




Abstract
The protein O6-methylguanine-DNA methyltransferase (MGMT) is able to repair the mutagenic O6-methylguanine (O6-MeG) adduct back to guanine. In this context, it may protect against colorectal cancer formation associated with N-nitroso compounds. Such compounds may be endogenously formed by nitrosylation of amino acids, which can give rise to mutagenic O6-MeG and O6-carboxymethylguanine (O6-CMG) adducts. It is well established that O6-MeG is repaired by MGMT. However, up to now, whether O6-CMG is repaired by this enzyme remains unresolved. Therefore, the aim of the present study was to analyze the fate of both types of O6-guanine adducts in the presence and absence of MGMT activity. To this end, MGMT activity was efficiently blocked by its chemical inhibitor O6-benzylguanine in human colon epithelial cells (HCECs). Exposure of cells to azaserine (AZA) caused significantly higher levels of both O6-MeG and O6-CMG adducts in MGMT-inhibited cells, with O6-CMG as the more abundant DNA lesion. Interestingly, MGMT inhibition did not result in higher levels of AZA-induced DNA strand breaks in spite of elevated DNA adduct levels. In contrast, MGMT inhibition significantly increased DNA strand break formation after exposure to temozolomide (TMZ), a drug that exclusively generates O6-MeG adducts. In line with this finding, the viability of the cells was moderately reduced by TMZ upon MGMT inhibition, whereas no clear effect was observed in cells treated with AZA. In conclusion, our study clearly shows that O6-CMG is repaired by MGMT in HCEC, thereby suggesting that MGMT might play an important role as a tumor suppressor in diet-mediated colorectal cancer.
Introduction
The human genome is constantly exposed to DNA alkylating and mutagenic agents threatening its integrity. For instance, N-nitroso compounds (NOC) such as N-alkylnitrosamines and N-alkylnitrosamides (formed in cured meat products) as well as tobacco-specific nitrosamines (occurring in cigarette smoke) are absorbed by different tissues and can trigger cancer driving mutations (1–3). Moreover, β-oxidized N-propyl-nitrosamines, several monoalkylnitrosoureas and dialkylnitrosoureas (4) as well as N-nitrososarcosine (5), the nitroso derivative of N-methylglycine, have been shown to induce tumors in the colon of rats.
NOC have also been shown to be formed in the human colon after consumption of cured foodstuffs, whereby the nitrate and nitrite salts contained in these food commodities were endogenously reduced to nitric oxide (NO) (6,7). As a highly reactive nitrosating agent, NO may induce the formation of N-nitrosoglycine from glycine, which originates from intestinal protein fermentation in the colon (8–10). Once formed, N-nitrosoglycine may give rise to the highly reactive SN1-alkylating agent diazoacetate, leading to the methylation of guanine and adenine at the N7, O6 and N3 positions (8,9,11). Although N7-guanine and N3-adenine mainly undergo methylation by diazoacetate, the O6 position of guanine can either be methylated, resulting in the formation of O6-methylguanine (O6-MeG) adducts, or be carboxymethylated, resulting in the formation of O6-carboxymethylguanine (O6-CMG) adducts (3,12,13). The assumption that NOC may actually induce pro-carcinogenic effects is supported by the finding that the number of exfoliated colonic cells positive for O6-CMG adducts correlated with the fecal NOC levels in humans (14). Furthermore, endogenous NOC formation and nitrosylated heme, the latter mainly occurring in cured meat products, are associated with an elevated risk of colorectal cancer (15,16).
Different repair pathways to remove DNA damage induced by alkylation are present in cells, thereby protecting them against the mutagenic and carcinogenic effects induced by such agents (17,18). For instance, N-alkylated purines such as N7-methylguanine and N3-methyladenine, which are the most abundant DNA alkylation adducts, are removed by base excision repair (19,20). Minor DNA alkylation adducts such as N3-methylcytosine and N1-methyladenine are repaired by the family of AlkB homolog (ALKBH) proteins via direct demethylation using oxygen and 2-oxoglutarate as substrates (21). In contrast, O6-alkylguanines are repaired by the enzyme O6-methylguanine-DNA methyltransferase (MGMT) in a single-step reaction, whereby the alkyl group is transferred to a cysteine residue in the active center of the enzyme (22). This reaction results in an irreversible inactivation of MGMT, followed by its ubiquitin-mediated degradation (23). One MGMT molecule is thus required for the repair of one O6-alkylated base. O6-CMG adducts may cause GC → AT transition or GC → TA transversion mutations due to the misincorporation of adenine and thymine during translesion DNA synthesis, whereas O6-MeG adducts solely result in GC → AT transition mutations (17,24,25). For instance, such mutations were detected in the tumor suppressing KRAS gene in 12 out of 26 analyzed colorectal adenoma samples, which were simultaneously strongly associated with a reduced MGMT activity (26). Thus, hypermethylation of the MGMT promotor, causing a reduced activity or inactivation of the repair protein, may predispose humans to colorectal cancer, so that this enzyme may be viewed as an important key factor in cancer prevention (3). In support of this view, studies using transgenic mouse models demonstrated the crucial role of MGMT as a barrier against NOC-triggered colorectal cancer formation (27).
However, until now, it is unclear whether MGMT actually repairs O6-MeG as well as O6-CMG adducts. On the one hand, the repair of O6-MeG and O6-CMG adducts induced by N-nitrosoglycocholic acid was analyzed using extracts of Escherichia coli or the human lung fibroblast cell line MRC-5 expressing MGMT (11). Although O6-MeG adducts were repaired by this enzyme, neither a transfer of the radiolabeled carboxymethyl group to the repair protein nor a loss of signal in high-performance liquid chromatography chromatograms was observed in the case of O6-CMG adducts, which indicates that MGMT does not repair such adducts (11). On the other hand, it has been shown that an O6-CMG-containing oligonucleotide duplex efficiently inactivates MGMT in a cell-free system, indicating that this type of adduct is indeed an MGMT substrate (28).
Considering the above-mentioned discrepancy, the aim of the present study was to analyze the fate of both types of O6-guanine adducts in the presence and absence of MGMT activity in human colon epithelial cells (HCEC).
Materials and methods
Chemicals
Azaserine (AZA; purity ≥ 98%; Merck, Darmstadt, Germany) was dissolved and diluted in ultrapure water, whereas O6-benzylguanine (O6BG; purity ≥ 98%; Merck) stock solutions were prepared in dimethyl sulfoxide (DMSO; purity ≥ 99.5%; Carl Roth, Karlsruhe, Germany), which was also used as solvent control in the in vitro assays. The solvent of temozolomide (TMZ; Sigma–Aldrich, Schnelldorf, Germany) consisted of 1/3 (v/v) DMSO and 2/3 (v/v) ultrapure water. tert-Butyl hydroperoxide (t-BOOH; purity ≥ 98%; Merck) was diluted with cell culture medium prior to application. The source of all further chemicals used in the present work is mentioned in the corresponding part of Materials and methods.
HCEC subclone 1CT cells
HCEC subclone 1CT (HCEC 1CT) cells were established and kindly provided by Prof. Jerry W. Shay (Department of Cell Biology, Southwestern Medical Center, University of Texas, Dallas, TX). Human colon epithelial cells were isolated from a normal tissue biopsy sample obtained during a routine screening colonoscopy and immortalized by expression of cyclin-dependent kinase 4 and the catalytic component of human telomerase as described previously (29). HCEC 1CT cells were tested and authenticated by DNA fingerprinting as well as by Evercyte (Vienna, Austria; http://www.evercyte.com/hcec-1ct-166.html) and were last tested in April 2018. Furthermore, HCEC 1CT cells were characterized by performing Affymetrix SNP, Array-CGH and SKY analyses. The cells were cultured in a mixed medium consisting of 20% (v/v) Medium 199 and 80% (v/v) Dulbecco’s minimal essential medium GlutaMAX™ supplemented with 2% (v/v) cosmic calf serum (all obtained from Thermo Fisher, Darmstadt, Germany), 25 ng/ml epidermal growth factor, 10 µg/ml bovine insulin, 2 µg/ml transferrin, 50 µg/ml gentamycin, 1 µg/ml hydrocortisone and 5 nM sodium selenite (all obtained from Sigma–Aldrich). The cells were cultured at 37°C in a hypoxic humidified atmosphere containing 2% O2 and 5% CO2.
Chemical exposure and subsequent processing of cell cultures
MGMT activity and protein level assays
In order to inhibit MGMT, HCEC 1CT cells were first incubated with O6BG, which is one of the most efficient and well-established MGMT inhibitors and effectively induces the ubiquitination and degradation of the protein in a similar way as O6-MeG (23,30,31). In the in vitro assays, cell culture medium only (AZA and TMZ) or 0.1% DMSO (O6BG) was used as solvent control. For the MGMT studies (radiolabeled DNA as well as western blotting analyses), 3 × 105 cells were plated in 75 cm2 Primaria™ cell culture flasks (Corning, Kaiserslautern, Germany; three flasks per sample). On the next day, HCEC 1CT cells were exposed to 0.1, 1 or 10 µM O6BG for 4 h in the case of the radiolabeled DNA analyses or 50 µM O6BG for 2, 4, 8, 16 and 24 h in the case of the western blotting analyses. The concentration range of 0.1–10 µM O6BG used in the radiolabeled DNA analyses was chosen based on a previous study by Kaina et al. (32), in which the IC50 of O6BG to inhibit the MGMT activity in HeLa S3 cells (588 fmol/mg protein, which is higher than that in HCEC 1CT cells; see Results for details) was 0.3 µM, whereas the concentration of 50 µM O6BG for the western blotting analyses was used to ensure an efficient long-term inhibition of MGMT. Thereafter, the cells were washed twice with phosphate buffered saline, trypsinized using trypsin/ethylenediaminetetraacetic acid (Merck), pooled in centrifuge tubes (Greiner Bio-One, Frickenhausen, Germany) and centrifuged at 1600g for 5 min at 4°C. Then, the supernatant was removed, the cells washed with phosphate buffered saline and centrifuged again. Finally, the supernatant was discarded and the cell extract was frozen in liquid nitrogen followed by storage at −80°C until further use.
Genotoxicity and cytotoxicity assays
In the case of the DNA adduct analyses, HCEC 1CT cells were plated (3 × 105 cells per 75 cm2 cell culture flask, three flasks per treatment) and incubated 24 h later with 50 µM O6BG or 0.1% DMSO for 4 h followed by a medium change and the addition of 0, 50 or 100 µM AZA or TMZ. In the case of the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) cytotoxicity assay, an additional concentration of 150 µM AZA and TMZ was tested. TMZ, a triazene derivative inducing the formation of O6-MeG but not O6-CMG adducts, is used as DNA alkylating agent in glioma therapy (30). AZA mainly induces the formation of carboxymethylated DNA lesions, e.g. O6-CMG and, less frequently, O6-MeG, as well as N-methylpurine adducts (12,33). Twenty-four hours after exposure to AZA or TMZ, cell samples were either prepared and assayed as described below or incubated again with O6BG. The supernatant of all samples was collected separately, mixed with O6BG or DMSO and the cells were re-exposed to the medium containing the test compound. Twenty-four hours after the second exposure to the MGMT inhibitor, the cells were collected, frozen in liquid nitrogen and stored at −80°C until further use. To determine the levels of DNA strand breaks induced by AZA and TMZ with or without MGMT inhibition in the alkaline comet assay, 1.5 × 105 cells were plated on 6 cm dishes (Corning) and incubated with the two above-mentioned compounds 24 h after seeding as described above.
Quantification of MGMT activity using radiolabeled DNA
The exposed cell extracts were lysed as described previously (34), followed by protein quantification using the Bradford-based Bio-Rad protein assay (Bio-Rad, Munich, Germany). For the MGMT activity assay, radiolabeled O6-MeG generated from the incubation of calf thymus DNA (Sigma–Aldrich) with 3H-MNU (18 Ci/mmol; Amersham Biosciences, Freiburg, Germany) was mixed with 100 µg of cell extract protein as described previously (35). The data were evaluated as the transfer rate of radiolabeled DNA to the MGMT protein within 90 min and expressed as fmol radioactivity per milligram protein. Extracts from MGMT-deficient HeLa MR cells were used as negative control. These cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in F12/Dulbecco’s minimal essential medium (Thermo Fisher) supplemented with 5% (v/v) fetal bovine serum (PAN Biotech, Aidenbach, Germany) at 37°C in a humidified atmosphere containing 7% CO2.
Quantification of MGMT protein levels by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and western blotting
The chemical exposure and harvest of the cells were performed as described above, and the whole cell extracts were prepared as described previously (36). Protein content was determined using the Bradford assay. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and western blotting were essentially conducted as reported previously (36). Briefly, equal amounts of protein were loaded onto 12.5% acrylamide gels, separated by SDS–PAGE and transferred onto a nitrocellulose membrane (PerkinElmer, Rodgau, Germany) by making use of Bio-Rad equipment and the wet blotting technique. Unspecific binding sites on the membrane were blocked with 5% skimmed dry milk in Tris-buffered saline/0.1% Tween 20 for 1 h at room temperature. The incubation with primary antibodies [Hsp90ɑ/β: mouse monoclonal; Santa Cruz (sc-13119); 1:1000 in Tris-buffered saline/0.1% Tween 20; MGMT: mouse monoclonal; Merck Millipore (MAB16200); 1:750 in Tris-buffered saline/0.1% Tween 20)] was performed overnight at 4°C. Following three washing steps with Tris-buffered saline/0.1% Tween 20, the membranes were incubated with the respective secondary antibody for 1 h at room temperature. Then, following three further washing steps, the proteins were detected with the Azure Biosystem c300 (Biozym, Hessisch Oldendorf, Germany) using the Western Lightning® Plus-ECL substrate (PerkinElmer). MGMT protein expression levels were normalized to the corresponding Hsp90 signal using the Image Lab Software (Bio-Rad).
DNA adduct analysis
HCEC 1CT cells were plated (3 × 105 cells per 75 cm2 cell culture flask, three flasks per treatment) and 24 h later exposed to the test compounds for 24 and 48 h as described in the cell culture protocol above. Cells were then harvested in the same way as for the MGMT activity assay, frozen in liquid nitrogen and stored at −80°C. For DNA adduct analysis, samples were thawed and DNA was isolated by using a DNA isolation kit for cells and tissues (Roche Diagnostics, Mannheim, Germany) according to the instructions of the manufacturer with a calculated cell number of 5 × 106 cells per sample. Finally, the resulting DNA pellets were air-dried and stored at −20°C until further analysis. The levels of O6-carboxymethyldeoxyguanosine (O6-CMdG) and O6-methyldeoxyguanosine (O6-MedG) were measured according to the method described recently by Geisen et al. (37). Briefly, DNA was resuspended in Tris buffer, spiked with 100 fmol D3-O6-MedG and 200 fmol 15N5-O6-CMdG per sample (200 µl final volume) and subjected to enzymatic digestion. Standards were synthesized by previously reported methods (38,39). A portion of the sample was removed to quantify dG by high-performance liquid chromatography and the rest of the sample was concentrated to dryness. Samples were resuspended in 200 μl mass spectrometry (MS) grade water + 0.1% formic acid and isolated after solid-phase extraction on a C18 cartridge, isolation of eluted fractions, drying under vacuum and dissolving in 10 μl MS grade H2O for MS analysis. Simultaneous quantification of O6-CMdG and O6-MedG was achieved on an Orbitrap Fusion Lumos (Thermo Scientific, Waltham, MA) equipped with a nano-electrospray ionization source and a nanoAcquity UPLC M-class system (Waters, Milford, MA). Nano-electrospray ionization was used in positive mode and MS-based detection was performed in parallel reaction monitoring with higher-energy-collisional-dissociation with Orbitrap detection (PRM HCD OT) mode. Extensive optimization of analysis parameters and validation of the method have been reported (37). The identity of the analyte peaks was confirmed by coelution with internal standards and verification of accurate precursor and fragment mass. The limit of detection and limit of quantification were defined as the amount of analyte generating a signal-to-noise ratio (S/N) of 3 or 10 in the extracted ion chromatograms, respectively. The limit of detection was 5 pM for O6-CMdG and 1 pM for O6-MedG. For both adducts, the limit of quantification was 10 pM for isolated standards and 50 pM in a matrix of cellular DNA digest. The relative standard deviation ranged from 1.7 to 25.6% for O6-CMdG and from 0.8 to 23.5% for O6-MedG. Analyte recovery ranged from 84 to 113% for O6-CMdG and from 92 to 123% for O6-MedG. The results are expressed as the number of O6-CMdG or O6-MedG adducts per 107 unmodified nucleotides (nt).
Alkaline comet assay
The alkaline comet assay was performed as described previously (40), with a cell lysis step (50 min) followed by a DNA unwinding step (20 min) taking place before electrophoresis. Exposure to the free radical generator t-BOOH (50 µM, 20 min) was performed as a positive control. Samples were analyzed by using an Axioskop 2 fluorescence microscope (Zeiss, Jena, Germany) as well as the Comet Assay IV software (Perceptive Instruments, Bury St Edmunds, UK), scoring 50 cells per slide.
MTS cytotoxicity assay
The cytotoxicity of O6BG, AZA and TMZ was measured separately after 24 and 48 h of exposure using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS; Promega, Mannheim, Germany) as reported previously (41). For this purpose, 5 × 103 cells per well (24 h) or 2.5 × 103 cells per well (48 h) were seeded in 96-well plates (Corning). One day after seeding, the cells were exposed to each of the compounds as described above. Thereafter, the assay was performed according to the instructions of the manufacturer. A final concentration of 4% (v/v) MTS was used, and the plates were incubated for 60 min at 37°C in an atmosphere containing 5% CO2 prior to the recording of absorbance.
Statistical analyses
All data sets were statistically analyzed with Prism (version 9.1.0; GraphPad, La Jolla, CA). Prior to the significance analyses, the data were examined regarding normal distribution by applying the Shapiro–Wilk test. Data sets that did not follow the normal distribution were analyzed with non-parametric tests. MGMT protein level analyses, comet assays, cytotoxicity assays and O6-MedG adduct level analyses up to 24 h were thereby subjected to a Mann–Whitney test in order to compare the effect of O6BG for each dose and point in time with the solvent control (DMSO), whereas MGMT activity level data were analyzed with a Kruskal–Wallis test followed by Dunn’s multiple comparison test. The normally distributed O6-CMdG and O6-MedG adduct level data as well as the cytotoxicity assay data (all after 48 h of incubation) were subjected to a two-way analysis of variance followed by Šídák’s multiple comparison test, whereby the effect of O6BG and the dose of AZA or TMZ were tested separately for each point in time and alkylating agent. Statistical significance was defined throughout as P < 0.05, and all data were evaluated for outliers with the Grubb’s test prior to significance testing.
Results
O6BG leads to the inhibition of MGMT activity and MGMT depletion in HCEC 1CT cells
The MGMT activity measured in untreated HCEC 1CT cells (236 fmol/mg protein) was in line with the average MGMT activity of ~50–200 fmol/mg protein determined previously in normal human colonic tissue (42,43). In order to address the role of MGMT activity in the repair of O6-CMG adducts, the potent MGMT inhibitor O6BG was used (31). In a first step, the efficiency of O6BG to inhibit MGMT activity and deplete MGMT protein levels in HCEC 1CT cells was analyzed. A time-dependent depletion of MGMT protein in HCEC 1CT cells exposed to 50 µM O6BG was observed, although protein bands were still detectable after 24 h (Figure 1A; western blots including molecular size markers are shown in Supplementary Figure 1). MGMT protein levels were reduced when compared with DMSO-treated cells after 4 h of exposure to O6BG, with the decrease being even stronger after 8 h of exposure (Figure 1B). However, after 16 and 24 h of exposure, MGMT expression slightly increased again (Figure 1B). The incubation of HCEC 1CT cells with O6BG led to a concentration-dependent decrease in MGMT activity, with the lowest concentration (0.1 µM) reducing MGMT activity by 50% (Figure 1C; Supplementary Table I). Moreover, a concentration of 10 µM O6BG resulted in total enzyme inhibition, and the detected radioactivity levels in exposed HCEC 1CT cells equaled those observed in MGMT-deficient samples (Supplementary Table I). Thus, O6BG was effective in completely inhibiting MGMT activity in HCEC 1CT cells despite the presence of residual MGMT protein. The above-mentioned findings are in agreement with those of a previous report (31), in which 5 µM O6BG led to a complete loss of MGMT activity in the human colon adenocarcinoma cell line HT29, a cell line known for its high MGMT activity (~800 fmol/mg protein) (44).
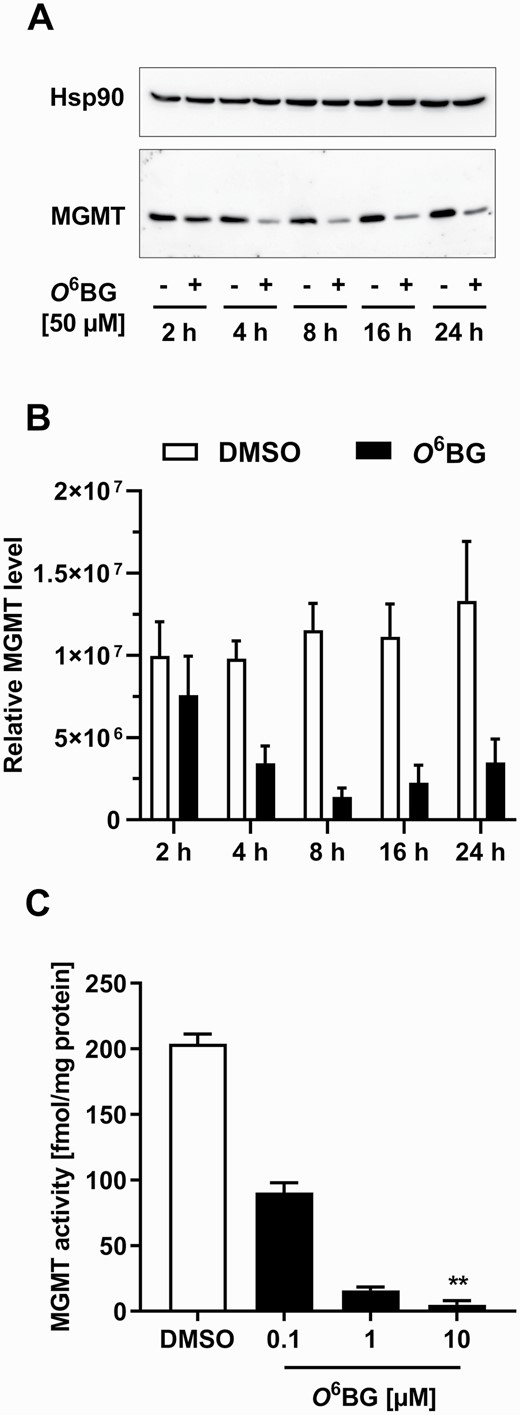
Inhibition of MGMT activity and MGMT depletion by O6-benzylguanine (O6BG) in HCEC 1CT cells. (A) Western blot images of MGMT and Hsp90 (loading control) protein levels in HCEC 1CT cells exposed to 0.1% DMSO (−) or 50 µM O6BG (+) for 2, 4, 8, 16 or 24 h. (B) MGMT protein levels after exposure to 0.1% DMSO or 50 µM O6BG (normalized to Hsp90 protein expression). (C) MGMT activity after incubation with increasing concentrations of DMSO (0.1%) or O6BG for 4 h. The measured activity of MGMT-deficient HeLa MR cells served as blank and was set to 0 fmol/mg protein. All results are shown as mean + SEM (n = 3). **P < 0.01.
MGMT contributes to the removal of O6-MedG and O6-CMdG adducts in HCEC 1CT cells
Having established conditions in cells to control MGMT activity, we aimed to determine if the inhibition of this enzyme had an impact on the removal of O6-CMdG adducts. Thus, HCEC 1CT cells were incubated with AZA in the presence or absence of the MGMT inhibitor O6BG, followed by DNA adduct quantification by MS. In MGMT-proficient cells, AZA induced a time but not concentration-dependent accumulation of very low levels of O6-MedG adducts (Figure 2A; targeted m/z with MSn and the chemical structure of the internal standard are shown in Supplementary Table II; corresponding chromatograms are shown in Supplementary Figure 2), which corroborates that the MGMT activity present in the cells efficiently removes O6-MedG adducts in the absence of the MGMT inhibitor. Pre-conditioning of the cells with O6BG prior to AZA exposure significantly and concentration-dependently increased (6–8-fold) the content of O6-methylated DNA lesions. The increase in O6-MedG levels is completely in line with the well-established role of MGMT in O6-MedG repair and provides an important positive control in our evaluation of the impact on O6-CMdG. Moreover, the number of O6-MedG adducts was 3.5-fold higher after 48 h when compared with the 24 h incubation. The O6-CMdG adduct level was ~18-fold higher than that of O6-MedG adducts in MGMT-proficient HCEC 1CT cells exposed to 100 µM AZA for 24 h (Figure 2A and B), whereas the O6-CMdG adduct levels were similar after 24 and 48 h of exposure to 100 µM AZA (Figure 2B; targeted m/z with MSn and the chemical structure of the internal standard are shown in Supplementary Table II; corresponding chromatograms are shown in Supplementary Figure 3). Finally, the levels of O6-CMdG adducts were 3.2- and 2.4-fold higher in MGMT-inhibited cells exposed to 100 µM AZA than in the corresponding control (i.e. MGMT-proficient) cells after 24 and 48 h, respectively (Figure 2B). The data show that inhibiting MGMT activity results in increased O6-MedG as well as O6-CMdG adduct levels.
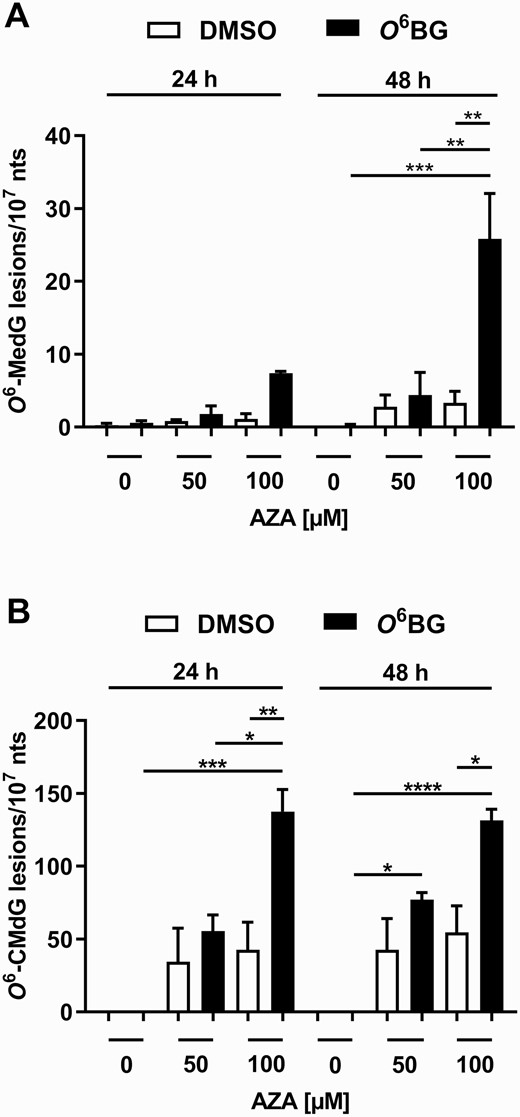
AZA-mediated induction of O6-methylated and O6-carboxymethylated deoxyguanosine adducts in HCEC 1CT cells upon MGMT inhibition. Induction of O6-MedG (A) and O6-CMdG adducts (B) by AZA in HCEC 1CT cells was quantified by nanoLC-ESI-HRMS2 after an exposure to 0.1% DMSO (white bars) or 50 µM O6BG (black bars) for 4 h. All results are shown as mean + SEM (n = 3) and expressed as number of adducts per 107 nucleotides (nt). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
MGMT does not inhibit DNA strand break induction by AZA in HCEC 1CT cells
Since MGMT was shown to be involved in the repair of O6-CMdG and O6-MedG adducts in HCEC 1CT cells exposed to AZA, we hypothesized that MGMT would also be able to inhibit DNA strand break induction following an exposure of cells to this compound. In order to test this assumption, HCEC 1CT cells were incubated with AZA and subsequently analyzed using the alkaline comet assay. HCEC 1CT cells exposed to increasing AZA concentrations showed higher Olive tail moment (OTM; a marker for DNA strand breaks induction) levels when compared with the negative control. Interestingly, O6BG pre-treatment led to an equally high or, in the case of the incubation with 50 µM AZA, to a significantly lower induction of DNA strand breaks (Figure 3A and B), although DNA adduct levels were significantly increased after MGMT inhibition (Figure 2A and B). HCEC 1CT cells were also exposed to TMZ, which only generates O6-MeG adducts, and then analyzed using the same experimental setup. Up to 100 µM TMZ did not result in an increase of the OTM in MGMT-proficient HCEC 1CT cells (Figure 3A and B). However, a significantly higher level of DNA strand breaks was detected in TMZ-treated cells after MGMT inhibition, whereby this effect was concentration-dependent. In summary, incubation of HCEC 1CT cells with TMZ resulted in an increased DNA strand break formation following MGMT inactivation, whereas DNA strand break levels induced by AZA remained unaffected or were even reduced in the above-mentioned experimental setting.
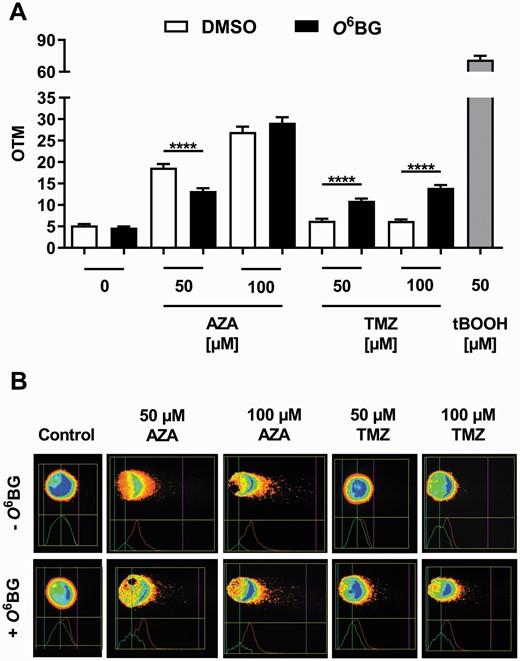
Genotoxic effects of AZA and TMZ in HCEC 1CT cells upon MGMT inhibition. Cells were incubated for 4 h with 0.1% DMSO (white bars) or 50 µM O6BG (black bars) following AZA or TMZ exposure. (A) DNA strand breaks, shown as Olive tail moment (OTM), induced by AZA or TMZ −/+ O6BG exposure. t-BOOH (50 µM, 20 min) served as positive control. All results are shown as mean + SEM (n = 3). ****P < 0.0001. (B) Representative images of comet assays forming the basis for results shown in (A).
MGMT protects HCEC 1CT cells to a very limited extent against the cytotoxicity of TMZ
Whether the MGMT status of HCEC 1CT cells had an influence on the cytotoxicity of AZA and TMZ was analyzed using the MTS cytotoxicity assay. The viability of MGMT-proficient and MGMT-deficient HCEC 1CT cells was not affected by AZA in concentrations of up to 150 µM after an incubation period of 24 and 48 h (Figure 4A and B). A concentration of 100 µM TMZ led to a slight decrease in the viability of MGMT-deficient HCEC 1CT cells after 24 h (Figure 4A), whereas concentrations of 100 and 150 µM TMZ decreased the viability of MGMT-deficient HCEC 1CT cells by up to 13% after 48 h (Figure 4B). Taken together, the results obtained show that AZA and TMZ lead, if at all, to a minor cytotoxic effect in HCEC 1CT cells and that the cytotoxicity of TMZ is only increased to a very limited extent when MGMT activity is inhibited.
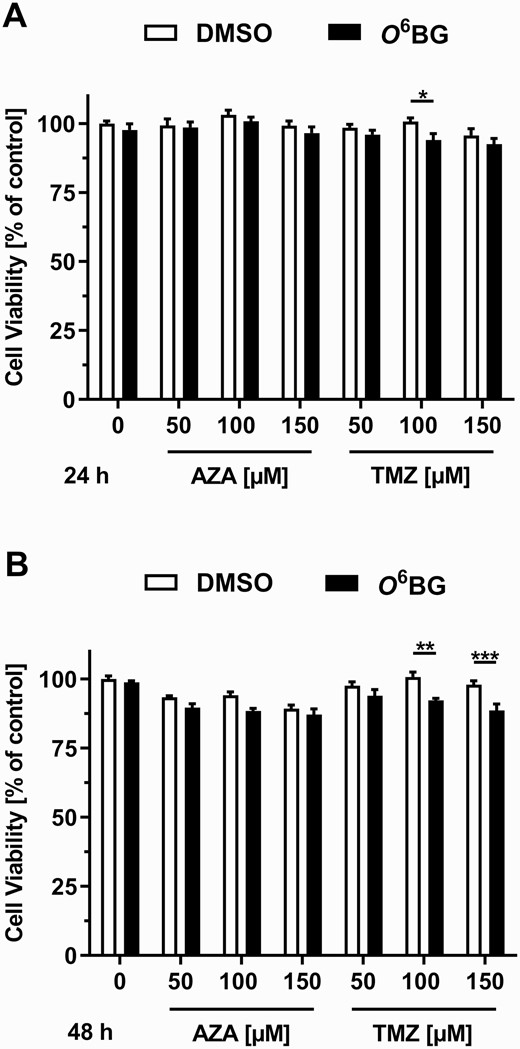
Cytotoxicity of AZA and TMZ in HCEC 1CT cells upon MGMT inhibition. Relative cell viability of cells exposed to 0.1% DMSO (white bars) or 50 µM O6BG (black bars) for 4 h, followed by the incubation with AZA or TMZ for 24 h (A) or 48 h (B). All results are shown as mean + SEM (24 h, n = 4; 48 h, n = 3). *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
The aim of the present study was to shed light on the ongoing discussion on whether MGMT is able to repair both O6-MeG and O6-CMG DNA adducts, or if this enzyme only repairs the former type of adduct. The experimental approach pursued in the present study to resolve this controversy was to inhibit MGMT activity in HCEC 1CT cells with O6BG, then expose the cells to AZA and finally quantify the AZA-induced O6-MedG and O6-CMdG adducts by nanoLC-ESI-HRMS2. In the present study, up to 138 O6-CMdG lesions/107 nucleotides were detected as a result of this exposure, whereas the level of O6-MedG lesions was 5–20 times lower and ranged from 7 to 26 lesions/107 nucleotides. Similar differences in the abundance of various O6-guanine adducts have also been described in previous studies (33,45). For instance, Yu et al. (45) reported an 18-fold higher O6-CMdG adduct level when compared with the O6-MedG adduct level in AZA-exposed HCT-116 cells, a human colon carcinoma cell line with a MGMT activity similar to that measured in the cells used herein (195 fmol/mg protein) (46). Moreover, the reaction of cell-free calf thymus DNA with AZA or potassium diazoacetate resulted in O6-CMG to O6-MeG ratios of 38 and 16, respectively (33). In the present study, the levels of both types of DNA lesions were significantly higher in MGMT-inhibited cells, but the effect of MGMT was higher with respect to the removal of O6-MedG adducts (6–8-fold) versus O6-CMdG adducts (2–3-fold). The reduced effect of MGMT inhibition on the abundance of O6-CMdG adducts suggests that another DNA repair mechanism specific for this type of lesion and not inhibited by O6BG might also be involved. Indeed, O’Driscoll et al. (12) have shown previously that the sensitivity of human lymphoma cells toward AZA was not affected by the activity of MGMT and, based on this finding, hypothesized that O6-CMG adducts may be repaired by nucleotide excision repair (NER; 19). A combination of NER- and MGMT-mediated repair may explain the relatively low yet significant increase in the O6-CMdG adduct levels in MGMT-inhibited HCEC 1CT cells. Nonetheless, to our knowledge, this is the first time that O6-CMdG adducts have been directly quantified in the presence or absence of MGMT, showing that this protein does in fact repair O6-CMG lesions in HCEC.
The basis of previous contradictory findings concerning whether MGMT is involved in O6-CMG adduct repair may be related to details regarding the different experimental models used. In the present study, we directly quantified DNA lesions in HCEC. In two previous studies, the repair of O6-CMG was analyzed in a cell-free system using indirect methods (11,28), whereby only the results obtained by Senthong et al. (28) are in agreement with the data shown in the present study. Furthermore, Shuker et al. (11) analyzed the activity-reducing effects of carboxymethylated DNA on a bacterial MGMT equivalent (ada ATase) and, although O6-MeG was a substrate for the ada ATase protein, it was unable to act on DNA containing O6-CMG adducts. However, as also discussed by Senthong et al. (28), the extrapolation of experimental findings obtained using a bacterial MGMT equivalent to those obtained using mammalian MGMT is difficult at best, whereas the results presented herein provide strong evidence for mammalian MGMT-mediated repair of O6-CMG adducts in HCEC.
Although the direct detection of O6-MedG and O6-CMdG adducts clearly shows that the latter lesions are indeed repaired by MGMT, the detection of NOC-induced DNA strand breaks using the alkaline comet assay delivered unexpected results. Although MGMT inhibition led to more DNA lesions after incubation with AZA, a similar increase in OTM levels was not observed. Generally, if MGMT is inhibited or the level of O6-methylations exceeds the repair capacity of the enzyme (i.e. its cellular concentration), persistent O6-MeG adducts may mispair with thymine, resulting in O6-MeG:T mismatches after the first round of replication, followed by the detection of these lesions by the mismatch repair system (MMR) (42,43,47,48). The MMR comprises the MutSα and MutLα complexes (49), which consist of the MSH2 and MSH6 (MutSα) as well as MLH2 and PMS2 (MutLα) subunits (50,51). The MutSα subunit can bind to O6-MeG and O6-MeG:T mismatches, whereby the latter are preferred (51,52). After binding, mispaired lesions first lead to DNA single strand breaks, which finally result in DNA double strand breaks after the second round of replication (47,49). The direct correlation of O6-MeG:T mismatches with DNA strand breaks is the basis for the use of the comet assay to assess O6-MeG adduct levels (47).
In the present study, TMZ induced less DNA strand breaks when compared with AZA, although the same concentrations of both alkylating agents were used. The toxicity of TMZ in cancer therapy is mainly a consequence of the formation of O6-MeG adducts, although these only constitute 5–10% of all DNA lesions induced by this compound, whereas more frequently N3-methyladenine and N7-methylguanine are formed (53,54). The lack of DNA strand breaks in MGMT-proficient cells exposed to TMZ may be related to the high cellular MGMT activity effectively repairing O6-MeG lesions and thus preventing the formation of DNA strand breaks. Moreover, the significant difference seen in the present study between samples harboring a functional and an inhibited MGMT strongly suggests that the MMR system may play a role in HCEC 1CT cells, whereby higher O6-MeG adduct levels in MGMT-inhibited cells result in more DNA strand breaks in the comet assay. This assumption is further supported by the results obtained in the cell viability assay, showing a higher cytotoxicity in cells treated with the MGMT inhibitor and TMZ, and by previous studies reporting the presence of the MMR subunits MSH2, MSH3 and MSH6 in HCEC 1CT cells (55,56).
Although O6-MeG lesions and DNA strand breaks induced by TMZ were efficiently repaired in MGMT-proficient cells, OTM levels induced by AZA were relatively high and increased concentration-dependently regardless of the MGMT status. The hypothesis that MGMT only plays a minor role in O6-CMG-mediated DNA strand break induction is supported by the missing increase of OTM levels in MGMT-deficient cells. As in the case of TMZ exposure, we expected AZA incubation to induce more DNA strand breaks in MGMT-inhibited cells as a result of the missing MGMT-mediated repair and an active MMR system (under the assumption that O6-CMG is recognized by MMR). However, the level of DNA strand breaks was similar or even lower when MGMT was inhibited. As shown by the DNA adduct analyses, AZA induced the formation of O6-CMdG as the major adduct and that of O6-MedG as a minor lesion. Nonetheless, it is still unknown if O6-CMG mispaired with T is actually recognized by the MMR system and if it gives rise to DNA strand breaks in a similar way as O6-MeG adducts (3). Moreover, an oligonucleotide duplex harboring a S6-CMG:T mispair, which is a chemical analog of O6-CMG:T, showed almost no complex formation with MutSα in a band shift assay (12). Therefore, the formation of DNA strand breaks by O6-CMG mediated by the MMR system seems less probable. Furthermore, the same authors observed cytotoxicity in AZA-treated Raji Burkitt’s lymphoma cell lines regardless of their MMR and MGMT status (12). If O6-CMG:T would be detected by the MMR system in a similar way as O6-MeG:T, this should result in DNA strand break formation, particularly upon MGMT inhibition. However, this was not observed in the comet assays presented herein, thus suggesting that another mechanism may be responsible for DNA strand break induction upon exposure of cells to AZA. For instance, it was shown that the mispair of O6-CMG with thymine could lead to a high-wobble O6-CMG:T pair, in which the carboxyl group forms a hydrogen bond with thymine, resulting in a chemical structure not described for other O6-alkylG:T mispairs (57). Moreover, it was hypothesized that this wobble structure is recognized and repaired by NER (57). If this high-wobble O6-CMG:T pair would be indeed responsible for the activation of an O6-CMG-specific DNA repair mechanism, it may have an influence on DNA stability or the efficient bypass by DNA polymerases and finally result in increased DNA strand break formation.
In conclusion, the results of the present study provide for the first time experimental evidence that MGMT repairs both O6-MeG and O6-CMG DNA lesions in HCEC and support the view that MGMT might play an important role in preventing NOC-triggered DNA alkylation and the potential formation of GC → AT transition mutations. This is particularly important in the case of genes such as KRAS, which is often affected by these types of mutations and has been shown to be present in the majority of colorectal cancer cases (26). By repairing the most abundant NOC-induced DNA lesions, MGMT may serve as a critical tumor suppressor in colorectal carcinogenesis.
Abbreviations
- AZA
azaserine
- DMSO
dimethyl sulfoxide
- HCEC
human colon epithelial cell
- HCEC 1CT
HCEC subclone 1CT
- MGMT
O6-methylguanine-DNA methyltransferase
- MMR
mismatch repair system
- MS
mass spectrometry
- NER
nucleotide excision repair
- NOC
N-nitroso compound
- O6BG
O6-benzylguanine
- O6-CMdG
O6-carboxymethyldeoxyguanosine
- O6-CMG
O6-carboxymethylguanine
- O6-MedG
O6-methyldeoxyguanosine
- O6-MeG
O6-methylguanine
- OTM
Olive tail moment
- SDS–PAGE
sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- TMZ
temozolomide
Funding
Deutsche Forschungsgemeinschaft (grant numbers STE 493/21-1 and FA 1034/3-3); the Schweizerischer Nationalfonds zur Förderung der wissenschaftlichen Forschung (grant numbers 185020 and 186332); Krebsliga Schweiz (grant number KFS-4443-02-2018-R).
Acknowledgement
The authors wish to thank Jutta Barras-Akhnoukh for her excellent technical assistance.
Conflict of Interest Statement: None declared.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
Author notes
TK, MTE and NS contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}