





Abstract
The reversible phosphorylation of serine, threonine and tyrosine hydroxyl groups is an especially prominent form of post-translational modification (PTM) of proteins. It plays critical roles in the regulation of diverse processes, and mutations that directly or indirectly affect these phosphorylation events have been associated with many cancers and other pathologies. Here, we describe the development of a new BioMart tool that gathers data from three different biological resources to provide the user with an integrated view of phosphorylation events associated with a human protein of interest, the complexes of which the protein (modified or not) is a part, the reactions in which the protein and its complexes participate and the somatic mutations that might be expected to perturb those functions. The three resources used are the Reactome, PRIDE and COSMIC databases. The Reactome knowledgebase contains annotations of phosphorylated human proteins linked to the reactions in which they are phosphorylated and dephosphorylated, to the complexes of which they are parts and to the reactions in which the phosphorylated proteins participate as substrates, catalysts and regulators. The PRIDE database holds extensive mass spectrometry data from which protein phosphorylation patterns can be inferred, and the COSMIC database holds records of somatic mutations found in human cancer cells. This tool supports both flexible, user-specified queries and standard (‘canned’) queries to retrieve frequently used combinations of data for user-specified proteins and reactions. We demonstrate using the Wnt signaling pathway and the human c-SRC protein how the tool can be used to place somatic mutation data into a functional perspective by changing critical residues involved in pathway modulation, and where available, check for mass spectrometry evidence in PRIDE supporting identification of the critical residue.
Database URL:http://www.reactome.org/cgi-bin/mart
Introduction
Covalent modification of amino acid residues (‘post-translational modification’, PTM) is an important part of the molecular toolkit used by many organisms to generate full sets of functional forms of proteins from the relatively limited sets of open reading frames specified in their genomes. The reversible phosphorylation of serine, threonine and tyrosine residues is the most frequent PTM in the mammalian proteome (1). Approximately 30% of all proteins in a human cell are phosphorylated at any given time (2). Reversible phosphorylation of proteins through the activity of protein kinases and phosphatases have been exploited by nature to modify the function of a protein in almost every conceivable way: increasing or decreasing its biological activity, stabilizing it or marking it for destruction, facilitating or inhibiting movement between cellular compartments and initiating or disrupting protein–protein interactions (2), thereby regulating almost all aspects of cell function and cell–cell interaction in processes including differentiation, proliferation and migration. The disruption of normal phosphorylation states of intracellular proteins by pathogens, toxins or mutations is associated with many diseases and disorders (2), and development of drugs that target specific protein kinases and phosphatases is the focus of active research.
Mass spectrometry (MS) is currently the most commonly used technology for the identification and quantification of proteins. Technological advances in proteomics have enabled identification of protein modifications as well as protein subcellular location(s) allowing researchers to gain an accurate picture of cellular processes (3). The resulting MS data can be deposited in proteomics databases such as PRIDE (PRoteomics IDEntifications database, http://www.ebi.ac.uk/pride) (4). PRIDE is a standards-compliant public repository of MS-derived data. It stores MS and MS/MS mass spectra as peak lists, the derived peptide and protein identifications, as well as PTMs and experimental metadata.
Biologists commonly represent the biochemical processes that take place at a subcellular level as networks of reactions, called pathways. A familiar example is the citric acid cycle. Reactome (http://www.reactome.org) (5) is a database of human pathways specified in molecular detail. The data are contributed by expert biologists, supported by literature and undergoes an iterative curation and peer-review process. It encompasses a wide variety of biological processes, such as metabolism, signaling, apoptosis, transcription and the cell cycle. Reactome makes extensive use of molecular complexes, particularly protein complexes. The website provides facilities for browsing and viewing the pathways, plus tools for analyzing user-supplied data in a pathway context. Various export formats are provided in order to make the content accessible to the systems biology community. All Reactome software and data are available under the terms of the Creative Commons Attribution 3.0 Unported License.
The catalog of somatic mutations in cancer (COSMIC) (http://www.sanger.ac.uk/genetics/CGP/cosmic/) (6) is a database that gathers, curates, organizes and presents somatic mutations in human cancer.
The Reactome, PRIDE and COSMIC databases all employ a BioMart interface (http://www.biomart.org) (7) on their websites for data integration. BioMart is an open source data management resource, which provides a variety of query interfaces enabling users to group and refine data, based upon many different criteria. It provides a web interface with a consistent look and feel and performs federated queries across different databases such as Ensembl (8) and UniProt (9) as well as PRIDE and Reactome—located on different servers or at different geographical locations. Federation utilizes shared common identifiers (IDs) e.g. Ensembl gene IDs, UniProt IDs.
Development of a new BioMart tool
BioMart has its own special data structure in order to allow very fast queries and efficient federation. It structures the user's data into databases, which contain data sets. This allows the BioMart database designer to separate data into different types. The standard Reactome BioMart (10) has four data sets, pathway, reaction, complex and interaction. The pathway data set allows users to retrieve ID, name, species and several other attributes from all of Reactome's pathways. It also uses dimension tables so that proteins, complexes and literature references associated with each pathway can also be retrieved. A large set of filters is provided, so that a user can, for instance, find all of the pathways associated with a given set of UniProt IDs. The reaction and complex data sets are structured very similarly to the pathway data set.
The PRIDE BioMart can be used to interrogate the three different kinds of information stored—peptide and protein identifications derived from MS or MS/MS experiments, MS and MS/MS mass spectra as peak lists and any associated metadata. It also provides filtering by experiment, sample details, peptide identification, protein identification, mapped protein IDs and protein modification.
The COSMICMart (11) consists of a single data set, which allows users to retrieve information about the sample, gene, mutation, site and histology, plus details such as Entrez, UniProt, Ensembl gene, PubMed and COSMIC study IDs. A large set of filters is provided, so that users can, for instance, find all the somatic mutations together with the sample details associated with a given set of UniProt IDs. In this article, we describe data integration from Reactome, PRIDE and COSMIC databases, based on updated data sets and newly created ‘canned’ queries.
The protein data set in Reactome
We have created a new data set in Reactome BioMart, ‘Protein’, to enable users to explore PTM details for a protein and retrieve information such as the cellular compartment of the protein, the name of the modification, the modified residue, the coordinate of the modified residue on the protein sequence and the start and stop positions of the protein sequence. In terms of data integration, the data set can be interrogated in conjunction with other pre-existing Reactome data sets e.g. pathway, complex and reaction or with federated data sets hosted elsewhere that share IDs e.g. UniProt, Ensembl and PSI-MOD IDs.
To federate two data sets, BioMart's MartEditor tool provides the means to edit each data set's configuration XML by defining an ordered list of attributes that are common between the two data sets. The attributes from a data set to be shared with the second data set are referred to as the exportables, while those from another dataset to be used as filters in querying the dataset are referred to as importables. For a data set to link to a second data set, an importable needs to be created in that data set with the same linkName setting as the exportable. The linkName can either use a single-filter value system for the importable e.g. UniProt ID, or a multifilter value system as required e.g. for a combination of UniProt and PSI-MOD (12) IDs, as long as the attribute value(s) of the corresponding exportable are specified in the same order as the importable.
We used BioMart's capability of federating between multiple data sets based on shared IDs to define importables and exportables available for this data set. To link to PRIDE's modification location table described below, a two-filter/attribute importable and exportable were defined using the shared UniProt and PSI-MOD IDs, while to link to the COSMICMart data set, the two-filter/attribute values were based on shared UniProt ID and modified/mutated residue position.
The Modification Location table in PRIDE
The ‘Mapped Protein Modification (PTM) to UniProt Proteins Attributes—Reactome Link’ table on PRIDE BioMart data set was specifically created to capture phosphorylated proteins and their associated details such as the location of the modification, name of the modification, sample information including species, tissue, sub-cellular location and/or disease state, the sample used, and the spectrum for each peptide that was used in identifying the protein. The Resid (13) and Unimod (14) ontology terms were manually mapped to PSI-MOD (12) terms by a PRIDE curator for integration with Reactome's ‘Protein’ data set, in order to link protein modification information (PSI-MOD) from a pathway context to MS data. All the low level PSI-MOD terms were also mapped to a top level PSI-MOD term i.e. all the serine, tyrosine and threonine phosphorylations have been mapped to modified residue terms, to simplify matching these terms across the two databases.
The COSMICMart data set
COSMICMart allows users to query genes, tissues and mutations information from the samples available in the COSMIC database and to federate these with other datasets such as Ensembl. An update has recently been made to enable the new Reactome protein data set to link to the COSMIC data set: a two-filter/attribute importable and exportable were defined based on the shared UniProt IDs and modified/mutated residue position, respectively. The single input value filter 'Swissprot ID' was changed to a multivalued input filter to allow users to query the data set using more than one UniProt ID. In addition, links were created from COSMICMart's AA Mutation Start and AA Mutation Stop position attributes to the COSMIC gene histogram page, giving the user a graphical view of the spread of mutations in their region of interest on the gene sequence. The gene histogram page also offers a number of options for filtering the data, as well as pie charts, accessible by clicking the distribution button, summarizing the data.
Database integration
Based on the updated data sets, the Reactome BioMart can retrieve and integrate protein phosphorylation data derived from expert manual curation of the research literature (from Reactome) with data from high-throughput MS experiments (from PRIDE), link these data to known mutations affecting the amino acid sequences of proteins (from COSMIC) and to the roles of these proteins in complexes and reactions (from Reactome). A user might approach this system from the point of view of a protein of interest, seeking information on its modifications and their effects on its functions, or might approach from the point of view of a process of interest, seeking information on the proteins and complexes involved in it. We have developed ‘canned’ queries to facilitate these approaches. The ‘canned’ queries are also starting points from which a user can develop customized queries. The examples described here illustrate useful ‘canned’ approaches and one way in which such an approach might be customized.
Queries to retrieve modified proteins from Reactome data sets
Users can retrieve modified proteins and their associated details from three Reactome data sets; pathway, complex and reaction.
Query: ‘Find all modified proteins in a pathway’—this query enables users to retrieve modified proteins associated with Reactome pathway(s) of interest for further analysis (Figure 1). After the user selects the query, a data input page appears where Reactome Pathway name and species can be selected (Figure 2). The query will be processed and results presented in a BioMart results page (Figure 3). The query spans the ‘Pathway’ and ‘Protein’ data sets.
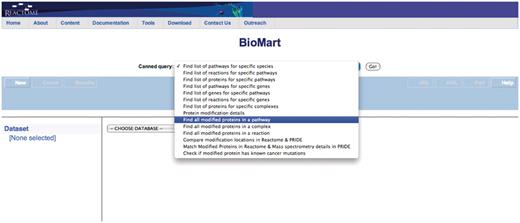
Select the appropriate canned query, in this case ‘Find all modified proteins in a pathway’.
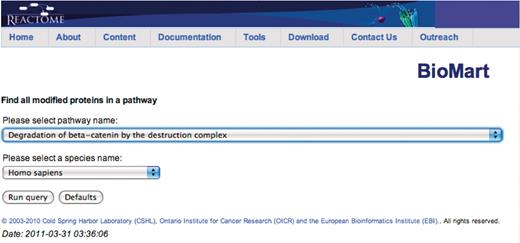
Canned query data input page. The user selects the pathway and species names from the respective drop down lists and then presses the ‘Run query’ button.
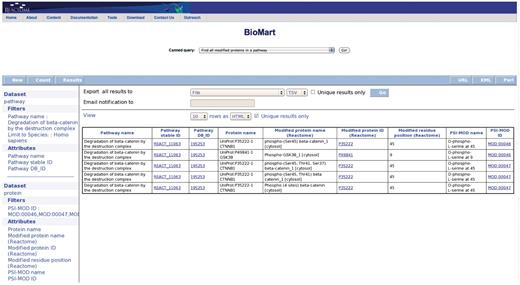
Results of finding all modified proteins in a pathway. The user is then able to inspect the retrieved proteins for the kinds of modification present on each protein (PSI-MOD), the modified residue in the protein, the cell compartment of the modified protein and the position of the modified residue on the protein if known (Modified Residue coordinate).
Similarly, finding all modified proteins in a complex or reaction can be retrieved with predefined queries.
Query: ‘Compare the modification locations in Reactome & PRIDE’—to be able to compare which residues in a protein are reported to be modified in Reactome and their corresponding positions with PRIDE MS experiments, the ‘Compare the modification locations in Reactome & PRIDE’ canned query can be used. The user enters the UniProt ID(s) in a data input page and the results are presented in a BioMart results page (Figure 4). This query spans the Reactome ‘Protein’ and ‘PRIDE’ data sets.
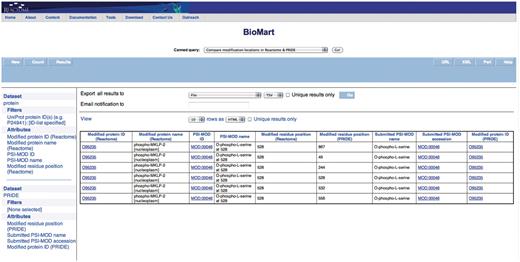
Results of comparing modification locations between Reactome and PRIDE. The modified protein's UniProt ID, its name, cellular compartment, the modified residue and its residue coordinate from Reactome are reported. PRIDE also reports corresponding details of the protein present in their database i.e. location of modification on protein, PSI-MOD name, ID and the UniProt Accession.
Query: ‘Match modified proteins in Reactome & MS details in PRIDE’—after finding a matching or conflicting modified residue position, one can then inspect the peptides and the spectrum used to identify the modification. The ‘Modified proteins in Reactome & Mass spectrometry details in PRIDE’ canned query can be used to retrieve this information. The user enters the UniProt ID(s) in a data input page and the results are presented in a BioMart results page (Figure 5) with the option to view the spectrum (Supplementary Figure S8). This query spans the ‘Protein’ and ‘PRIDE’ data sets.
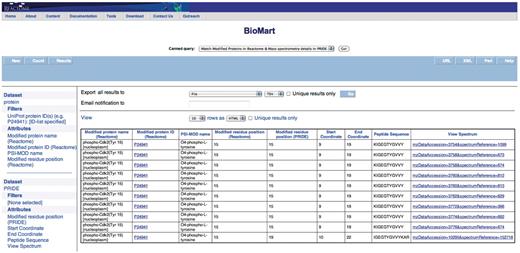
Results of matching modified proteins in Reactome with the MS details available in PRIDE. The peptide sequence and the option to view the spectrum (Supplementary Figure S8) are returned in addition to the columns returned in Figure 4.
Query: ‘Check if modified protein has known cancer mutations’—users can also check if their protein of interest has any known somatic mutations that are linked to cancer through COSMIC by using the ‘Check if modified protein has known cancer mutations’ canned query. The user enters the UniProt ID(s) in a data input page and the results are presented in a BioMart results page (Figure 6). This query spans the ‘Protein’ and ‘COSMIC’ data sets.
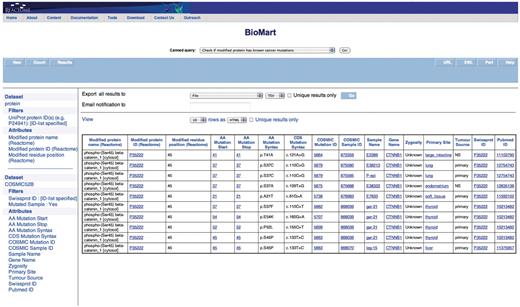
Results of checking if the modified protein has known cancer mutations. The modified protein's UniProt ID, the COSMIC Sample ID and name, the gene name, the nucleotide and residue changes caused by the mutation, the zygosity, primary histology, tumor source and the reference paper for the study are returned. By further choosing more attributes finer details can be retrieved. The query spans the ‘Protein’ and ‘COSMIC’ data sets.
Biological examples
Using two case studies, we illustrate the new Reactome, COSMIC and PRIDE data integration that provides a new way of looking at known information residing in different databases.
Wnt Signaling
The canonical Wnt signaling pathway (doi: 10.3180/REACT_11045.1) regulates the cytoplasmic levels of the β-catenin protein that is involved in the activation of the Wnt target genes in the nucleus thus playing an important role in regulating the balance between stemness and differentiation in adult stem cell niches (15) such as skin and hair follicle (16), the mammary gland (17) and hematopoietic tissues (18). This pathway is regulated by the β-catenin destruction complex, which comprises of the adenomatous polyposis coli (APC), Axin 1/2, casein kinase 1 (CK1) and glycogen synthase kinase-3β (GSK-3β) proteins. In the absence of Wnt signaling, this complex controls the levels of cytoplasmic β-catenin by amino-terminal serine/threonine phosphorylation of β-catenin at Serine 45 by CK1-α, which primes the subsequent sequential GSK-3-mediated phosphorylation at threonine 41, serine 37 and serine 33 (19). Phosphorylated β-catenin is recognized and ubiquitinated by the SCF-β TrCP ubiquitin ligase complex and is subsequently degraded by the proteasome (20) (doi: 10.3180/REACT_11063.1). It is therefore not surprising that carcinogenesis is likely to be initiated in tissues with aberrant Wnt signaling activation due to mutations in genes encoding its downstream components such as β-catenin and the APC genes.
The β-catenin protein (UniProt Accession: P35222) presents an instance where critical residues that are normally phosphorylated as part of the Wnt signaling pathway modulation and reported in the Reactome database (serine 45 and threonine 41), are mutated, leading to aberrant signaling associated with cancer as identified using Query 4: ‘Check if modified protein has known cancer mutations’. Each of the reported mutated residues in COSMIC has a link to the gene histogram page (Figure 7) by clicking either the value of AA Mutation Start or AA Mutation Stop column. The gene view histogram presents a graphical view of the spread of mutations in a region of interest, in this case showing that the phosphorylated residues serine 45 and threonine 41 on the β-catenin protein are lost.
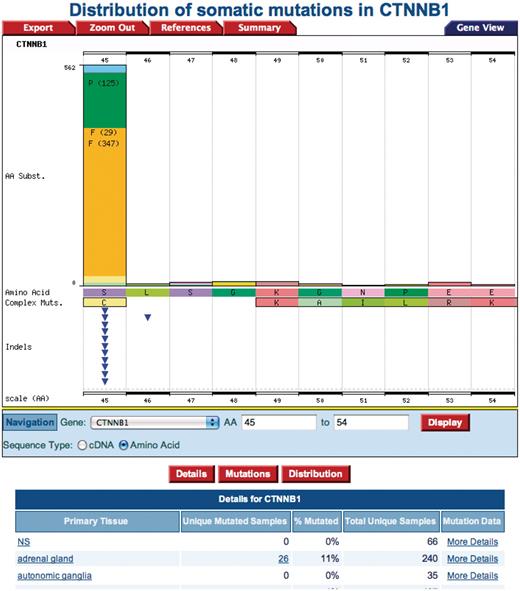
A COSMIC Gene View histogram from clicking the ‘AA mutation start or AA mutation stop’ position. The amino acid serine at position 45 is mutated to a phenylalanine.
The human c-SRC protein
The human cellular SRC (c-SRC), a nonreceptor tyrosine kinase, is involved in numerous cellular processes such as cell proliferation, regulation of cell adhesion, invasion and motility, which are thought to contribute to tumor progression and metastasis, although this role is not yet fully understood (21, 22). It has an N-terminal SH4 domain that contains the myristoylation site needed for membrane localization, a variable ‘unique’ region, SH3 domain for binding to specific proline-rich sequences, SH2 domain that binds to specific tyrosine phosphorylation, SH1 catalytic tyrosine kinase domain, which is the autophosphorylation site and a short C-terminal region containing a conserved tyrosine residue. The two known functionally important tyrosine residues in the c-SRC protein are tyrosine 419 located in the SH1 catalytic tyrosine domain that is the autophosphorylation site, and tyrosine 530 contained in the short C-terminal region that is known to be the principal regulatory phosphotyrosine. The phosphorylation of c-SRC at C-terminal regulatory tyrosine 530 facilitates intramolecular binding to the SH2 domain causing the protein to be inactivated, whereas the dephosphorylation of tyrosine 530 disrupts this binding with the SH2 domain facilitating the autophosphorylation of tyrosine 419 leading to c-SRC activation (23). A truncating mutation at codon 531 (Glutamine to stop) occurs directly at C-terminal regulatory tyrosine 530 and appears to activate the tyrosine 530 in some cancers e.g. advanced colon cancers (22) and in some cases of endometrial carcinoma (24). Reactome reports both tyrosine 419 and tyrosine 530 as functionally important residues, PRIDE reports tyrosine 419, whereas COSMIC reports the truncating mutation at codon 531.
Comparison of curated and high-throughput phosphorylation data
Direct comparison of Reactome (Release 34) functional annotation with PRIDE (core version: 2.8.6, February 2011) high-throughput phosphoproteomic data reveal independent confirmation and highlights the potential for further investigation in cases of differing observations (Supplementary Table S1). A total of 13 unique protein accessions were retrieved from a Reactome-PRIDE BioMart database match based on the two-filter uniProt/modified residue position filter system. From these, PRIDE reports a total of 43 phosphorylation sites occurring on either serine or tyrosine residues, while Reactome reports 14 phosphorylation sites on the same residues. The two databases corroborate on five modified residue positions (Supplementary Figure S9) derived from five UniProt protein accessions (Supplementary Table S2).
The canned query ‘Compare the modification locations in Reactome & PRIDE’ shows instances where PRIDE reports more modified residue positions than those reported by Reactome for a given protein. For example, Figure 4 shows where PRIDE reports six phosphorylated serine residues (at positions 49, 244, 528, 532, 556, 867) on kinesin-like protein KIF20A (UniProt Accession: O95235), whereas Reactome annotates the only modification with known function (serine 528). This integration presents an easy and systematic platform to compare critical residue positions identified through traditional biochemical techniques that are mainly limited to investigating how phosphorylation at specific sites affect a single protein of interest (25), with high-throughput phosphoproteomics approaches that have the ability to identify novel in vivo phosphorylation sites on a large scale but with the potential risks of averaging over multiple cell types and states. Modifications data, like all of Reactome's data, are obtained from literature, expert-authored, manually curated and peer-reviewed before being added into the database by a Reactome curator. The experimental evidence of the reported modifications in PRIDE is obtained from high-throughput MS proteomics experiments. In these approaches, proteins are digested with a single protease, typically trypsin, and the resulting peptides—modified or not—are analyzed using a mass spectrometer (26). This experimental workflow has strengths and limitations that have been widely described before (27–29). This data will serve as a tool for validation and the potential addition of functional modification sites in Reactome.
Discussion
We have applied a key feature of BioMart, namely its capability to formulate queries across multiple independent BioMart installations, to link manually curated pathway data in Reactome to high-throughput phosphoproteomics data in PRIDE and somatic mutation data in COSMIC.
Five of the 13 identified proteins that overlap between Reactome and PRIDE matched their phosphorylation positions perfectly, with PRIDE having more phosphorylations sites identified through high-throughput experiments. Once these additional phosphorylation sites are validated independently, they may or may not be functionally relevant and might then be added to Reactome annotation. The traditional concept of a functional active site is based on the assumption that protein phosphorylation modulates protein function through the specific position of the phosphorylation in the protein sequence. However, it has been recently argued that nonpositionally conserved phosphorylation sites may very well be functional, as the authors speculate that the nonpositionally conserved phosphorylation sites could be modulating biomolecular association of phosphorylated proteins possibly by fine-tuning the protein's bulk electrostatic charge and by creating binding sites for phospho-binding interaction domains (30). This resource could be used to identify and differentiate the nonpositionally conserved modified sites.
Annotated, functionally relevant phosphorylation sites in Reactome might be used as independent confirmation for a test set of high-confidence phosphoproteomics spectra, which is highly valuable for MS methods development. Preliminary results in Wnt signaling link known somatic mutation to annotated, functionally relevant phosphorylation sites in the Wnt pathway representation in Reactome, and thus to possible interpretation of carcinogenic effects of these mutations. We plan to extend our approach to other functionally relevant PTMs such as ubiquitination, acetylation and myristoylation.
Funding
National Human Genome Research Institute at the National Institutes of Health, Development of the Reactome Knowledgebase (P41 HG003751); European Union 6th Framework Programme ‘ENFIN’ (LSHG-CT-2005-518254). Development of PRIDE has been supported by BBSRC “Tools and Resources”, the Wellcome Trust, EMBL Core funding, European Union 6th Framework Programme ‘ProDac’ (LSHG-CT-2006-036814), EU FP7 grants ‘LipidomicNet’ (grant number 202272) and ‘ProteomeXchange’ (grant number 260558). Funding for open access charge: National Institutes of Health (P41 HG003751).
Conflict of interest. None declared.
Acknowledgements
The authors are grateful to Rebecca Shepherd from the Cancer Genome Project at the Sanger Institute, UK for creating the required links from COSMICMart to Reactome and for her comments on COSMICMart.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}