





Abstract
Veterinary diagnostic laboratories derive thousands of nucleotide sequences from clinical samples of swine pathogens such as porcine reproductive and respiratory syndrome virus (PRRSV), Senecavirus A and swine enteric coronaviruses. In addition, next generation sequencing has resulted in the rapid production of full-length genomes. Presently, sequence data are released to diagnostic clients but are not publicly available as data may be associated with sensitive information. However, these data can be used for field-relevant vaccines; determining where and when pathogens are spreading; have relevance to research in molecular and comparative virology; and are a component in pandemic preparedness efforts. We have developed a centralized sequence database that integrates private clinical data using PRRSV data as an exemplar, alongside publicly available genomic information. We implemented the Tripal toolkit, a collection of Drupal modules that are used to manage, visualize and disseminate biological data stored within the Chado database schema. New sequences sourced from diagnostic laboratories contain: genomic information; date of collection; collection location; and a unique identifier. Users can download annotated genomic sequences using a customized search interface that incorporates data mined from published literature; search for similar sequences using BLAST-based tools; and explore annotated reference genomes. Additionally, custom annotation pipelines have determined species, the location of open reading frames and nonstructural proteins and the occurrence of putative frame shifts. Eighteen swine pathogens have been curated. The database provides researchers access to sequences discovered by veterinary diagnosticians, allowing for epidemiological and comparative virology studies. The result will be a better understanding on the emergence of novel swine viruses and how these novel strains are disseminated in the USA and abroad.
Database URLhttps://swinepathogendb.org
Introduction
The diversity of endemic viruses that circulate in swine continues to increase (1). Many of these viruses cause disease that adversely affect health through morbidity and mortality and impact production through increased costs associated with vaccination, treatment and increased biosecurity programs (1, 2). In addition, novel viruses periodically emerge in swine populations (e.g. Senecavirus A (SVA), porcine epidemic diarrhea virus (PEDV) (3, 4)) that can lead to the establishment and persistence of antigenically distinct viruses that lack appropriate vaccines due to limited information from which to derive rational formulations. Given that the genetic makeup of viruses continually changes, monitoring the patterns of evolution of viruses in swine is needed to identify possible emerging threats and to help control endemic viruses.
Veterinary diagnostic laboratories in the USA sequence thousands of clinical samples or isolates of porcine reproductive and respiratory syndrome virus (PRRSV), SVA, PEDV and other coronaviruses annually (3, 5–9). The advent and broad use of next generation sequencing platforms has also resulted in the production of full-length genomes. Unfortunately, these pathogen sequences are rarely publicly available, as the samples may be associated with sensitive information that veterinarians and pork producers may not want to disclose, or the process of annotating, validating and sharing the genomic sequence information is burdensome. Currently, public genomic information is housed in NCBI GenBank (10), a comprehensive sequence database. Submitted data may only receive cursory curation and metadata may not be accurate, and although identifying and downloading annotated sequence data are possible, there is a steep learning curve for new users. Ideally, each nucleotide sequence would be annotated with start and stop sites for translation, the species of virus, potential open reading frames, along with the inclusion of data useful for genomic epidemiology studies, e.g. age of pig, collection location and collection date. Thus, swine disease researchers, outside of diagnostic laboratories, have limited means to identify novel isolates, where pathogens emerged, re-emergence of an identical but previously detected pathogen and other knowledge that may be applied to improve animal health.
To address this problem for PRRSV, a producer funded initiative was implemented and maintained from 2005 to 2008 (porcine reproductive and respiratory syndrome virus database—prrsvdb). The prrsvdb archived over 13 000 PRRSV open reading frame 5 (ORF5) sequences from both Type 1 (European) and Type 2 (North American) isolates from predominantly regional veterinary diagnostic laboratories and deposited over 8200 unique sequence submissions to GenBank. The sequences generated and shared in this three-year period represent 25% of all available PRRSV data and included many critical PRRSV index strains derived from early 1990 field isolates. These data have been heavily used by molecular epidemiologists and other researchers worldwide and are still being accessed (11–15).
We developed the United States Swine Pathogen Database (US-SPD) to provide a mechanism for incorporating veterinary laboratory diagnostic data with sequence data for swine pathogens. The database was designed for the exploration of carefully curated genetic sequences to allow researchers and stakeholders the ability to determine how genetic diversity of swine pathogens is changing spatially and temporally. Sequences in the database are derived from public data in NCBI GenBank and clinical samples submitted to the Iowa State University Veterinary Diagnostic Laboratory, the Kansas State University Veterinary Diagnostic Laboratory and the South Dakota Animal Disease Research & Diagnostic Laboratory at South Dakota State University. The core function of this database is to collect, store, view, annotate and query genomic data for swine pathogens, including PRRSV, SVA, PEDV, porcine deltacoronavirus (PDCoV), foot-and-mouth disease virus (FMDV), African swine fever virus (ASFV), classical swine fever virus (CSFV) and others (18 swine pathogens are currently included with a listing provided at https://swinepathogendb.org/sample_search). The apparent utility of genomic data in epidemiological analyses and control strategies (e.g. (16)) has facilitated a proliferation of genomic databases. Projects such as the Influenza Research Database (17) and ISU FLUture (18) implement curation and analytical tools for single pathogens. The comprehensive NCBI viral genome resource provides reference sequence annotation information for viral pathogens and associated search tools (19). Some databases ingest and visualize public data for swine pathogens such as the ASFVdb (20), the Disease BioPortal (https://bioportal.ucdavis.edu/) or the Swine Disease Reporting System (21). Our approach complements these by including customized curation pipelines for swine pathogens, additional curation by subject-area experts (22) and by providing diagnostic laboratory data alongside public data. We additionally introduce a pathogen agnostic analysis pipeline, the swine pathogen analysis resource, that accurately annotates sequence data with genomic features and exports files that may be ingested into the US-SPD or NCBI GenBank.
Materials and methods
Database construction and implementation
The US-SPD operates as a web-based, curated, relational database as part of infrastructure developed by the United States Department of Agriculture, Agricultural Research Service (USDA-ARS SCINet). The web interface of the US-SPD was developed using Tripal v3 (23–25) that builds upon the open source Drupal content management system and the Chado database schema (26). Tripal extension modules that incorporate NCBI BLAST sequence similarity search (27) and JBrowse genome browser (28, 29) were implemented, providing tools for genetic data analysis and visualization. We modified the Tripal BLAST UI module to classify and visualize PRRSV by restriction fragment length polymorphism (RFLP) patterns within ORF5 genes; this module can be modified for additional classification tasks for other pathogens. Additionally, we developed a custom Drupal module for the search interface that incorporates the ability to search genomic information derived from publications accessible via NCBI PubMed (30) or through metadata provided by sequence submitters in NCBI GenBank: these workflows and the search interface (e.g. search by gene name, date of collection, or sequence submitter) are in query pipelines in SQL. The database web server is hosted on an Amazon EC2 instance, currently a Linux server (Ubuntu 16.04 LTS), with Apache v2.4.18, PostgreSQL v9.5.23, PHP v7.0.33 and Drush v5.10. The US-SPD is updated monthly, and the curation pipeline is scheduled to run automatically when new data are acquired from diagnostic laboratories or public databases.
Data collection and processing
All US-SPD sequence data, provided by veterinary diagnostic laboratories or sourced from NCBI GenBank, are processed by a genome annotation pipeline (available at https://github.com/us-spd) capable of identifying any kind of continuous genomic feature that can be translated into an amino acid (AA) sequence (Figures 1, 2). In brief, the process can be broken down into the following two sections: pre-processing and query curation. Pre-processing consists of designing reference files necessary for annotating query sequences. For each feature, a python script builds a reference scaffold multiple sequence alignment (MSA) using MAFFT (31), companion profile hidden Markov model (HMM) and general feature format (GFF3) templates. The MSAs are then used to derive a JSON file containing regular expression patterns that represent spaced fragments of genomic features. When the requisite files have been generated, query sequences may be submitted for curation. Firstly, regular expressions from the JSON reference are used to determine position and reading frame of genomic features. If the regular expressions are unable to definitively annotate submitted sequences, profile HMM alignment is performed to supplement pattern matching to reliably find feature terminal ends and/or frameshift locations (Figure 2). The curation pipeline can also use BLAST to identify query species. Deployment of the annotation pipeline occurs in conjunction with parsing scripts that derive genomic information and metadata such as: sample source; collection date; collection location; NCBI PubMed ID; Authors; and strain name (Figure 2).
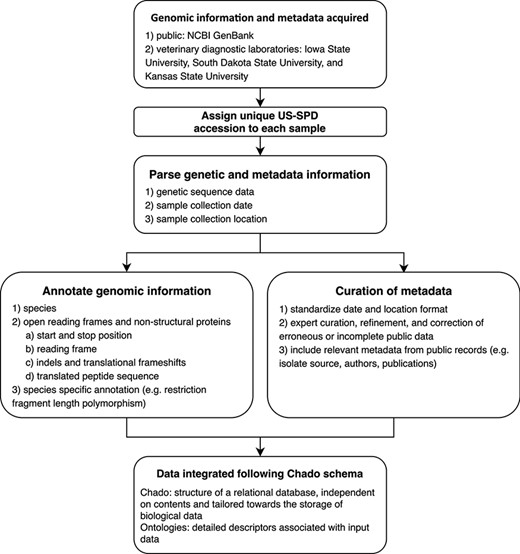
Conceptual model describing the automated pipeline implemented in the US Swine Pathogen Database that takes raw sequence data to fully anonymized and annotated virus sequence record in the relational database.
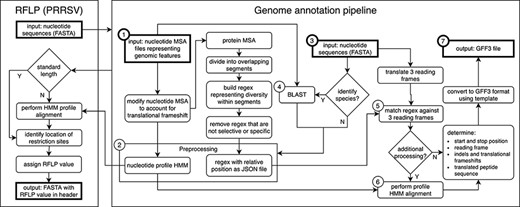
Genome annotation for the United States Swine Pathogen Database. Genome annotation begins with preprocessing, which requires nucleotide multiple sequence alignment files (MSA) representing genomic features as input (①). The products of preprocessing (②) are a nucleotide profile hidden Markov model (HMM) and a structured file containing regular expression patterns representative of diversity within sections of translated input MSAs. Following preprocessing, query nucleotide sequences in FASTA format (③) may be supplied to the annotation pipeline. If species identification is necessary (e.g. differentiating type 1 and 2 PRRSV), BLAST is performed (④) using preprocessing input files (①) as a reference. Once the query sequence species is known, relevant preprocessing files (②) are selected. Regular expressions are matched against three reading frames (⑤) of query sequence (③) to determine the location of genomic features. If more processing is necessary due to frame changes or uncertainty in the start or stop position, a profile HMM alignment is performed (⑥). These steps produce genome annotation and additional information with high confidence. The output produced by the pipeline is general feature format (GFF3) file, a standard nucleic acid or protein feature file format (⑦). The annotation pipeline is available at https://github.com/us-spd/.
At the core of the annotation pipeline are template nucleotide MSAs that are representative of the genetic data of each swine pathogen. The MSAs are used to generate AA regular expressions and nucleotide profile HMM templates and through this process are robust and prevent curation failure by capturing as much genetic variation as possible. A consequence is that the AA regular expressions within the pipeline may become non-specific in areas where homology is low. For example, in PRRSV it was not uncommon for 10+ unique residues to be observed at any given position, which produces a pattern that may not match selectively. To avoid erroneous matching, low occurrence residues may be removed from patterns until the expression is passably unique.
The outcome is highly curated genomic information that allows users to easily search and download data for additional user applicable analyses (e.g. fine-scale analysis of the spatial dissemination of viruses within the USA). For data provided by the veterinary diagnostic laboratories, the annotation tool produces a GFF3 formatted file that can be uploaded to NCBI GenBank for public dissemination of the data with appropriate recognition given to the individual laboratory.
Customized search interface for genomic epidemiology and comparative virology
Genomic epidemiology and comparative virology rely on four core data items: genomic information; the date of collection; the US state or country the collection was made; and a unique identifier. The US-SPD system standardizes data from three major veterinary diagnostic laboratories for swine and NCBI GenBank. Newly ingested data from the diagnostic laboratories curated by the US-SPD has this information, and sequences that do not meet these criteria may be optionally excluded from downstream analyses (Figure 1). In the interests of diagnostic laboratory confidentiality issues, diagnostic laboratory data that has been submitted to the US-SPD has an anonymized unique ID, i.e. only the submitting laboratory retains the ability to track a sample and its genomic information and metadata to a producer or veterinarian. The US-SPD has also ingested and curated publicly available sequence data; in these cases, custom parsers have extracted available metadata, but these records may not have information beyond sequence.
The US-SPD custom search interface allows users to query and retrieve data in standard formats, e.g. FASTA sequence file, comma separated text files with associated metadata. The search interface is split by pathogen, and queries may be based on virus; gene; date of collection; state of collection; isolation source; host; sequence length, completeness, with duplicate records identified; author or submitting laboratory; PubMed ID; and isolate strain name. This returned information is standardized and with additional analyses can be applied for vaccine design; determining when and how fast pathogens are spreading across the landscape; genomic structure determination; and translational and post-translational protein analyses. Importantly, by integrating large-scale genomic information, representative virus genes or genomes may be selected, potentially sourced from regional diagnostic laboratories, and used in comparative approaches to determine phenotypic consequences of genetic diversity, e.g. whether specific nucleotide and/or AA changes affect phenotype (32). Whole genome strain data is searchable using similar strategies. For viruses such as PRRSV, this provides the ability to study the role of recombination in evolutionary dynamics, and identify vaccine targets, e.g. ORF5, nonstructural protein 2 (33–35).
Identifying homologous sequence data, PRRSV ORF5 classification, and gene visualization
The US-SPD includes an implementation of the Tripal BLAST UI extension module (https://github.com/tripal/tripal_blast/). This module is an intuitive implementation of the NCBI Blast+ tools (27) and relies upon the Tripal interface with Drupal integration for easy deployment and management. As applied in the US-SPD, the tool provides access to nucleotide- and protein-based BLAST functionality (e.g. blastn, blastx, blastp, tblastn) and is integrated to facilitate rapid queries, job submission and background execution for all pathogens. The US-SPD currently allows queries against all data in the database, or specific subsets of that data, i.e. a user is able to query the aggregated database, or a specific pathogen, or annotated sequences. The utility of querying annotated sequences is most apparent with PRRSV, whereby the similarity of a query ORF5 gene may be determined against an established ORF5 genetic nomenclature that assigns lineage information (15). The module produces a graphical exploration of genetic similarity, including the reference sequence location where the query gene is most similar. These data can be used to locate similar genes within the database for further comparative analyses or if there are no similar sequences within the US-SPD, it would indicate a query virus that may not have been previously detected in US swine herds, e.g. (36).
Custom analysis scripts have been deployed through modification of the Tripal BLAST UI extension module. Specifically, a script rapidly classifies PRRSV-2 strains by RFLP patterns from user submitted query sequences and outputs corresponding RFLP assignment. RFLP values have been extensively used to quantify the diversity of ORF5 genes (e.g. (13, 14)). The approach uses cuts created by three restriction enzymes (MluI, HincII and SacII) to classify strains based on the cut site location of each restriction enzyme (37). Although the application of this tool has limitations as diversity in ORF5 may not reflect diversity across the genome or the phenotype of a virus, it has utility in assessing, categorizing, and linking contemporary genetic signatures to archived sequence records. The implemented tool takes user-submitted ORF5 nucleotide data, checks the length of the sequence and then aligns with HMMER3 (38). Subsequently, the tool identifies restriction sites and classifies the viral sequence with the appropriate three-digit numeric code. RFLP patterns for PRRSV-2 ORF5 sequences have been determined and stored within the database as a searchable field, allowing users to quickly screen their data and identify similar sequences for analyses.
To further facilitate comparative analyses of genomes, we have implemented the Tripal JBrowse extension module (https://github.com/tripal/tripal_jbrowse). The module embeds JBrowse (28, 29), an interactive, client-side genome browser, into a Drupal webpage and provides a simple interface for managing and creating JBrowse instances. We have deployed seven JBrowse instances covering ASFV, CSFV, FMDV, PDCoV, PEDV, PRRSV and SVA with annotations of coding sequence, genes, mature peptides and untranslated regions of reference sequences from the NCBI RefSeq database (http://www.ncbi.nlm.nih.gov/genome). This feature remains in development and may be refined as more information is derived during sequence annotation, incorporated through subject-area expert input or requests, and these metadata may be captured and shared within the JBrowse interface. As this tool is integrated within the Tripal toolkit, future versions of the US-SPD have the potential to integrate user submitted information from each of the JBrowse genome instances into the US-SPD database, or users can save and share their JBrowse annotations with the community.
Utility and discussion
Overview of the genomic data in the US-SPD
The US-SPD now includes 86 096 validated virus genomes, including 82 540 sourced from NCBI GenBank and an additional 3556 from regional diagnostic laboratories. There are 18 viruses currently in the database, each of which represents an agricultural pathogen (1), with priority derived from the Swine Health Information Center ‘Swine Viral Disease Matrix’ (https://www.swinehealth.org/swine-disease-matrix/). The pathogens include 11 virus genera (Alphacoronavirus, Apthovirus, Asfivirus, Betaarterivirus, Circorvirus, Deltacoronavirus, Enterovirus, Orthohepevirus, Pestivirus, Senecavirus and Varicellovirus). Each gene record includes a link to the original GenBank record when available, and the information parsed from this record can be annotated onto the FASTA definition line or in a metadata download for additional analyses. Genes and/or strains that have been used in empirical studies have been incorporated for all viruses and are linked via the PubMed ID to the publication. In total, there are 205 001 validated viral and genome segments within the US-SPD database. Unfortunately, while some of the virus strains in the US-SPD are well studied in the laboratory, many other sequenced viruses and strains are not. The establishment of the US-SPD highlights the diversity of these viruses and provides an interface to study their phenotype.
Cocirculation and turnover of multiple genetic clades of PRRSV
PRRSV represents the single most significant threat to the health and economic productivity of swine in the USA today. Since its discovery in the late 1980s, PRRSV has caused devastating production losses in herds throughout the USA and the world (39–41). Despite significant advances in our understanding of PRRSV biology (42), the disease has remained very difficult to control. There are two reasons for the remarkable resistance of PRRSV to control through vaccination: firstly, PRRSV is immunomodulatory and immunoevasive; and secondly, PRRSV has extraordinary genetic and antigenic variability (11). PRRSV may suppress early events in the activation of host cellular immunity and misdirects or delays the production of neutralizing antibodies (42–44). Underlying this property is an extraordinary genetic and antigenic variability and a consequent ability to evolve rapidly due to the low fidelity of the viral RNA polymerase (45).
Using the US-SPD, we generated a sequence dataset of PRRSV ORF5 sequences that capture the extent of genetic diversity from 2000 to 2021. These data include all sequences from isolates in GenBank (n = 29 325), alongside previously unpublished data derived from participating regional diagnostic laboratories (n = 3078). As of March 2021, these sequences were comprised of 32 403 worldwide samples with most viruses from the USA, but viruses from Canada, Mexico, China, Korea, Japan, Thailand, Austria, Denmark, Italy and Poland were also present. Using the RFLP-typing tool on the US-SPD, we quantified RFLP values for all complete sequences from 2000 to 2021 as a means to quantify temporal changes in the diversity of ORF5 genes. We document concurrent circulation of the 15 major RFLP patterns consistently across all years, i.e. patterns that were the most frequently detected in total across the time period, and an additional 138 ‘Other’ patterns that represent ∼10 to ∼25% of detections each year representing 1991 total detections of 16 403 total patterns (Figure 3). The predominance of certain RFLP patterns rapidly changes from year to year, e.g. 1-4-4 was ∼50% of detections in 2011–12 but declined to ∼10% by 2015, superseded by 1-7-4 as the dominant type, which then rapidly declined to ∼15% of detections in 2020. In the last two years, these data revealed that no single RFLP pattern became predominant, with almost even detections of 1-7-4, 2-5-2, 1-8-4, 1-2-2 and ‘Other’.
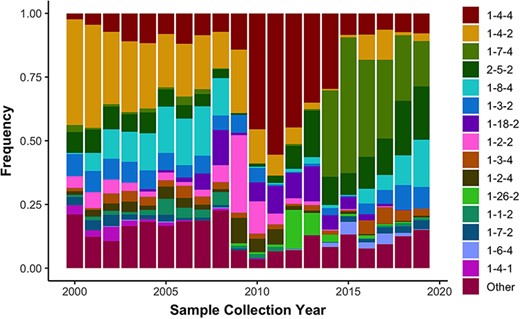
Observed frequency of the top 15 most commonly detected restriction fragment length polymorphism (RFLP) patterns in Type 2 porcine reproductive and respiratory syndrome virus sampled in the USA from 2000 to present (n = 16 403). Less common RFLP patterns were grouped together and labeled as “Other”.
Recombination and PRRSV evolution in the USA, 2014 to 2021
We demonstrate the utility of our search interface to conduct genomic analyses by conducting a phylogenetic network analysis of a field-relevant PRRSV dataset. This is motivated by our prior empirical study (13) that demonstrated recombination in PRRSV resulted in differential evolutionary dynamics that were associated with unique patterns of pathogenesis and transmission. Importantly, recombination in PRRSV has two biological outcomes: it increases genetic diversity; and it increases the likelihood of the emergence of a virus that is antigenically novel via a ‘sampling effect’, e.g. (46, 47). In support of this hypothesis are data that demonstrate that following recombination, novel viruses with unique phenotypes have emerged in China, Europe and the USA (13, 48–50).
For these analyses, we searched for all PRRSV complete strains (n = 1389) in the US-SPD. Completeness was determined by searching for the presence of all full open reading frames and non-structural proteins. We then restricted and identified strains that were collected in the USA from 2014 to present and downloaded the individual annotated genes (n = 253; data available at https://github.com/us-spd/database-manuscript). The genomes were separated into the constituent genes, and a single outgroup was included, VR-2332 (GB000137/DQ217415). We apply a recent algorithm, RF-Net, that was built to infer virus networks accommodating for influenza A virus reassortment (51, 52). To generate the required input trees for RF-Net, genes were aligned using default parameters in MAFFT (31), and maximum likelihood phylogenetic trees were inferred following automatic model selection using IQ-TREE (53, 54). We explored virus networks in RF-Net with reticulation events, r, ranging from 0 to 20.
The inferred phylogenetic network (Figure 4) with a minimum of three reticulation events was the best of those we explored (data at https://github.com/us-spd/database-manuscript). We visualize the network with r = 5 to demonstrate three previously reported signals of recombination (55) alongside additional two recombination events to highlight how unique strains may be identified through the US-SPD search interface with subsequent phylogenetic analysis. Firstly, strain IA30788-R (GB043798, Figure 4b) was recently described as a putative recombinant between a wildtype strain (IA76950-WT) and a commercial vaccine (55). Similarly, additional recombination signals were detected in 23 199-S4-L001 (GB043498, Figure 4c) and 7705R-S1 (GB043536, Figure 4d) strains that have also been reported as, or associated with, recombination (50). Typically, PRRSV phenotypic studies focus on reference strains because of ease-of-access, publication history and evaluation of whether the variant represents a particular geographic location or biological property (56). Consequently, an application of the US-SPD is to search data, conduct phylogenetic analyses using algorithms such as RF-Net (51, 57), RDP5 (58) or BEAST 2 (59) to identify putative recombinants, and viruses with recombination signals may be identified or ranked (60, 61) for additional phenotypic characterization.
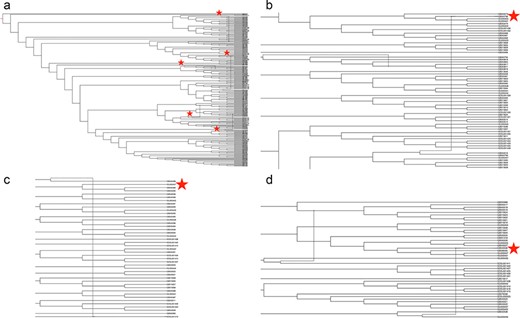
Phylogenetic network of porcine reproductive and respiratory syndrome virus collected in the Midwest of the USA from 2014 to present (a) Putative recombination events are indicated by verticle lines within the network, annotated by red stars. Panel (b) strain IA30788-R (GB043798), Panel (c) strain 23199-S4-L001 (GB043498) and Panel (d) strain 7705R-S1 (GB043536) are visualized separately to demonstrate evolutionary relationships and recombination nodes. Phylogenetic network with tip labels, recombination layers 1 through 20 and gene tree embeddings is provided at https://github.com/us-spd/.
Conclusions
The database and curation pipeline provide a recording system that will standardize data from swine veterinary diagnostic laboratories alongside public data deposited to NCBI GenBank. Further, the search interface allows the dissemination of these data in standard format (e.g. FASTA sequence file) based upon user queries. This is a simple interface, but because it allows researchers to screen data it has the power to provide information for vaccine design (62–64), determining when and how pathogens are spreading across the landscape, identifying transmission hotspots at a coarse spatial scale and generating datasets appropriate for comparative virology. Additionally, through the standardized availability of date collection on all data, users may create time-scaled phylogenies allowing the time of emergence for novel viral isolates to be determined, and these data can be used to determine viral spread across the landscape using the state level geographical information provided by the diagnostic laboratories, e.g. (50, 65, 66). Another benefit of these data is that genomic information covering thousands of sequences allows for the identification of AA substitutions associated with particular genetic clades of viruses. Understanding these substitutions can be used to inform vaccine updates and composition, e.g. (11, 67).
Future directions
As viral genome sequencing in swine virology increases, e.g. (68), databases are required to link those in the diagnostic community with researchers to improve animal health and economic productivity. This process should include careful curation, with standardized sequence annotation methods that ensure data quality to enhance our ability to make sound inference from the data, i.e. linking nucleotide/AA changes across disparate parts of the genome (epistasis), where insertions and deletions are evolving, where other key translation events are located and linking genetic diversity to antigenic phenotype. Once annotated, large-scale genome sequence data needs to be available in ways that facilitate discovery. This requires automated metadata capture and data standardization, as well as interfaces that leverage the annotated metadata for scientific discovery. Many different approaches are possible (e.g. NCBI Virus Variation Resource (69)), but the US-SPD achieves this by providing search fields that are specific for the swine health community and will allow for access to sequences discovered by regional veterinary diagnostic laboratories. While currently limited to 18 swine pathogens, our intent is to expand the automated US-SPD data annotation pipeline to include more viruses and also be flexible enough to account for the emergence of novel pathogens. As a public resource, the US-SPD serves a range of users in the animal health community (to date, registered users include regulatory agencies, industry and academia) who conduct work in diagnostic laboratories developing assays, to those who conduct basic research in molecular virology to understand the biology, evolution and transmission of viruses (e.g. (70)). Our mission is informed by their use, and by engaging our stakeholders and working together on shared goals we can provide the rigorous resources necessary to support a comprehensive swine pathogen database. Collectively, reducing the impact of viral pathogens in swine requires a fundamental knowledge of what viruses are circulating in the population and the US-SPD achieves this by providing a tool for investigators to develop rational and representative vaccines which will reduce viral burdens, decreasing the economic burden of viral disease and improving animal health through targeted interventions and surveillance.
Availability
The code and data used in this manuscript are available in the GitHub repository (https://github.com/us-spd/); the database implements the open source Tripal project that is available at the following website https://tripal.info/. Restrictions apply to the immediate availability of diagnostic laboratory data due to client confidentiality: these data are embargoed from 12 months from receipt from the diagnostic laboratory and then become publicly available. Data may be available upon reasonable request and with permission of the contributing diagnostic laboratories.
Acknowledgements
The authors gratefully acknowledge pork producers, swine veterinarians and laboratories for participating in surveillance for viral pathogens infecting swine and publicly sharing sequences in the United States Swine Pathogen Database and NCBI GenBank. The authors acknowledge the efforts in pathogen sequence generation and reporting by all technical personnel at Iowa State University Veterinary Diagnostic Laboratory, Kansas State University Veterinary Diagnostic Laboratory and the South Dakota Animal Disease Research and Diagnostic Laboratory. The authors thank Alexey Markin for providing a pre-release version of RF-Net-2 and discussing implementation and visualization of inferred virus networks.
Funding
This work was supported by the U.S. Department of Agriculture (USDA) Agricultural Research Service (ARS project number 5030-32000-230-000-D); the USDA Animal and Plant Health Inspection Agency (USDA ARS project numbers 5030-32000-108-21I and 5030-32000-108-37I); the USDA Agricultural Research Service Research Participation Program of the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the U.S. Department of Energy (DOE) and USDA Agricultural Research Service (contract number DE-AC05-06OR23100 to B.I.); the SCINet project of the USDA Agricultural Research Service (ARS project number 0500-00093-001-00-D); and the National Pork Board (NPB project number 16-222). Funding for open access charge from the U.S. Department of Agriculture (USDA) Agricultural Research Service (ARS project number 5030-32000-230-000-D). The funders had no role in study design, data collection and interpretation or the decision to submit the work for publication. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA, DOE or ORISE. USDA is an equal opportunity provider and employer.
Conflict of interest
The authors report no conflicts of interest.
References
Author notes
Tavis K. Anderson, Blake Inderski1 contributed equally to this work.
Present address: Diego G. Diel, Department of Population Medicine and Diagnostic Sciences, College of Veterinary Medicine, Cornell University, 240 Farrier Road, Box 47, Ithaca, NY 14853, USA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}