) | 42.0 (35.0–48.5) | <0.001 |
| Plasma TF (pg/mL)b | 21.5 (15.0–29.2) | 36.0 (28.2–45.5) | 41.5 (27.7–58.0) | <0.001 |
| Serum TNFα (pg/mL)b | 28.5 (25.0–39.2) | 59.0 (45.0–70.2) | 65.0 (50.0–87.2) | <0.001 |
| Serum IL1-β (pg/mL)b | 10 (8–14) | 24 (18–28) | 31 (18–39) | <0.001 |
hs-CRP, high-sensitivity C-reactive protein; IL1, interleukin 1; LPS, lipopolysaccharide; LVEF, left ventricular ejection fraction; PLT, platelets; TF, tissue factor; TNF, tumour necrosis factor; WBC, white blood cells.
Data are represented as media ± standard deviation.
Data are represented as median and interquartile range.
Overall, smokers presented higher levels of serum LPS [median values and interquartile range, 32.0 (28.4–38.3) vs. 25.0 (14.0–33.2) pg/mL, P < 0.001], plasma sP-selectin [29.0 (23.0–34.5) vs. 21.0 (14.0–27.0) ng/mL, P < 0.001], and serum zonulin [4.02 (2.79–5.31) vs. 2.16 (1.13–3.87) ng/mL, P < 0.001] compared to non-smokers. After log-transformation, factorial ANOVA analyses showed that these biomarkers remained significantly different among the three groups (STEMI, SA, and control patients) also after adjusting for smoking habit (F = 19.9, P < 0.001 for LPS; F = 34.5, P < 0.001 for sP-selectin; and F = 5.0, P = 0.008 for zonulin).
Peripheral blood concentration of oxLDL, TF, TNFα, and IL1-β were higher in STEMI than in SA and controls (Table 1, Figure 1 D–G). Thirty-four percent of STEMI patients were positive for 16SrDNA vs. 12% of SA patients and 4% of controls (Figure 1H).
In the whole cohort, serum LPS correlated with zonulin (Rs = 0.585; P < 0.001), plasma sP-selectin (Rs = 0.501; P < 0.001), hs-CRP (Rs = 0.420; P < 0.001), WBC count (Rs = 0.421 P < 0.001), oxLDL (Rs = 0.281; P < 0.001), TF (Rs = 0.537; P < 0.001), TNFα (Rs = 0.314; P < 0.001), and IL1-β (Rs = 0.276; P < 0.001). Moreover, sP-selectin correlated with TNFα (Rs = 0.462; P < 0.001).
Significant correlations were also found between LPS and zonulin within each group, i.e. controls, SA, and STEMI patients (Rs = 0.657, P < 0.001; Rs = 0.484, P < 0.001; and Rs = 0.448, P = 0.001, respectively) (Figure 2).
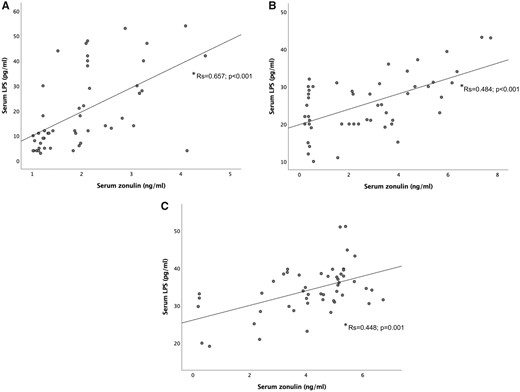
Correlation between LPS and zonulin in peripheral blood of control subjects, stable coronary disease and ST-elevation myocardial infarction patients. (*Spearman rank correlation tests).
Lipopolysaccharide and markers of platelet activation, oxidative stress, tissue factor and inflammatory cytokines in coronary thrombi and intracoronary blood of ST-elevation myocardial infarction and stable angina patients, respectively
Lipopolysaccharide was higher in coronary thrombi of STEMI patients than in intracoronary blood of SA patients [39.2 (29.1–43.3) vs. 20.0 (11.7–22.0) pg/mL; P < 0.001] (Figure 3A). Similarly, sP-selectin was higher in coronary thrombi of STEMI patients than in intracoronary blood of SA patients [45.0 (34.0–62.0) vs. 26.0 (20.5–29.5) ng/mL; P < 0.001] (Figure 3B). No significant difference of oxLDL was detected in coronary thrombi of STEMI and intracoronary blood of SA patients [27.0 (22.5–34.2) vs. 26.0 (23.0–36.5) μg/mL; P = 0.629] (Figure 3C).
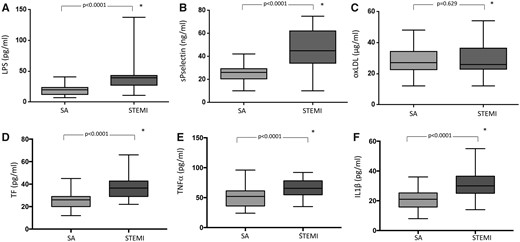
(A) LPS, (B) soluble P-selectin (sP-selectin), (C) oxLDL concentration, (D) tissue factor (TF), (E) TNFα, and (F) IL1-β in intracoronary blood of patients with stable coronary disease and in intracoronary thrombi of patients with ST-elevation myocardial infarction. Data are expressed as median and interquartile range (boxes) and non-outlier range (whiskers) *Mann–Whitney U test.
Tissue factor levels were higher in coronary thrombi of STEMI patients than in intracoronary blood of SA patients [36.5 (29–42.7) vs. 26.0 (20.5–29.0) pg/mL; P < 0.001] (Figure 3D). Similarly, proinflammatory cytokines were higher in coronary thrombi of STEMI patients than in intracoronary blood of SA patients [TNFα: 65.5 (54.7–78.9) vs. 52.0 (36.0–61.2) pg/mL; P < 0.001; IL1-β: 30.0 (25.0–36.5) vs. 21.0 (15.7–25.2) pg/mL; P < 0.001] (Figure 3 E and F).
Serum zonulin vs. lipopolysaccharide and soluble P-selectin in coronary thrombi and blood
To analyse the relationship between zonulin, endotoxaemia, and platelet activation, we divided the population according to serum zonulin levels. Overall, median zonulin levels were 2.85 ng/mL. Zonulin levels were above the median values in 80% of STEMI patients, 46% of SA patients, and 22% of controls (P < 0.001). Coronary thrombi from STEMI patients with higher serum zonulin (i.e. above the median) had higher concentration of LPS and sP-selectin than thrombi from patients with lower serum zonulin [39.2 (38.0–45.4) vs. 24.4 (18.9–34.1) pg/mL; P = 0.006 and 45.5 (34.0–62.0) vs. 38.0 (31.0–56.0) ng/mL; P = 0.024, respectively].
Similarly, coronary blood from SA patients with higher serum zonulin had higher concentration of LPS and sP-selectin than coronary blood from patients with lower serum zonulin [21.6 (15.0–28.0) vs. 17.0 (10.0–21.0) pg/mL; P = 0.005 and 28.0 (24.0–30.1) vs. 23.0 (20.0–28.0) ng/mL; P = 0.018, respectively].
Histologic and immunohistochemical analyses
On histologic examination, all the samples aspirated from the coronary arteries were composed of fibrin, platelets, and intact granulocytes, consistent with fresh thrombus (i.e. less than 1 day old) (Figure 4A). On immunohistochemistry, neutrophilic granulocytes were diffusely and intensely stained with antibodies to cathepsin G and TLR4 (Figure 4 B and C). All thrombus samples were positive for cathepsin G (Figure 4B), 10 out 12 samples (80%) were positive for TLR4 (Figure 4C) and 3 out of 12 (25%) were positive for E. coli-LPS (Figure 4D).
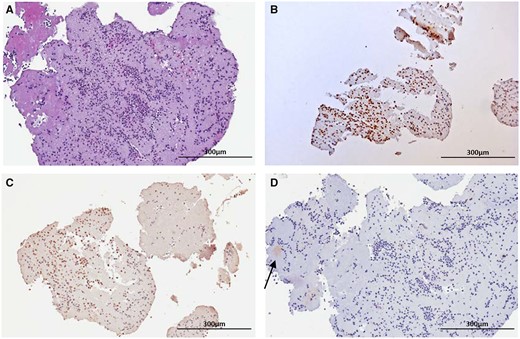
Histologic and immunohistochemical analyses of thrombi. Representative picture of a fresh thrombus aspirated from a patient. (A) On haematoxylin–eosin stain there are abundant neutrophils admixed with platelets and fibrin. (B) On immunohistochemistry neutrophilic granulocytes are diffusely and intensely stained with antibodies to cathepsin G or (C) with antibodies to Toll-like receptor 4. (D) The thrombus shows a weak and focal positive stain for LPS (arrow). Original magnification, 10×.
In vitro study
We performed in vitro experiments to explore if LPS, at the concentration detected in coronary thrombi, was biologically active via interaction with its specific receptor TLR4. Moreover, we investigated if oxLDL, that is also able to bind TLR4,9 could stimulate polymorphonuclear leukocytes (PMNs)/platelets aggregate. Thus, we incubated whole blood from healthy subjects with LPS (at concentration detected in the coronary thrombi, i.e. 50 and 100 pg/mL) or oxLDL (at concentration detected in the coronary thrombi, i.e. 10–20 µg/mL), in presence or less of TLR4 or cathepsin G inhibitors to evaluate PMNs/platelets aggregates. Lipopolysaccharide yielded PMNs/platelets aggregation in a dose-dependent manner, an effect blunted by TLR4 or cathepsin G inhibitors (Figure 5A). OxLDL was able to induce PMNs/platelets aggregates only at the highest concentration, which were not affected by TLR4 or cathepsin G inhibitors (Figure 5B).
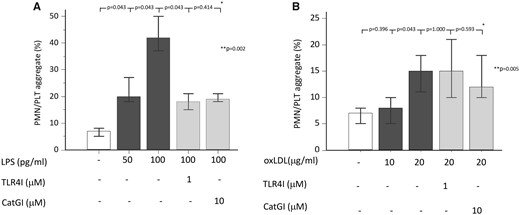
Lipopolysaccharide promotes polymorphonuclear leucocytes/platelets aggregate in vitro. (A) Percentage of polymorphonuclear leucocytes/platelets aggregates in whole blood after pre-incubation with cathepsin G inhibitor (10 μM) or Toll-like receptor 4 inhibitor (1 μM) and stimulation with lipopolysaccharide (50–100 pg/mL). (B) Percentage of polymorphonuclear leucocytes/platelets aggregates in whole blood after pre-incubation with cathepsin G inhibitor (10 μM) or Toll-like receptor 4 inhibitor (1 μM) and stimulation with oxLDL (10–20 μg/mL). Data are expressed as median and min-max range. *Wilcoxon test. **Friedman test (overall analysis).
In vivo model of thrombosis
After 5 h from artery lesion, mice treated with LPS (0.5 mg/kg) showed a significant reduction of peak blood flow velocity in femoral arterial (FA) of operated limb compared to controls (Figure 6 A); conversely, no significant changes of blood velocity in FA of non-operated limbs was detected in LPS-treated mice vs. untreated ones [115 (111–129) vs. 153 (123–174) m/s, P = 0.114, respectively]. Histological analyses also showed that the vessels of mice treated with LPS displayed a higher occlusive thrombosis with respect to untreated mice (Figure 6B) with a significant increase in the percentage of FA lumen occlusion (Figure 6C). Furthermore, animals treated with LPS (0.5 mg/kg)+TLR4 (2 mg/kg) inhibitor displayed a normal blood flow velocity in FA as compared to mice treated with LPS alone (Figure 6D). Of note, no significant differences between mice treated with LPS and those treated with LPS+TLR4 inhibitor were observed regarding blood flow velocity of FA of non-operated limbs [125 (102–110) vs. 127 (103–124) m/s, P = 0.215, respectively].
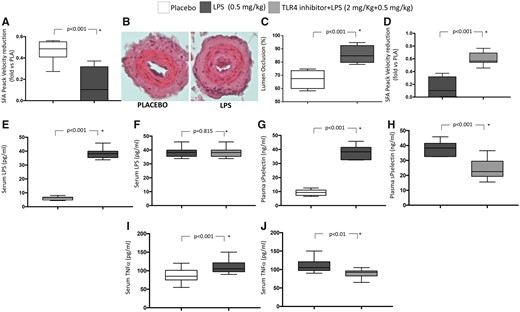
Lipopolysaccharide on thrombosis formation in a mouse model of wire-induced endothelial damage. Wild-type C57BL6J mice were subjected to LPS (n = 10), LPS+TLR4 inhibitor injection (n = 10), or saline administration (n = 10) (i.p. injection) and then to wire-induced endothelial damage of femoral artery. Data are expressed as median and interquartile range (boxes) and non-outlier range (whiskers) *Mann–Whitney U test. (A) Peak blood flow velocity in femoral artery of operated limb after wire-induced endothelial damage from control mice and mice given intraperitoneal lipopolysaccharide injection. (B) Femoral artery from a control mouse after wire-induced endothelial damage. A thin rim of mural thrombus is present, without significant lumen stenosis. Femoral artery from a mouse subject to the same procedure after intraperitoneal lipopolysaccharide injection. The vessel shows occlusive thrombosis. Original magnification 20×. (C) Lumen occlusion (%) after placebo and lipopolysaccharide injection. (D) Peak blood flow velocity in superficial femoral artery of operated limb after wire-induced endothelial damage from mice after intraperitoneal lipopolysaccharide injection and from mice after intraperitoneal LPS+TLR4 inhibitor injection. (E) Serum lipopolysaccharide concentration after placebo and lipopolysaccharide. (F) Serum lipopolysaccharide concentration after lipopolysaccharide and LPS+TLR4 inhibitor injection. (G) Plasma soluble P-selectin after placebo and lipopolysaccharide. (H) Plasma soluble P-selectin after lipopolysaccharide and LPS+TLR4 inhibitor injection. (I) Serum tumour necrosis factor alpha after placebo and lipopolysaccharide. (J) Serum tumour necrosis factor alpha after lipopolysaccharide and LPS+TLR4 inhibitor injection.
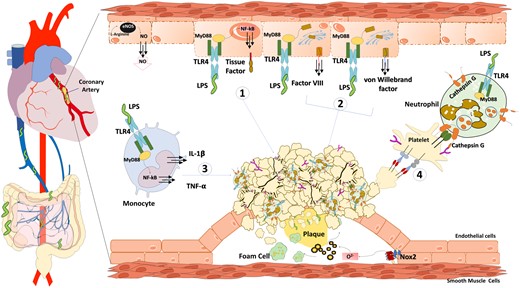
Low-grade endotoxaemia deriving from gut microbiota allows the growth of the thrombus at the site of coronary plaque rupture via several mechanisms such as (1) clotting activation by endothelial cells/monocytes tissue factor up-regulation; (2) endothelial activation via release of von Willebrand factor and factor VIII; (3) tumour necrosis factor alpha-mediated platelet activation by activated monocytes; (4) leucocytes cathepsin G-mediated platelet activation.
As expected, LPS blood levels were significantly higher in animals given LPS compared to controls [38.2 (35.6–40.5) vs. 6.1 (4.9–6.9) pg/mL; P < 0.001] (Figure 6E). No difference of serum LPS was found in mice treated with LPS+TLR4 inhibitor as compared to mice given LPS alone [36.8 (34.7–41.7) vs. 36.8 (33.9–40.0) pg/mL; P = 0.815] (Figure 6F). Furthermore, LPS-treated animals showed a significant increase of sP-selectin compared to controls [38.3 (32.6–41.5) vs. 9.8 (7.5–11.8) ng/mL; P < 0.001] (Figure 6G). In animals given LPS+TLR4 inhibitor a significant decrease of sP-selectin was detected [38.3 (32.6–41.5) vs. 22.0 (20.8–29.5) ng/mL; P < 0.001] compared to animals given LPS alone (Figure 6H).
Tumour necrosis factor alpha blood levels were significantly higher in animals given LPS compared to controls [105 (97.5–121.3) vs. 85.0 (75.0–101.3) pg/mL P < 0.001] (Figure 6I). In animals given LPS+TLR4 inhibitor a significant decrease of TNFα blood levels was detected [92.5 (82.5–96.5) vs. 105 (97.5–121.3) pg/mL; P < 0.01] compared to animals given LPS alone (Figure 6J).
Discussion
This is the first study reporting that low-grade endotoxaemia concurs to thrombus growth at site of artery lesion, suggesting that a similar mechanism may occur in patients with MI at site of coronary plaque rupture.
About 100 trillion of gut bacteria contribute to an enteric reservoir of >1 g LPS, which circulate in the blood of healthy subjects in a range of approximately 1–200 pg/mL.10 Low-grade endotoxaemia has been detected in healthy subjects after a high-fat diet11 or in patients at risk of cardiovascular disease such as those with type I and II diabetes12 or in patients with acute MI in whom it correlated with systemic inflammation and poor cardiovascular outcomes.13 LPS may exert a pro-atherogenic role as suggested by the association between high systemic levels of LPS and risk of carotid atherosclerosis.8 Other evidence in favour of the pro-atherogenic role of LPS includes a reduction of atherosclerotic plaque in animals with genetic deletion of TLR4 and acceleration of plaque formation in animals injected with LPS.14
While the role of LPS in triggering thrombosis in septic patients is well-established,15 less is known about the impact of low-grade endotoxaemia on coronary thrombosis in vivo. A novelty of the present study is that we found a progressive increase of low-grade endotoxaemia from controls to SA and to STEMI patients, who displayed the highest blood levels of LPS.
In addition to LPS, bacterial-derived DNA fragments, stable components of bacteria, were also detected in human serum in numerous pathologies associated with altered gut permeability and can be considered a surrogate marker of bacterial translocation.16
Microbiome analysis is usually performed by amplification of the highly conserved 16S rRNA gene region followed by next-generation sequencing for taxonomic classification. In this study, we used primers that amplify the V3 hypervariable region of 16S rRNA gene-specific for E. coli 17 and we found that patients with STEMI showed the highest percentage of E. coli DNA positivity compared to the other two groups.
Data regarding LPS were paralleled by the elevation of sP-selectin, which progressively increased from SA to STEMI patients and correlated with circulating LPS. Moreover, samples taken from coronary thrombi showed higher levels of LPS and sP-selectin compared to intracoronary blood of SA patients. Based on these findings, we speculated that LPS may behave as a molecule amplifying platelet aggregation at site of plaque rupture and, to address this issue, we performed immunohistochemical analysis of thrombi from a subgroup of STEMI patients. In coronary thrombi, we found positivity for leucocyte TLR4 and cathepsin G in 80% and 100% of samples, respectively, leading to speculate that, at site of plaque rupture, LPS could amplify platelet aggregation via TLR4-mediated leucocyte activation of cathepsin G, a powerful platelet stimulus.18 To investigate the validity of such hypothesis, we performed in vitro experiments demonstrating that E. coli-LPS, at concentrations similar to those detected in thrombi of patients with STEMI, enhanced leucocyte/platelet aggregates in a dose-dependent manner, a phenomenon blunted either by TLR4 or cathepsin G inhibitors. Taken together, these findings lead us to hypothesize that, at site of coronary plaque rupture, stimulation of leucocyte TLR4 by biologically active LPS elicits cathepsin G and in turn platelet activation so contributing to thrombus growth. Other circulating molecules such as oxLDL, which could induce platelet activation via TLR4,9 seemed to be excluded, since oxLDL at concentration detected in coronary thrombi, enhanced PMNs/platelets aggregates in vitro but with a TLR-4-independent mechanism.
To substantiate our hypothesis on the potential role of low-grade endotoxaemia as mechanism amplifying thrombus growth we developed an experimental mice model, where the circulating levels of LPS after E. coli injection mimicked the LPS concentrations detected in the coronary thrombi of STEMI patients, i.e. around 40 pg/mL. Animals given E. coli-LPS showed a higher occlusion rate as compared to controls suggesting a LPS-dependent mechanism of thrombus growth. In this context, it is noteworthy that animals given E. coli-LPS displayed enhanced circulating levels of sP-selectin suggesting that low-grade endotoxaemia is associated with platelet activation in vivo. The role of LPS as amplifier of thrombus growth was further corroborated by experiments in LPS-treated animals given a TLR4 inhibitor, which, in fact, showed a lower occlusion rate and platelet activation compared to LPS-treated ones, so reinforcing the hypothesis that LPS elicits thrombus growth via interaction with its specific receptor TLR4 and eventually platelet activation.
The increased concentration of LPS in the intracoronary blood and coronary thrombi of patients with coronary heart disease may have different explanations. A coexistent infection by Gram-negative bacteria could be a possible cause but, excluding one patient with acute cystitis, none of the patients enrolled had clinical or serological evidence of infections. There is a growing body of evidence indicating that gut microbiota may contribute to systemic inflammation and atherosclerosis and its complications.19 In fact, gut microbiota may contribute to atherosclerosis and its complications via formation of atherogenic and pro-thrombotic products such as trimethylamine-N-oxide, which is associated with cardiovascular risk.20 Enhanced gut permeability leading to LPS translocation into systemic circulation may be another mechanism accounting for elevated circulating levels detected in patients with coronary heart disease. Translocation of bacterial LPS from gut to systemic circulation is being considered a new factor accounting for chronic inflammation detectable in patients at risk of atherosclerosis. For example, LPS translocation into systemic circulation activates macrophages, so favouring inflammation of visceral adipose tissue and hepatic Kupfer cells resulting in non-alcoholic fatty liver disease.21 The higher E. coli-DNA positivity in serum of STEMI and SA patients compared to healthy controls might be a further element in support of a bacterial translocation from the intestinal lumen in patients with cardiovascular disease.13 To explore if patients with coronary heart disease had enhanced gut permeability, we measured serum zonulin, which modulates gut permeability by disassembling the intercellular tight junctions.22 Experimental and clinical studies demonstrated that zonulin up-regulation plays a role in increasing gut permeability and that serum level of zonulin correlates with enhanced intestinal permeability.22 Serum zonulin was increased in patients with coronary heart disease, particularly in those with STEMI, and independently correlated with serum LPS suggesting that changes in gut permeability could be a potential mechanism responsible for enhancing circulating LPS in patients with coronary heart disease.
The study has implications and limitations. Previous study demonstrated that E. coli-LPS localizes into carotid plaques close to activated macrophages, suggesting its role in the atherosclerotic process.8 The present study showing higher levels of LPS in coronary thrombi of STEMI patients provides further support to the role of LPS as a factor potentially implicated in the atherothrombotic process. A recent study by Amar et al.23 demonstrated that bacterial-derived DNA fragments are elevated in patients with MI. Our study reporting positivity of E. coli-DNA in the blood of STEMI patients corroborates the Amar’s et al. report and suggests E. coli as a bacterium potentially responsible for LPS elevation and eventually thrombus growth. However, we cannot exclude that other gut bacteria may translocate in human circulation in patients at risk of cardiovascular disease.
While LPS was systematically detected in the fresh thrombus by ELISA method, immunohistochemical analysis showed LPS positivity in only 25% of coronary thrombi; this could depend on the extracellular location of LPS, predisposing to its dilution during the sample processing for paraffin embedding.
Lipopolysaccharide could contribute to thrombus growth via leucocyte-induced platelet activation24 but we cannot exclude its direct interaction with platelet TLR4 receptors. In this context, the experiments showing that in mice thrombus growth is lowered by TLR4 inhibitor injection may open new avenues to counteract thrombosis in patients with STEMI. Furthermore, we cannot exclude that platelet activation may also occur as consequence of systemic inflammation. For instance, LPS may act as proinflammatory molecule to elicit TNFα production25 and eventually favour thrombosis as recently suggested by experimental and clinical studies showing an important role for TNFα as trigger of platelet activation26; in this context, the relationship between TNFα and sP-selectin may support such hypothesis. Finally, we cannot exclude that LPS contributes to thrombus growth with a mechanism involving clotting as well as endothelial activation as shown by previous study suggesting a role for LPS as promoter of TF up-regulation in monocytes or release of von Willebrand factor and factor VIII by endothelial cells27 (Take home figure).
We have only an indirect measure of gut permeability, thereby further study is necessary to support the interplay between gut permeability and coronary heart disease. Also, further study is necessary to establish if other mechanisms including changes of gut microbiota or enhanced LPS translocation via chylomicrons absorption28 contribute to enhancing circulating LPS in patients with cardiovascular disease. Serum LPS could be a marker to stratify the risk of atherothrombosis as suggested by previous study in patients with atrial fibrillation29 but further prospective study is necessary to validate this hypothesis. Furthermore, we found a significantly correlation between serum LPS and C-reactive protein suggesting a potential role for LPS as trigger of systemic inflammation and eventually thrombosis in patients with cardiovascular disease. Accordingly, we also found a significant correlation between LPS and inflammatory cytokines such as TNFα and IL1-β.
In conclusion, LPS from E. coli is increased in coronary thrombi of STEMI patients coincidentally with enhanced platelet activation, suggesting E. coli-LPS as a novel trigger for platelet activation at site of coronary plaque rupture. Experimental model of low-grade endotoxaemia supports this hypothesis and suggests that TLR4 inhibition may represent a novel tool to counteract coronary thrombosis in patients with cardiovascular disease.
Funding
This study was supported by the Sapienza University of Rome 2018 (RM118164366B89BD) to F.V. and in part by grants from the Sapienza University of Rome to (RM11715C62C29647) to R.C.
Conflict of interest : none declared.
References
Author notes
Roberto Carnevale and Sebastiano Sciarretta contributed equally to the study.
Gaetano Tanzilli and Francesco Violi declare joint seniorship.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}