





Abstract
The set of annotations at the Saccharomyces Genome Database (SGD) that classifies the cellular function of S. cerevisiae gene products using Gene Ontology (GO) terms has become an important resource for facilitating experimental analysis. In addition to capturing and summarizing experimental results, the structured nature of GO annotations allows for functional comparison across organisms as well as propagation of functional predictions between related gene products. Due to their relevance to many areas of research, ensuring the accuracy and quality of these annotations is a priority at SGD. GO annotations are assigned either manually, by biocurators extracting experimental evidence from the scientific literature, or through automated methods that leverage computational algorithms to predict functional information. Here, we discuss the relationship between literature-based and computationally predicted GO annotations in SGD and extend a strategy whereby comparison of these two types of annotation identifies genes whose annotations need review. Our method, CvManGO (Computational versus Manual GO annotations), pairs literature-based GO annotations with computational GO predictions and evaluates the relationship of the two terms within GO, looking for instances of discrepancy. We found that this method will identify genes that require annotation updates, taking an important step towards finding ways to prioritize literature review. Additionally, we explored factors that may influence the effectiveness of CvManGO in identifying relevant gene targets to find in particular those genes that are missing literature-supported annotations, but our survey found that there are no immediately identifiable criteria by which one could enrich for these under-annotated genes. Finally, we discuss possible ways to improve this strategy, and the applicability of this method to other projects that use the GO for curation.
Database URL:http://www.yeastgenome.org
Introduction
The integration and comparison of biological information can be complicated by the human tendency to express the same concept in multiple ways. The Gene Ontology (GO) addresses the difficulty of functional classification for gene products, and has become the main resource for capturing such information in a controlled format that can be effectively used for search and computational analysis (1,2). GO annotations are comprised of a gene product; a structured vocabulary term that represents a molecular function, a biological process or a cellular component; the literature reference for the assignment; and an evidence code that indicates how the reference supports the annotation (3,4).
Annotations based on manual curation of the published literature are generally considered to be the gold standard. These are derived from published sources by highly trained scientific biocurators, who annotate gene products with the current and most direct information, considered in the context of all available experimentally defined knowledge (5,6). As of Fall 2011, the Saccharomyces Genome Database [SGD; http://www.yeastgenome.org, (7)] had manually assigned nearly 38 000 GO annotations. The goal of creating GO annotations at SGD is to provide a summary of the biological role of a given gene product. This requires reviewing the entire body of literature for a gene and synthesizing that knowledge to provide a concise and accurate presentation of the role of that gene product in the cell.
In addition to the literature-based set of annotations, SGD also provides a large set of annotations automatically generated by in silico methods (8). These predictive computational methods use single or multiple inputs—for example, protein sequence signatures, protein–protein and genetic interactions or mutant phenotypes—for algorithms that generate annotations for gene products in an unbiased manner. These predictions can complement existing manual annotations, and provide clues about the functions of uncharacterized proteins. Among the computationally predicted annotations are those provided by the GOA project at UniProtKB (9–11), including InterPro to GO, which is based on protein sequence signatures (12,13); and SwissProt Keywords (SPKW) to GO (http://www.geneontology.org/external2go/spkw2go). SGD also includes annotations from methods presented in publications, which, at the time of this analysis, included two sophisticated algorithms that integrate multiple data sets to automatically assign annotations: bioPIXIE/MEFIT and YeastFunc. BioPIXIE/MEFIT considers relationships between genes inferred from different types of high-throughput data sets, such as protein localization, interactions and genomic expression data, to generate predicted GO annotations in Biological Process (14–16). The YeastFunc method (17) integrates large-scale data sets with sequence-based inferences to make predictions in all three GO aspects (Molecular Function, Biological Process, Cellular Component). The GO annotations in SGD that are assigned via all of these computational methods carry either the IEA (Inferred from Electronic Annotation) or RCA (Reviewed Computational Analysis) evidence codes (http://www.geneontology.org/GO.evidence.shtml). SGD works with authors of publications to determine the most appropriate cut-off in order to provide the best representation of the predictive method while maintaining a high level of confidence in the computational annotation. All computationally predicted annotations are maintained in SGD for up to 1 year, after which time the annotations are removed unless the providing source has refreshed the analysis based upon the latest GO structure and data available in the literature, since both are constantly changing.
Among the tasks that are considered highest priority at SGD are the annotation of genes for which a novel function has been identified, and the review of annotations that could be incorrect. The challenge is to identify these genes in an efficient manner. Because manual curation requires significant effort and our resources are limited, we need to define a pipeline that will support this type of prioritization of our curatorial tasks (18,19). To begin addressing this issue, we previously explored whether computational predictions can be used as an indicator for identifying genes with ‘unknown’ annotations that need review (20). ‘Unknown’ annotations are created by manually assigning the root term of a GO aspect, which are the broadest terms that exist: ‘molecular_function’ (GO:0003674), ‘biological_process’ (GO:0008150), and ‘cellular_component’ (GO:0005575). These annotations indicate that at the time of curatorial review no evidence is present in the literature that would allow a more specific annotation to be made for the gene product (2,21). We presented a method by which we paired a manual literature-based annotation with a computationally predicted annotation and looked for correspondence between the two, a method that in this article we will refer to as ‘Computational vs Manual GO annotations’ (CvManGO). In the previous study, we found that when an InterPro prediction existed for a gene that was manually annotated to ‘unknown’, we were often able to find evidence in the literature to assign a biological function to that gene (20). Here we extend this analysis by considering additional prediction methods. To represent a broad range of methods used to generate computational annotations, in addition to the sequence-based method InterPro, we chose SPKW, a method based upon curated associations, bioPIXIE/MEFIT, a Bayesian method and YeastFunc, a guilt-by-association/profiling method.
In addition to exploring more computational sources, we also extended the analysis to include manually assigned annotations other than ‘unknown’. For a given gene, annotations and predictions were sorted into pairs and categorized by the relationship between the paired GO terms. We present data on pairs of annotations we categorized as ‘mismatches’, indicating that the paired terms are not in the same lineage (path to the root node) of the GO ontology, or ‘shallow’, indicating that while the literature-based annotation and computationally predicted annotations are in the same lineage, the literature-based annotation provides less detailed information. We show here that both of these categories of annotation pairs allow us to flag genes whose manually curated annotations need to be reviewed and updated. In particular, we hope to find an efficient way by which these computational predictions can help us identify our highest-priority set of genes for review: those that are under-annotated and missing annotations from their manually curated set (i.e. cases where experiments supporting functional annotations exist in the literature but have not yet been captured by SGD). We also discuss factors that contribute to the effectiveness of this method, and ways in which it may be more efficiently applied.
Methods
Sources of data
All annotations were derived from the SGD gene association file dated 11 October 2009 (gene_association.sgd Revision 1.1460, available at http://cvsweb.geneontology.org/cgi-bin/cvsweb.cgi/go/gene-associations/gene_association.sgd.gz). We chose this date because this set of SGD GO annotations included recently updated computational predictions generated by the four different methods selected for this study. Literature-based annotations were considered to be all annotations not bearing the IEA and RCA evidence codes. Computationally predicted annotations used in this study included all annotations from the sources ‘YeastFunc’ and ‘bioPIXIE_MEFIT’ (SGD gene association file column 15) and annotations from source ‘UniProtKB’ bearing the evidence code IEA and with ‘Interpro’ or ‘SP_KW’ in column 8 of the gene association file. Annotations with the NOT qualifier, indicating evidence for the negative annotation, were excluded from this analysis.
A contemporaneous version of the GO file, version 5.1097 dated 13 October 2009, (http://cvsweb.geneontology.org/cgi-bin/cvsweb.cgi/go/ontology/gene_ontology_edit.obo) was used to evaluate the relationship between GO terms used in the computationally predicted and the literature-based annotations. Using a version of the ontology that is current with respect to the annotations ensures that the differences between the literature-based and computationally predicted annotations will not be based on changes to the ontology, such as merged or obsoleted GO terms.
Process for flagging annotations for review
If a gene product had multiple manual annotations in the same direct lineage of the ontology (i.e. in the same path leading up to the root of the ontology), the manual annotations were filtered in order to keep only the most specific annotations. In cases where both a literature-based and computationally predicted annotation existed for the given gene and GO aspect (Molecular Function, Biological Process, Cellular Component), the filtered granular manual annotations were compared against all of the computational predictions for that gene to create annotation pairs. Each annotation pair was evaluated in order to classify the relationship in the ontology between the two terms. Genes with annotations with the following relationships were flagged for further review: (i) the GO term used for the literature-based annotation is in the same lineage of the ontology but the literature-based annotation is closer to the root than the computational prediction and (ii) the GO term used for the literature-based annotation is in a different GO lineage from the computationally predicted annotation.
Process for selecting genes to review
To generate a representative set of 336 genes with literature-based annotations flagged by CvManGO to review, we began with a random set of genes from each class (see below for descriptions of the classes) and supplemented with additional genes to obtain similar coverage for each class, computational source, and GO aspect. This representative set of genes minimizes the numbers of genes needing review while providing equivalent representation of different attributes we consider and examine in this study. A control set of 70 genes to review was randomly selected from those genes that had computational annotations but had no literature-based annotations flagged by CvManGO. This sample size would provide statistical power to detect a difference of 20% with 90% confidence (α = 0.05).
Gene scoring methodology
For each of the flagged genes, we reviewed the body of literature published before January 2011 and assessed whether the set of annotations for that gene was in need of updating. Any gene needing no change to its current annotations was scored as ‘no change’, while those needing additional information or corrections to the existing set of annotations were scored ‘updatable’ and the type of update made based on each flagged annotation was noted (Supplementary Tables S1 and Supplementary Data). For scoring computational predictions, each prediction was examined in light of the published literature for the gene product and current SGD standard annotation practice. If we were able to find evidence in the literature supporting the computational prediction, such that we were able to add a manual annotation using either the same term as the prediction or a term in the same branch of the ontology, then the computational annotation was scored as ‘helpful’. If no evidence supporting the computational prediction was found in the literature or the predicted term did not comply with SGD's annotation standards, then the prediction was scored as ‘not helpful’. Review of the annotations, genes, and their associated literature presented in this study required ∼1000 person-hours and was conducted over 7 months.
Results and Discussion
Additional sources of computationally predicted annotations
Previously, we performed a feasibility study in which we presented a method that paired a manual literature-based annotation with a computationally predicted annotation and looked for correspondence between the two (20). For ease of reference we will herein call this method CvManGO (Computational versus Manual GO annotations). In the feasibility study we looked at instances where the CvManGO method found disparities for literature-based annotations designated as ‘unknown’. ‘Unknown’ annotations are created by manually assigning the root term of a GO aspect, which are the broadest terms that exist: ‘molecular_function’ (GO:0003674), ‘biological_process’ (GO:0008150) and ‘cellular_component’ (GO:0005575). They indicate that at the time of curatorial review no evidence is present in the literature that would allow a more specific annotation to be made for the gene product. When the CvManGO method found a computational prediction paired with a manually assigned ‘unknown’ annotation, we considered that this might indicate that there is evidence in the literature to support a non-‘unknown’ annotation, and we therefore reviewed the body of literature for those genes to see if there were annotations missing from our manual set. We previously performed this CvManGO analysis with literature-based ‘unknown’ annotations in the October 2009 SGD gene association file compared to computational predictions made by the GOA group at UniProtKB based on InterPro sequence signatures (12,13,20). In this study, we extended this analysis to include additional sources of computational predictions, in order to determine whether this would increase our coverage across the genome and help enrich for those genes which could be updated from an ‘unknown’ to a more informative annotation.
For our additional sources of computationally assigned annotations, we sought to use annotations based on methods that differed from the InterPro sequence-signature based technique. We chose to use annotation outputs based on SPKW, bioPIXIE/MEFIT and YeastFunc. SPKW, also provided by the GOA project, is an automated method based on curated associations (9–11), while bioPIXIE/MEFIT from the Troyanskaya group at Princeton University is a Bayesian analysis that integrates biological data sets (14–16), and the YeastFunc algorithm from the Roth group at the University of Toronto is a guilt-by-associative/profiling method that also integrates multiple types of biological data (17).
Each of these sources individually provides computational predictions for only a fraction of the ‘unknown’ annotations (Table 1). SPKW provided the best coverage, having computational predictions corresponding to 15.4% (637/4129) of all ‘unknown’ annotations, followed by InterPro with 14.7% (608/4129), YeastFunc with 1.9% (79/4129) and bioPIXIE/MEFIT with 1.3% (52/4129). While no single computational source provided predictions for more than ∼15% of the total number of unknown annotations, the combination of all four sources provided a computational prediction for 24.4% of all ‘unknown’ annotations. So, even though there does exist some overlap between the sources, meaning that a given ‘unknown’ annotation may have a corresponding prediction from more than one source, inclusion of multiple sources does allow more annotations to be analyzed when applying CvManGO. Although the number of ‘unknown’ annotations with corresponding predictions by either bioPIXIE/MEFIT or YeastFunc was small, these methods do provide predictions for genes not covered by the InterPro or SwissProt methods. The small number of ‘unknowns’ paired with bioPIXIE/MEFIT predictions probably results from the fact that this method only provides computationally predicted annotations for the Biological Process aspect of GO.
Number of biocurator-assigned ‘unknown’ annotations (to the root terms) that have a corresponding computational prediction, by source. Data are from the SGD gene association file dated October 11, 2009
| Number of annotations | |
|---|---|
| Total ‘unknowns’ | 4129 |
| ‘Unknowns’ with InterPro predictions | 608 |
| ‘Unknowns’ with SwissPro Keyword predictions | 637 |
| ‘Unknowns’ with bioPIXIE/MEFIT predictions | 54 |
| ‘Unknowns’ with YeastFunc predictions | 79 |
| ‘Unknowns’ with a prediction from any source | 1011 |
| Number of annotations | |
|---|---|
| Total ‘unknowns’ | 4129 |
| ‘Unknowns’ with InterPro predictions | 608 |
| ‘Unknowns’ with SwissPro Keyword predictions | 637 |
| ‘Unknowns’ with bioPIXIE/MEFIT predictions | 54 |
| ‘Unknowns’ with YeastFunc predictions | 79 |
| ‘Unknowns’ with a prediction from any source | 1011 |
Number of biocurator-assigned ‘unknown’ annotations (to the root terms) that have a corresponding computational prediction, by source. Data are from the SGD gene association file dated October 11, 2009
| Number of annotations | |
|---|---|
| Total ‘unknowns’ | 4129 |
| ‘Unknowns’ with InterPro predictions | 608 |
| ‘Unknowns’ with SwissPro Keyword predictions | 637 |
| ‘Unknowns’ with bioPIXIE/MEFIT predictions | 54 |
| ‘Unknowns’ with YeastFunc predictions | 79 |
| ‘Unknowns’ with a prediction from any source | 1011 |
| Number of annotations | |
|---|---|
| Total ‘unknowns’ | 4129 |
| ‘Unknowns’ with InterPro predictions | 608 |
| ‘Unknowns’ with SwissPro Keyword predictions | 637 |
| ‘Unknowns’ with bioPIXIE/MEFIT predictions | 54 |
| ‘Unknowns’ with YeastFunc predictions | 79 |
| ‘Unknowns’ with a prediction from any source | 1011 |
In addition to examining whether inclusion of additional sources for computational predictions would provide improved coverage of the annotations, we sought to explore whether flagging of an ‘unknown’ annotation by more than one computational source would indicate an increased likelihood that an experimentally based manual annotation could be made from the literature to replace the ‘unknown’. We reviewed 50 ‘unknown’ annotations that were flagged by only one of any of our four sources, and 50 ‘unknown’ annotations that were flagged by two or more of the sources in our CvManGO comparison. For ‘unknown’ annotations that were flagged by predictions from a single source, 24% of the cases reviewed (12/50 annotations) could be updated, while 38% of the annotations (19/50 annotations) that were flagged by more than one source of computational predictions could be updated (Figure 1). However, this difference is not significant, as determined by the χ2 test (P-value = 0.13). Therefore, performing the CvManGO analysis using computational predictions from multiple sources does not seem to significantly enrich for genes that can be updated, as compared to using computational predictions from a single source. However, since including all sources of predictions allowed us to perform the comparison using a larger set of both ‘unknown’ and non-‘unknown’ literature-based annotations (data not shown), we included all sources in our further analyses.
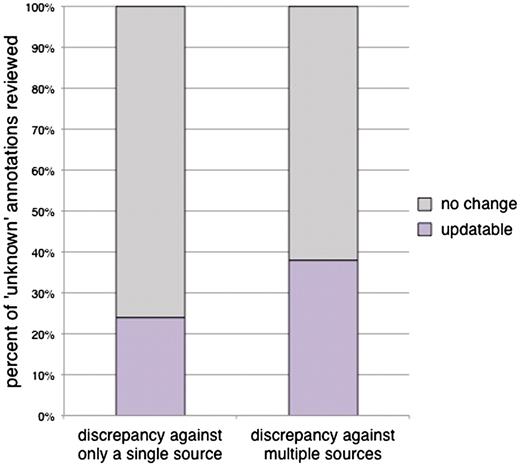
The percentage of annotations that could be updated from ‘unknown’ with predictions from either a single or from multiple computational methods. Fifty ‘unknown’ annotations were selected from each of two categories: a computational prediction existed for the ‘unknown’ annotation from the output of only one computational source, or computational predictions existed from two or more sources. Each of the predictions was evaluated against the existing body of literature for the associated gene to determine whether a more meaningful manual annotation could be assigned. In cases were the literature supported a novel function, the annotation was scored ‘updatable’. Annotations that remained ‘unknown’ after review by a biocurator were scored ‘no change’. Twenty four percent of the ‘unknowns’ flagged by a single source (12/50) were updatable to a literature-supported functional annotation upon review. For ‘unknown’ annotations that had predictions from two or more computational sources, 38% (19/50 annotations) were updatable. The slight increase in the number of updatable genes with additional computational sources is not statistically significant (χ2P-value = 0.13).
The remaining annotations: non-‘unknowns’
In the October 2009 set of literature-based annotations, ‘unknowns’ comprised only 13% of the total number of annotations (4129 of the total of 31 977). So, while replacing an ‘unknown’ annotation with any functional information is of great benefit to our scientific community, these types of annotations and situations represent only a small fraction of the total annotations at SGD. In light of this, we wanted to apply CvManGO to the remaining 87% of our literature-based annotations to see if the method could help identify further curation needs for this larger set of annotations.
When considering the effectiveness of the CvManGO method for identifying non-‘unknown’ annotations that might be updatable, we made the decision to shift from looking at the fate of individual annotations, to evaluating the entire annotation set for a gene. ‘Unknown’ annotations are often present as the sole annotation for a GO aspect for a gene, but non-‘unknown’ annotations are typically part of a set containing multiple annotations for a gene. Since SGD's GO annotation practice is to present a complete, summarized view of the functional role of a gene product, the whole annotation set for a gene must be reviewed in order to determine whether the CvManGO method resulted in an improvement to this summarized view. In reviewing the annotation set for a gene, the entire body of literature relevant to that gene must be considered.
Genes potentially needing review were flagged based on the results of the CvManGO comparison applied to the non-‘unknown’ annotations. Each literature-based annotation, when compared to a computational prediction, was sorted into one of four classes based on the relationship between the GO terms. After each literature-based annotation was compared to all computational predictions for that gene, the literature-based annotation was assigned to one and only one of the following four classes based on the following order of priority: Annotation pairs were classified into the first appropriate category, as the categories were considered in the order shown above. For example, a literature-based annotation that exactly matched one of the computational predictions and was a parent of another computational annotation would be classified only as ‘shallow’ and not as both ‘exact match’ and ‘shallow’. The number of genes flagged in each of these classes is presented in Table 2; although the annotations themselves are in disjoint sets, a gene can be present in more than one category. Out of a total of 6375 total features in the October 2009 SGD gene association file, 3032 and 4203 genes fell into the ‘exact match’ and ‘deep’ classes, respectively. If a gene had annotations only in these two classes, it was not flagged as needing review since we deemed that no additional information about a gene was indicated by any of its computational predictions. The ‘shallow’ class contained 646 genes, and the ‘mismatch’ class contained 3733 genes. Genes in both of these classes were reviewed, since the computational predictions provided additional or different information from the existing manual annotations, suggesting that updates to the manual annotations might be necessary.
‘Shallow’: For these annotation pairs, the literature-based annotation is in the same lineage of the ontology but closer to the root than the computational prediction. In this case the computational prediction provides more specific information than the literature-based annotation with which it is paired, potentially indicating that a more granular manual GO annotation can be made. An example of this would be if the literature-based annotation were to ‘mitochondrion’ (GO:0005739) and the computational prediction to ‘mitochondrial membrane’ (GO:0031966). Genes with annotation pairs in this class were flagged as needing review.
‘Exact match’: In these pairs the literature-based annotation exactly matches a computational prediction. Because there was no discrepancy between the two annotations, annotation pairs in this class were not considered as flags for review.
‘Deep’: Here the literature-based annotation is in the same lineage, or path to the root node, of the ontology but farther from the root than the computational prediction. Since the existing literature-based annotation is more specific than the paired computational annotation, there is no additional information provided by the discrepant computational prediction. An example of this would be a literature-based annotation for a gene to ‘protein serine/threonine kinase activity’ (GO:0004674) paired with a computationally predicted annotation to ‘kinase activity’ (GO:0016301). Annotation pairs in this class were not considered as flags for review.
‘Mismatch’: In this class the literature-based annotation is in a different GO lineage from the computationally predicted annotation. One example of this would be if the literature-based annotation were to the term ‘pseudohyphal growth’ (GO:0007124) and the computational prediction were to ‘proteasomal protein catabolic process’ (GO:0010498). The discrepancy could indicate several possibilities, such as a novel potential annotation missing from the literature-based set; an incorrect annotation in the literature-based set; or an incorrect computational prediction. Genes with annotation pairs in this class were flagged as needing review.
Pairs of annotations, comprised of one biocurator-assigned literature-based annotation and one computationally predicted annotation, were evaluated for the type of relationship they had to each other in the Gene Ontology directed acyclic graph structure
| Total number of genes | Number of genes reviewed | |
|---|---|---|
| All genes | 6375 | 336 |
| ‘Unknown’ with a computational prediction | 815 | 77 |
| Flagged by ‘exact match’ | 3032 | N/A |
| Flagged by ‘deep’ discrepancy | 4203 | N/A |
| Flagged by ‘shallow’ discrepancy | 646 | 264 |
| Flagged by ‘mismatch’ discrepancy | 3733 | 265 |
| Total number of genes | Number of genes reviewed | |
|---|---|---|
| All genes | 6375 | 336 |
| ‘Unknown’ with a computational prediction | 815 | 77 |
| Flagged by ‘exact match’ | 3032 | N/A |
| Flagged by ‘deep’ discrepancy | 4203 | N/A |
| Flagged by ‘shallow’ discrepancy | 646 | 264 |
| Flagged by ‘mismatch’ discrepancy | 3733 | 265 |
‘Unknown’ indicates that a gene has been manually annotated to the root node of the ontology. ‘Exact match’ refers to pairs where the manual annotation and computational prediction use the same term. ‘Deep’ and ‘shallow’ are instances where the GO term used by the literature-based annotation is in the same path to the root as the GO term used in computational prediction, but the literature-based annotation is either farther from or closer to the root, respectively, than the computational annotation. ‘Mismatch’ discrepancies are those where the two annotations have no relationship to each other in the GO hierarchy.
Pairs of annotations, comprised of one biocurator-assigned literature-based annotation and one computationally predicted annotation, were evaluated for the type of relationship they had to each other in the Gene Ontology directed acyclic graph structure
| Total number of genes | Number of genes reviewed | |
|---|---|---|
| All genes | 6375 | 336 |
| ‘Unknown’ with a computational prediction | 815 | 77 |
| Flagged by ‘exact match’ | 3032 | N/A |
| Flagged by ‘deep’ discrepancy | 4203 | N/A |
| Flagged by ‘shallow’ discrepancy | 646 | 264 |
| Flagged by ‘mismatch’ discrepancy | 3733 | 265 |
| Total number of genes | Number of genes reviewed | |
|---|---|---|
| All genes | 6375 | 336 |
| ‘Unknown’ with a computational prediction | 815 | 77 |
| Flagged by ‘exact match’ | 3032 | N/A |
| Flagged by ‘deep’ discrepancy | 4203 | N/A |
| Flagged by ‘shallow’ discrepancy | 646 | 264 |
| Flagged by ‘mismatch’ discrepancy | 3733 | 265 |
‘Unknown’ indicates that a gene has been manually annotated to the root node of the ontology. ‘Exact match’ refers to pairs where the manual annotation and computational prediction use the same term. ‘Deep’ and ‘shallow’ are instances where the GO term used by the literature-based annotation is in the same path to the root as the GO term used in computational prediction, but the literature-based annotation is either farther from or closer to the root, respectively, than the computational annotation. ‘Mismatch’ discrepancies are those where the two annotations have no relationship to each other in the GO hierarchy.
The CvManGO method considers the relationship between the GO terms used in the literature-based and computationally predicted annotations. While most previous studies only consider exact matches in their accuracy scores and analysis (17,22–25), we also consider annotations that are in close proximity to each other along the same path leading up to the root as concordant with each other and only consider whether the prediction provides additional information not already inherent in the GO term used by the manual annotation. Since the ‘deep’ class indicates that the term used by the computational prediction represents the same biology as the existing literature-based annotation but at a more general level, we did not consider this class of discrepancies as needing further review. By excluding the ‘deep’ flagged genes from our analysis we were able to increase the efficiency of our method by reducing the number of genes that require review.
We included additional genes from the ‘shallow’ and ‘mismatch’ discrepancy sets along with those evaluated in the ‘unknown’ analysis, giving us a total of 336 genes to review. When reviewing the annotation sets of these genes, we reviewed the entire body of literature for each gene to determine whether changes needed to be made to any of the annotations for that gene. If the review indicated that any change needed to be made to an annotation set, that gene was scored as ‘updatable’ and the type of update was recorded for the flagged annotation. If all the annotations were unchanged because they still were the best representation of the biological summary for that gene, the flagged gene was scored as ‘no change’.
Of the genes whose GO annotations were flagged for review, 77.4% (260/336) were found to require updates (Figure 2, Supplementary Table S1). In order to determine if annotations being flagged for review were significantly helpful in identifying genes whose GO annotations needed to be updated, we compared these results to a comparable set of genes whose GO annotations were not flagged for review. This control set was randomly chosen from the set of genes whose GO annotations were exclusively in the ‘exact match’ or ‘deep’ classes. We found that 48.6% of our control set (34 out of 70 genes reviewed; Figure 2) could be updated after review of the literature, suggesting that GO annotations flagged by CvManGO are significantly helpful (P < 0.001, χ2 test) in identifying genes whose set of GO annotations need to be reviewed.
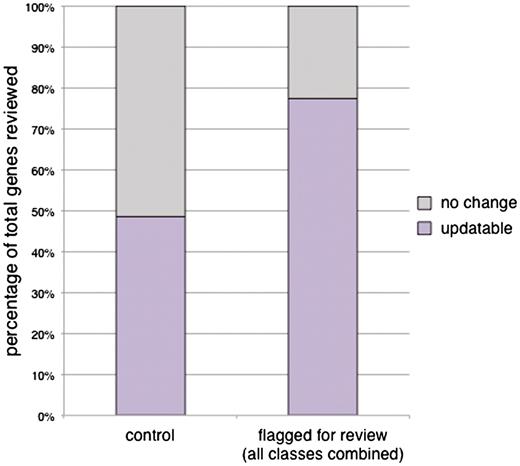
Efficacy of CvManGO as measured by percentage of gene annotation sets updated after literature review. A representative subset of genes was given full literature review and the set of annotations for those genes examined for their accuracy. Any change to the annotation set as determined by a biocurator resulted in an ‘updatable’ score for a gene. Genes with no changes to their annotation sets after review by a biocurator were scored ‘no change’. We observed that 77.4% (260/336) of the reviewed genes were updatable. This is a significant improvement over the 48.6% (34/70) updatability rate of the control set (χ2P < 0.001). The control set of genes was randomly selected from those genes that had computational annotations but had no literature-based annotations flagged for review.
Surveying the attributes of flagged genes
We explored several attributes of the genes whose annotations were updated in order to identify factors that will help pinpoint additional genes whose annotations will need review. We assessed the contribution of each discrepancy class towards the improvement of a gene's annotation set by separating the annotations flagged by CvManGO into their respective classes. We compared the flagged annotation sets from the ‘mismatch’ and ‘shallow’ classes to the results from the ‘unknown’ data discussed previously, which have been summarized to reflect updates at the gene level (Figure 3A). For both the ‘mismatch’ and ‘shallow’ classes, we saw an increase in the percentage of genes whose literature-based annotations were updated, compared to what was seen for the ‘unknown’ class (40.3%, 31/77 genes). Genes flagged by ‘mismatch’ annotation pair discrepancies could be updated 59.2% (157/265) of the time, while 78.8% (208/264) of genes flagged by ‘shallow’ annotation pair discrepancies could be updated.
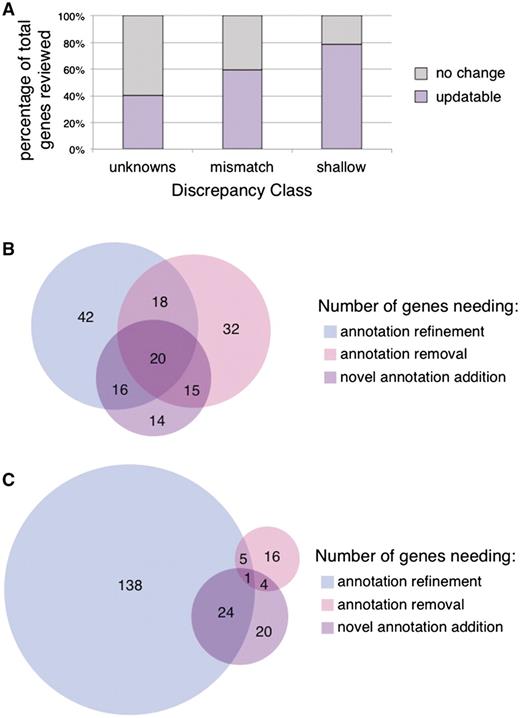
Gene update rates and type of updates by discrepancy class. (A) The updatability of reviewed genes as suggested by the flagged annotations from a given discrepancy class was examined. ‘Unknown’ genes had an updatable rate of 40.3% (31/77), ‘mismatch’ genes a rate of 59.2% (157/265), ‘shallow’ genes a rate of 78.8% (208/264). Genes that were scored as ‘updatable’ in Figure 3A were further evaluated for the type of update that a biocurator would determine was necessary for the annotation set. (B) Mismatch class genes. (C) Shallow class genes.
To further assess the improvements that were made to the annotation set, the annotations for the genes that were updated were classified for the type of update that was made. A gene was deemed ‘Refine’ if one of its existing annotations was technically correct, but evidence was found in the literature to annotate the gene product to a more specific term. ‘Remove’ indicated those genes for which an existing annotation should be removed, either because it was refuted by the literature or because it did not adhere to current annotation standards. Genes that were under-annotated, for which review of the literature revealed evidence to support novel GO annotations, were marked as ‘Add’.
We found that the distribution of these three types of improvements varied between the discrepancy classes evaluated (‘unknowns’ were not included). As might be expected, the distribution of types of updates is different between the two discrepancy classes, with most ‘shallow’ discrepancies leading to annotation refinement while a larger proportion of ‘mismatch’ discrepancies indicating incorrect or missing annotations. Figure 3B shows that for the mismatch class of 157 genes whose manual GO annotations could be improved, roughly equivalent numbers of genes required each of the three types of improvement, with the ‘Add’ category being the smallest (65 genes). For the ‘shallow’ class, the number of genes in the ‘Add’ category was even smaller (49 genes), with most genes in this class (159) only needing annotation refinement and/or removal (Figure 3C).
We also explored whether the source of the computational prediction, the GO aspect of the flagged annotations, or the number of publications for a gene would have any correlation to the rate and type of updates to its annotation set. None of these attributes on their own showed remarkable differences in identifying genes that needed to be updated (data not shown), suggesting we will need to look for additional factors or evaluate these factors in combination in order to enrich for genes whose annotations need review.
These data also show that even though CvManGO is very successful at identifying annotation sets that need updating, most of these improvements are still instances where an existing annotation is correct but could be annotated one or two levels deeper in the ontology. Examining the annotations that should be removed for a gene, the majority of these are not cases where the annotation is incorrect in terms of the biology of that gene product; rather, they are cases where the annotation is not compliant with the GO annotation standards of SGD. In particular, the majority of the annotations to be removed were instances where a downstream phenotype was captured using a GO annotation instead of through the SGD phenotype curation system (26). While the annotation is supported by evidence in the literature, it is SGD's policy not to capture a downstream phenotype when more specific information about a gene product's role in the cell is known. Since most of the ‘Refine’ and ‘Remove’ types of updates were found to be instances where the existing information is not likely to give the user an incorrect view of the biological picture of the given gene product's role in the cell, we would categorize these updates as lower priority than those genes scored as ‘Add’. As with the ‘unknown’ class, for which any improvement is through replacement of the ‘unknown’ annotation with a more informative annotation, we place a high priority on curation of those gene sets where addition of a missing annotation is required. In light of this, we will most likely focus future efforts on updating these types of genes and annotations from the ‘mismatch’ class.
Utility of computational annotations for a gene
While the simple existence of a computational prediction is useful in the CvManGO method to help flag genes, it is also of interest to know if the actual term suggested by the computational prediction could be applied when improving the literature-based annotation set. We reviewed all of the computational predictions from the annotation pairs of the already selected subset of flagged literature-based annotations. We scored each computationally predicted annotation as either ‘helpful’, meaning that the GO term used by the prediction or a GO term in the same branch of the ontology were directly applicable when making a literature-based update for that gene, or as ‘not helpful’ when we could not apply the GO term in updating the annotation set for the flagged gene. The ‘not helpful’ category included instances in which use of the term would not be consistent with SGD annotation practices and standards. For example, annotations to ‘intracellular’ (GO:0005622) or ‘binding’ (GO:0005488) were scored as ‘not helpful’: since the information conveyed by these terms is too general to be meaningful, these terms are not used for annotation at SGD. The ‘not helpful’ category also included instances where the body of literature for that gene refuted an association with the predicted term, or where no literature existed associating the particular gene product with the biological process represented by the prediction.
Examining the percentage of helpful predictions by class, we found that the predictions were useful for 32.5% (25/77) of the genes in the unknown class, 57.2% (147/257) of the mismatch class, and 75.0% (198/264) of the shallow class (Figure 4). For the shallow class, the computational predictions directly led to manual annotations using that term for 50% of genes (132/264) (Supplementary Table S3). These results are not an indication of the accuracy of the computationally predicted annotations as previously studied by Camon et al. in 2005 (27), but rather an evaluation of whether these predictions can be used as a curation aid in adding value to a manual set of annotations for a gene. It is interesting to note that for each of the classes the percentage of genes with helpful computational predictions closely matches that of the percentage of genes that were updatable in their literature-based set (Figure 3A).
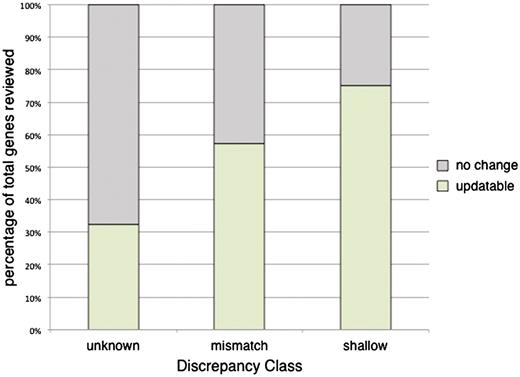
Evaluation of computational annotations for their utility in assigning literature-based annotations. The set of computationally predicted annotations was reviewed for each of the previously selected flagged genes. Each of the annotation sets was scored as either ‘helpful’ or ‘not helpful’. ‘Helpful’ annotation sets were those that had at least one computational prediction that was directly applicable in making an update to the literature-based manually curated set of annotations. Conversely, ‘not helpful’ annotation sets were those where none of the computational predictions aided in updating the manual annotations for a gene. The percentage of helpful annotation sets within each of the discrepancy classes are as follows: unknown 32.5% (25/77), mismatch 57.2% (147/257) and shallow 75.0% (198/264).
Conclusions
We have successfully shown that comparing computationally predicted versus manually curated literature-based GO annotations (CvManGO) for a gene is a measurably viable method for identifying genes that are in need of updating. We find that we can apply this method to any type of literature-based annotation and create pairings against all computational predictions for a gene. By sorting paired manual-computational annotations into classes based on the relationship between the two annotations, we can determine which annotations, and by association which genes, show lack of concordance, indicating that the set of annotations for a gene should be reviewed. Review of these genes showed that one could refine the existing annotations for the majority of them, remove inappropriate annotations, and even find novel/missing annotations for a proportion of them. Of the 336 genes that were reviewed, ∼77% required an update to the annotation set, ranging from a refinement of an annotation, removal of an annotation or an addition of a new annotation (Figures 2 and 3). Extrapolating these results to the entire set of genes in SGD, we estimate that the CvManGO method will help us update the GO annotations for a little over half of Saccharomyces cerevisiae gene products (Figure 5).
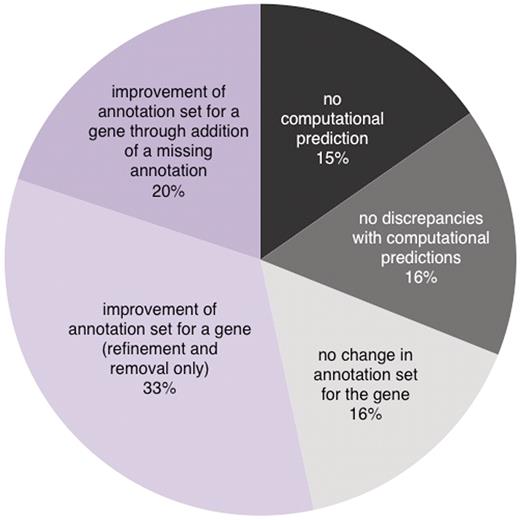
A projection of the fate of all genes in SGD when their annotations are analyzed by CvManGO. Based upon the rate and type of updates seen for the subset of genes reviewed in this study, we extrapolated our results to all of the genes in the SGD October 2009 gene associations file. A fraction (15%) of the genes would not have any computationally predicted annotations from any of the four sources we evaluated while roughly the same proportion (16%) would not need to be reviewed because they would have annotation pairs only in the ‘exact match’ and ‘deep’ classes. Of the 69% genes that CvManGO would flag for review, most of them would be expected to result in some sort of improvement in their annotation sets. Of the genes that are improved, more than half would only require annotation refinement or removal (33% of the total genes in SGD), while a smaller fraction (20% of the total) would require the addition of novel/missing annotations.
This method could be applied by other annotation groups and model organism databases looking to prioritize their genes for literature-based curation. The GOA project uses an automated annotation pipeline to provide predictions for over 120 000 species using multiple computational methods (http://www.ebi.ac.uk/GOA/faq.html). It is likely that most groups will be able to find computational predictions from at least one source for their organism of interest. While we do not find statistically significant evidence to support the idea that combining multiple sources of computational annotations can improve the ability of CvManGO to identify ‘unknown’ annotations that need to be updated, combining multiple sources provides greater coverage for the literature-based annotations [Table 2 and (20)]. While SGD prioritizes addition of novel or missing annotations, other groups may find that annotation refinement is helpful, depending on their annotation philosophy. SGD uses GO annotations to represent the biological summary of a gene rather than to present a comprehensive survey of the literature for a given gene. However, for groups that do generate GO annotations from all relevant literature, CvManGO could be an efficient way to prioritize curation needs and keep current with the literature, especially for genes flagged by the ‘shallow’ class.
Although a significant number of genes could potentially be updated at SGD, applying CvManGO alone may not be the most efficient method for SGD to use in prioritizing genes for curation. More than half of those genes updated were improved by only a refinement of an existing annotation or by removal of an experimentally supported and biologically correct annotation that does not comply with SGD standards. The 336 genes we reviewed for this study are associated with over 16 600 publications. Updating each gene required an average of 2.4 hours to review ∼50 publications. We feel that the time spent curating is disproportionately large for updates that only refine an existing annotation a level or two further in granularity in the GO structure. While these types of updates are useful in improving a gene's annotation set, we prefer to prioritize adding novel annotations as opposed to refining existing ones.
Preliminary data indicated that there was no simple or straightforward way to discern whether the annotations for a gene required updating. Here we explored attributes such as computational source, discrepancy class, GO aspect, and the amount of literature for a gene. While none of these factors alone proved to be a bellwether indicator for genes missing annotations, it is possible that a combination of two or more of these features plus additional ones could be more effective. Additional attributes for further consideration include number of discrepant annotations per gene, further analysis of the types of computational predictions that proved helpful, and inclusion of other computational sources for predicting GO annotations such as the GO Consortium's PAINT project, a method that transfers annotations between organisms based on homology (28). In addition to exploring the contribution of these attributes on the gene level, we can apply them to analyze the data at the annotation level. Exploring the data on a per annotation basis also allows us to consider factors such as the distance in the ontology between a discrepant annotation pair (the node distance in the GO hierarchy) and the date an annotation was made. Investigating these and other attributes in combination with each other may help to identify specific annotations that need to be updated, and to increase the specificity of CvManGO for finding genes that can be updated with novel functions.
We not only intend to explore additional attributes but we plan to pursue more sophisticated means to identify characteristics of flagged annotations that need to be updated. To further increase the efficiency of literature-based curation, the results of CvManGO could be combined with natural language processing or other text-mining strategies (29,30). This would identify literature containing uncurated or novel annotations and reduce the amount of literature that needs to be reviewed.
Rather than considering manual annotations and computational predictions as separate sets with little relevance to each other, the challenge for biological curation is to find efficient ways to compare them in order to ensure that the set of annotations for each gene is as high-quality, complete, and current as possible. We attempted to leverage the computational predictions as a curation aid to help us improve our set of manual annotations. The importance of high-quality GO annotations, particularly for a model eukaryote such as yeast, in combination with large quantities of published data and finite resources, make it imperative to develop efficient ways of identifying and prioritizing annotations for review and updating. By using both literature-based and computationally predicted annotations and leveraging the strengths of each against the other, we hope to improve the efficiency of our curation efforts in order to provide scientists with the most up-to-date, complete, and accurate biological information.
Funding
The National Human Genome Research Institute, National Institutes of Health: Saccharomyces Genome Database project [P41 HG001315] and the Gene Ontology Consortium [P41 HG002273]. Funding for open access charge: National Human Genome Research Institute at the National Institutes of Health [P41 HG001315].
Conflict of interest. None declared.
Acknowledgements
We thank Jodi Hirschman and Samuel Ieong for critically reading the article and all members of the SGD project for helpful discussions and support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Human Genome Research Institute or the National Institutes of Health.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}