





Abstract
Modeling of signaling pathways is an important step towards the understanding and the treatment of diseases such as cancers, HIV or auto-immune diseases. MaBoSS is a software that allows to simulate populations of cells and to model stochastically the intracellular mechanisms that are deregulated in diseases. MaBoSS provides an output of a Boolean model in the form of time-dependent probabilities, for all biological entities (genes, proteins, phenotypes, etc.) of the model.
We present a new version of MaBoSS (2.0), including an updated version of the core software and an environment. With this environment, the needs for modeling signaling pathways are facilitated, including model construction, visualization, simulations of mutations, drug treatments and sensitivity analyses. It offers a framework for automated production of theoretical predictions.
MaBoSS software can be found at https://maboss.curie.fr, including tutorials on existing models and examples of models.
Supplementary data are available at Bioinformatics online.
1 Introduction
Chemical kinetics and logical formalism have already been extensively used for modeling signaling pathways that are implicated in pathologies such as cancer, HIV, or auto-immune diseases. On one hand, models using chemical kinetics are more precise as they can output the variation of gene or protein concentrations over time, but to do so, speed of reactions and relative entity concentrations need to be known a priori. On the other hand, logical models can include a high number of variables at the expense of precision. Many tools and algorithms have been developed for logical modeling; and a community effort has been made to facilitate the exchange and the comparison of models among these tools (http://www.colomoto.org/; Naldi et al., 2015). With the framework of MaBoSS, we are able to describe temporal behaviors of these signaling pathways while maintaining the simple description inherent to Boolean models (Stoll et al., 2012). Since MaBoSS provides probabilistic results of all model entities (e.g. gene/protein/phenotype), theoretical results can be confronted to many types of experiments, including single-cell behaviors. MaBoSS has already proved its efficiency in biological interpretation of cancer-related models (Calzone et al., 2015; Cohen et al., 2015; Remy et al., 2015; Verlingue et al., 2016) and is constantly evolving.
In addition to the initial core C ++ script (MaBoSS 1.2.2), the new version of MaBoSS (2.0) includes an environment that facilitates the elaboration of a project in MaBoSS, in particular: model construction, visualization of solutions, simulations of mutations, drug treatments and sensitivity analyses (Fig. 1). The environment is accessible through separated scripts in Perl and Python, or in an integrated library at https://maboss.curie.fr/. A tutorial offers an example of the practical use of the functionalities presented here (Supplementary Material and https://maboss.curie.fr/).
2 Constructing a Boolean model using MaBoSS
MaBoSS is a simulation software for continuous time Boolean modeling. The model is described by a network, where nodes have Boolean states. Transitions between the node states are governed by logical equations, with an associated rate (a real number ). The model is written in a specific language and saved in a ‘.bnd’ file (for Boolean Network Description). Modeling parameters, including initial conditions, maximum time, etc. are gathered in a ‘.cfg’ file (for ConFiGuration file). The idea is to have a single.bnd file, and several.cfg files that account for different experimental situations (initial conditions, mutations, drug treatments, etc.). That way, ‘external variables’, noted by the name of the variable with a preceding $ and representing real numbers, are defined in the.bnd file and can be set and modified in the associated.cfg file.
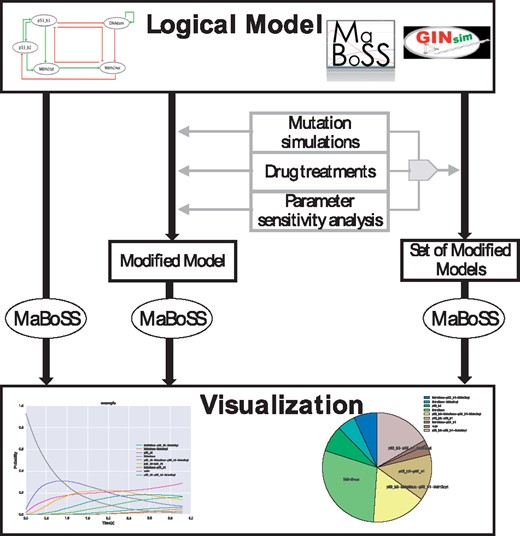
Pipeline of the use of MaBoSS 2.0 functionalities. A logical model can be simulated directly, modified by mutations, drug treatments or sensitivity analyses and all modifications can be gathered into a single command and simulated at once
Writing a MaBoSS model can be facilitated by creating and exporting it using the user-friendly tool GINsim (Naldi et al., 2009) (from http://ginsim.org/dev/). This export results in the creation of a.bnd file and a.cfg file with default parameters. Thus, any model in SBML qual standard format (Chaouiya et al., 2013) (including multi-valued models), can be exported in MaBoSS format. Similarly, it is possible to export a MaBoSS model (.bnd and.cfg files) into a set of logical rules written in a text format. The output is a text format that is supported by the library bioLQM (available at https://github.com/colomoto/bioLQM), which allows the export in SBML qual or other formats for other modelling tools such BoolNet (http://www.colomoto.org/software/boolnet.html; Müssel et al, 2010). Note that with a minor modification (a change of, into,), the output format can be imported directly in BoolNet.
One important asset of MaBoSS 2.0 is the possibility of using any probability distribution as an initial condition. For instance, for a set of 2 genes (A, B), the following probabilities can be assigned: and , thus offering the possibility to characterize an heterogeneous cell population where each probability of network state corresponds to a subpopulation.
3 Running MaBoSS and processing the output files
MaBoSS core script is accessible through command line. It estimates time dependent probabilities of ‘network states’, a network state is a vector of Boolean values associated to node states. The outputs of MaBoSS contain three files: a file for time dependent probabilities of network states (probtraj.csv file), a file for asymptotic probability decomposition in ‘indecomposable stationary distribution’ (statdist.csv file) and a file for the fixed points and their associated probability (fp.csv file). This latter file allows to quickly verify the existence of a limit cycle: when the sum of all fixed point probabilities is less than 1, it can be concluded that there exists at least one limit cycle that cannot be computed exactly but remains to be characterized.
3.1 Formatting of results
The outputs of MaBoSS are compact text files that are difficult to exploit as such. With practice, we have developed a script that extract specific information from these files: MBSS_FormatTable runs MaBoSS in a specific subfolder, and generates formatted tables for the trajectories, easily manageable in a spreadsheet.
3.2 Vizualization of results
The script MBSS_TrajectoryFig can be used for plotting trajectories, and MBSS_PieChart exports the asymptotic distribution in the form of a pie chart.
4 Modifications of a master model
One purpose of mathematical modeling is to predict the effect of specific perturbations (e.g. losses or gains of function, or combinations thereof). For instance, cancers can be considered as mutants of a model in normal conditions, which we refer to as ‘master model’. To do so, some of the following scripts create new ‘external variables’ and vary them according to the task the modeler wishes to perform. Recall that external variables of MaBoSS correspond to parameters in the.bnd file that can be modified in the.cfg file (see reference card).
Mutant simulations. Simulating gene alterations can be done by over-writing the rules of the corresponding components. The script MBSS_MutBnd allows the use of a single.bnd file for all different model variants (including wild type and mutant conditions). The script adds two external variables to each gene that the user wishes to mutate: the first one representing a possible deletion or a loss of function, the other reproducing amplifications or gains of function. These external variables can then be modified and set as desired in specific.cfg files. Once several.cfg files have been constructed, the use of FormatTable is particularly suited, because it generates a folder with the name of the.cfg file in order to keep track of each mutant simulation.
Parameter sensitivity analysis. The transition rates for each node can be varied to assess their importance in some outputs. For that, it is important to control their value through external variables. The script MBSS_SensitivityAnalysis allows to vary the value of external variables separately by creating a.cfg file per modification. Then, a shell script is automatically created and can run the simulations for all the newly created.cfg files.
Drug treatments. For the simulation of drug treatments, the script MBSS_DrugSim generates new nodes that will be used as drugs. They act as inhibitors of the specified targets. The strength of the inhibitor can be modulated to account for intermediate drug dosages. Also, in the course of a simulation, a drug treatment can be applied and removed after a certain time. For that, we have developed a script, MBSS_InitCondFromTrajectory, with which one can select the final probability distribution of a simulation and use it as the initial condition for another simulation of the model with the new drug condition.
5 Automation of multiple simulations
In order to optimize Boolean model analyses, MBSS_MultipleSim offers the possibility to generate multiple.cfg files and run the simulations at once with a single command.
6 Integrated functionalities
In order to run a complete project including all needed perturbations into a single command, MBSS_PrepareProjectFile prepares a folder containing all.cfg files corresponding to all the modifications without launching the simulations. In addition, it creates a shell script for running MaBoSS for all possible.cfg files and processing the outputs files. For running multiple simulations on a server, the folder can be imported and the shell script can be adapted to the server environment. Note that the definition of multiple runs is more straightforward in MBSS_MultipleSim, but is more flexible in MBSS_PrepareProjectFile.
7 Conclusion
MaBoSS 2.0 and its associated environment have been developed to answer specific needs of modelers. They can be combined and used into the following pipeline (see Supplementary Material for an example): (i) Construct a logical model, by writing directly the.bnd and.cfg files or by exporting them from GINsim; (ii) Run the model, analyze the results within a spreadsheet, and visualize the results; (iii) Test parameter sensitivity; (iv) Confront the model to known experimental observations: mutations, drug effects and (v) Set up a protocol for comparing experiments to model results for new biological insights.
In the future, we plan to construct a library version of MaBoSS in Python and in R to facilitate its implementation in these environments. We welcome the contribution of all future users to extend MaBoSS applications.
Acknowledgements
We thank Denis Thieffry, Loic Verlingue, Samuel Collombet, Loredana Martignetti and Claudine Chaouiya for their valuable contributions.
Funding
GK is supported by the Ligue Nationale contre le Cancer (Equipes labellisées), Site de Recherche Intégrée sur le Cancer (IRIC) Socrates, the ISREC Foundation, Agence Nationale pour la Recherche (ANR AUTOPH, ANR Emergence), the European Commission (ArtForce), a European Research Council Advanced Investigator Grant, the Fondation pour la Recherche Médicale (FRM), the Institut National du Cancer (INCa), the Fondation de France, Cancéropôle Ile-de-France, the Fondation Bettencourt-Schueller, the LabEx Immuno-Oncology and the Paris Alliance of Cancer Research Institutes. LC is partially funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement 668858.
Conflict of Interest: none declared.
References


{kind=link}