





The soy phytoestrogen, genistein, has an array of biological actions, including weak estrogenic effects, inhibition of tyrosine kinase, and cellular antioxidant activity. Recent studies showed that genistein may improve vascular function, but the mechanism is unclear. We show that genistein stimulates intracellular cAMP accumulation in intact bovine aortic endothelial cells and human umbilical vein endothelial cells over an incubation period of 30 min. Increases in intracellular cAMP are evoked by as low as 10 nm genistein but not by estrogen. These increases in cAMP may result primarily from enhanced adenylate cyclase activity by a mechanism that does not involve genomic actions or estrogen receptors. The cAMP induced by genistein activates cAMP-dependent protein kinase (PKA) in bovine aortic endothelial cells. The activation of PKA phosphorylates and activates cAMP response element-binding protein, leading to up-regulation of cAMP response element-containing gene expression. In addition, activation of PKA protects thrombin-induced endothelial monolayer permeability, a novel cardioprotective effect of genistein mediated by the cAMP/PKA cascade. These findings demonstrate that a nongenomic action of genistein leads to activation of the cAMP/PKA signaling system to protect the vascular barrier function and alter the expression of cAMP-regulated genes, thereby providing a novel mechanism underlying some of the cardiovascular protective effects proposed for soy phytoestrogens.
RECENTLY SOY PHYTOESTROGENS have drawn wide attention due to their potentially beneficial effects on some human degenerative diseases. Genistein, the primary soy derived phytoestrogen, has various biological actions including a weak estrogenic effect (1) by binding to estrogen receptors (ERs), and inhibition of tyrosine kinase (2). Studies demonstrate that genistein has antiatherogenic effects by inhibiting proliferation of vascular endothelial (3) and smooth muscle cells (4). Data from animal and in vitro studies suggest a protective role of genistein in the vasculature (5–13). However, genistein has only a moderate positive effect (5, 14–16) or a neutral effect (17–20) on plasma lipid profiles, suggesting alternative mechanisms may exist. Recent human intervention studies using isoflavones suggest a beneficial effect on atherosclerosis (21), markers of cardiovascular risk (22, 23), vascular motor tone (20, 24), vascular endothelial function (25), and systemic arterial compliance (17). Although these data indicate a protective role of genistein in the vasculature, the cellular or molecular mechanisms underlying these beneficial effects is still unknown.
A recent study suggests that genistein-induced vascular relaxation may be at least partially mediated by the cAMP-dependent mechanisms (26). Other reports demonstrate that genistein may increase cAMP levels in neural (27) and epithelial cell lines (28). We have also shown that genistein, at the concentrations achievable by dietary means, directly acts on vascular endothelial cells (ECs), leading to activation of endothelial nitric oxide synthase (eNOS) and nitric oxide production in vascular ECs that is mediated through the cAMP/protein kinase A (PKA) pathway (29). cAMP signaling plays a very important role in maintaining normal vascular function by inhibiting vascular endothelial (30) and smooth muscle cell proliferation (31), depressing leukocyte adhesion to ECs (32), and maintaining normal endothelial barrier function (33, 34). In the present study, we investigated whether genistein activates the cAMP-dependent signaling system in vascular ECs and, subsequently, whether genistein could protect against endothelial barrier dysfunction by activation of the cAMP/PKA pathway.
Materials and Methods
Materials
Bovine aortic endothelial cells (BAECs) were kindly provided by Dr. Joseph Dillon (University of Iowa, Iowa City, IA) and primary human umbilical vein endothelial cells (HUVECs) were obtained from the Cardiovascular Research Cell Culture Core at the University of Iowa. M199 media, fetal bovine serum (FBS), l-glutamine, and penicillin-streptomycin were purchased from Gibco-BRL (Gaithersburg, MD); vascular endothelial growth factors were obtained from Clonetics (San Diego, CA). cAMP response element-binding protein (CREB) and phospho-CREB antibodies were purchased from Upstate Biotechnology (Lake Placid, NY); nitrocellulose membranes were from Schleicher & Schuell (Keene, NH); superSignal chemiluminescence detection system, stripping buffer, and avidin-conjugated fluorescein [avidin-fluorescein (FITC)] were purchased from Pierce (Rockford, IL); protein assay kits were from Bio-Rad Laboratories (Hercules, CA); pRL luciferase control vector, PKA, and dual luciferase reporter assay kits were from Promega (Madison, WI); fibronectin-coated Transwell plates were obtained from BD Bioscience Labware (Boston, MA); cAMP enzyme immunoassay (EIA) kit was from Assay Design Inc. (Ann Arbor, MI); Fugene-6 transfection reagent was from Roche (Indianapolis, IN); Rp-cAMP and the cell permeable PKA inhibitor peptide (PKI) were obtained from Calbiochem (San Diego, CA); β-gal staining kit was from Invitrogen (Carlsbad, CA); ICI 182,780 (ICI) was purchased from Tocris Cookson (Baldwin, MO); genistein, daidzein, 17β-estradiol, forskolin, ATP, GTP, pyruvate kinase, myokinase, phosphoenolpyruvate, isobutylmethylxanthine (IBMX), H89, actinomycin D, cycloheximide, α-thrombin, protease inhibitor cocktail, phosphatase inhibitor cocktail I, and general chemicals were from Sigma Chemical Co. (St. Louis, MO); all other reagents were detailed as indicated in the text.
Cell cultures
BAECs and HUVECs were grown in M199 medium supplemented with 20% FBS, 50 U/ml penicillin, and 0.05 mg/ml streptomycin and incubated at 37 C in a 5% CO2-95% air environment. For culture of HUVECs, the medium was supplemented with endothelial cell growth factor. Medium was changed every second day until confluence. BAECs or HUVECs were serially passaged after 0.05% trypsin treatment and passages 4–8 (BAECs) or 3–4 (HUVECs) were used in all experiments. Before experiments investigating nongenomic effects of genistein on cAMP signaling, cells were cultured for 24 h in phenol-red-free M199 medium containing 10% charcoal-stripped FBS, and subsequently BAECs were serum starved in phenol-red-free M199 medium, and HUVECs were cultured in phenol-red-free M199 containing 1% FBS and 0.5% BSA for 12 h. The residual concentration of estrogen in charcoal-stripped serum is less than 0.1 nm (less than 0.01 nm in the culture medium). For endothelial barrier permeability study, BAECs were cultured on fibronectin-coated polycarbonate filters (0.6 cm, 3 μm pore size) of the Transwell plate in M199 medium with 20% FBS for 3–4 d until a tight monolayer formed. Before the experiment, cells were kept in phenol-red-free M199 medium containing 10% charcoal-stripped FBS for 12 h and then washed and incubated in phenol-red-free medium containing 1% charcoal-stripped serum for 2 h.
Intracellular cAMP assay
The accumulation of cAMP in BAECs or HUVECs under basal or stimulated conditions was determined by a specific EIA assay kit. BAECs cultured in phenol-red-free M199 medium were washed with Hank’s balanced salt solution (HBSS) and incubated with 5 μm genistein for various time periods or with different concentrations of genistein for 30 min in phenol-red-free medium at 37 C. HUVECs were exposed to 5 μm genistein or vehicle for 30 min. In some experiments, cells were preincubated with the specific ER antagonist ICI, mRNA transcription inhibitor actinomycin D, or the protein synthesis inhibitor cycloheximide for 30 min before addition of genistein, 17β-estradiol, or vehicle. After incubation, the supernatant was rapidly aspirated ,and the intracellular cAMP extraction and quantification were performed according to the manufacturer’s instructions. Data were normalized to the protein concentration in samples as determined by a protein assay kit.
Adenylate cyclase (AC) assay
Membrane preparations isolated from BAECs exposed to various concentrations of genistein, 20 μm forskolin, or vehicle were tested for AC activity. Plasma membrane preparations were isolated by differential centrifugation. Briefly, cells were homogenized with a Dounce homogenizer in 20 mm HEPES, 0.25 m sucrose, 1 mm EDTA, 5 mm benzamidine, and a protease inhibitor cocktail (1:500). Membranes were collected by two steps of differential centrifugation (1,000 × g for 5 min at 4 C and 40,000 × g for 20 min at 4 C) (35), and the protein concentrations in the samples were determined. For AC assay, 30 μg of membrane protein was added to reaction buffer [50 mm Tris-HCl (pH 7.4), 5.0 mm MgCl2, 0.5 mm EDTA, 1 mm ATP, 0.1 μm GTP, 0.2 IU pyruvate kinase, 0.1U myokinase, and 2.5 mm phosphoenolpyruvate] (36) and incubated for 15 min at 37 C. The converted cAMP from ATP in the supernatant of the samples was determined with an EIA assay kit.
cAMP-specific phosphodiesterase (PDE) assay
BAECs were scraped into lysis buffer [20 mm HEPES (pH 7.4), 1 mm EDTA, 1 mm dithiothreitol, and 1:500 protease inhibitor cocktail], Dounce homogenized, and then sonicated for 30 sec on ice. Cells lysates were centrifuged (14,000 × g for 5 min at 4 C), and 30 μg protein of cell lysates was incubated in reaction buffer [20 mm HEPES (pH 7.4), 90 mm KCl, 5 mm MgCl2, 0.75 mm CaCl2, and 100 nm cAMP] in the presence of increasing concentrations of genistein, 0.2 mm IBMX, or vehicle for 30 min at 30 C (27). The reaction was terminated by the addition of 0.1 N HCl and centrifuged. The cAMP remaining in the supernatants was measured by an EIA kit. The PDE activity was determined as the amount of cAMP hydrolyzed during the reaction time.
PKA-specific kinase assay
BAECs treated with genistein, forskolin, or vehicle were scraped and collected in PBS supplemented with protease and phosphatase inhibitor cocktail. Cytoplasmic and nuclear proteins were harvested by sonication and centrifugation. The enzymatic activity of PKA in lysates was assessed by measuring phosphorylation of kemptide, a highly specific peptide substrate for PKA. Briefly, 25-μl reaction mixtures in PepTag PKA kinase assay buffer containing 10 μg of cell lysates, 2 μg fluorescent-labeled kemptide, and 1 μl of peptide protection solution were incubated for 10 min at room temperature. The reaction was terminated by boiling for 5 min, and then 1 μl of 80% glycerol was added to each sample. Phosphorylated kemptide was separated from unphosphorylated substrate on a 0.8% agarose gel by electrophoresis as described (37) and visualized under UV light using an AlphaImager imaging system (Alpha Innotech Co., San Leandro, CA). The images of the fluorescent gels were photographed, and the amount of substrate phosphorylation was determined by quantifying the fluorescence intensity of the peptide bands.
Western blot analysis
After an experimental treatment, BAECs were harvested by scraping in lysis buffer [20 mm Tris/HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm Na4P2O7, 1 mm β-glycerolphosphate, and 1 mm Na3VO4] supplemented with protease inhibitor cocktail (1:500) and phosphatase inhibitor cocktail I (1:100). The extracts were sonicated and centrifuged at 10,000 × g for 5 min. Protein levels were measured using a Bio-Rad assay kit. Detergent-extracted proteins were mixed with Laemmli sample buffer and heated for 5 min at 95 C, and equal amounts of cell lysate proteins (50 μg) were resolved on 10% SDS-PAGE gels. The gels were blotted onto nitrocellulose membranes, probed with rabbit phospho-CREB antibody overnight at 4 C, and incubated with secondary antibody conjugated to horseradish peroxidase for 1 h at room temperature. The immunoreactive proteins were detected by superSignal chemiluminescence. Membranes were stripped and reprobed with CREB antibody. The protein bands were digitally imaged for densitometric quantitation with a software program (Silk Scientific, Inc., Salt Lake City, UT). CREB phosphorylation was expressed relative to total CREB from the same membrane.
Transfection for plasmid cAMP response element (CRE)-luciferase reporter construct
A reporter plasmid containing multiple copies of a consensus CRE-binding sequence fused to a TATA-like promoter region upstream of the gene for firefly luciferase (BD Clontech, Palo Alto, CA) was used to monitor the cAMP-dependent signaling pathway. BAECs were grown in 24-well plates in M199 medium until 60–70% confluence. Before transfection, the medium was changed to 1% FBS. BAECs were cotransfected with 0.25 μg of pCRE-Luc vector and 5 ng of pRL reporter control plasmid per well by using Fugene-6 transfection reagent according to the manufacturer’s protocol. Transfection efficiencies were determined by cotransfecting the cells with a pcDNA 3.1/His/lacZ control vector (Invitrogen). After transfection, cells were incubated with complete medium containing 20% charcoal-stripped FBS for 24 h before being serum starved for 12 h. The transfected cells were then treated with genistein (5 μm), 17β-estradiol (10 nm), or vehicle for various time periods in phenol-red-free M199 medium. Cells were harvested in reporter lysis reagent. Luciferase activity, normalized to pRL activity in the cell extracts was determined by using the dual luciferase reporter assay system.
Evaluation of barrier function in vitro
Passage of avidin-FITC through BAEC monolayers, as an index of barrier function, was assessed essentially as previously described (38, 39). Cells cultured on fibronectin-coated polycarbonate filters were treated with genistein or vehicle in phenol-red-free medium containing 1% serum for 30 min at 37 C. In some experiments, cells were preincubated with the PKA inhibitors for 30 min before the addition of genistein. Afterward, thrombin (2 U/liter) and avidin-FITC (1 μm/liter) were added to the upper compartment of the Transwells in the continued presence of genistein or vehicle. Samples were taken from the bottom compartment at various time intervals, and the volume was adjusted by adding the same volume of medium. The avidin-FITC concentration in each sample was determined using a fluorometer plate reader (BMG Labtech. Inc., Durham, NC). Passage rates were expressed as nanogram per square centimeter per hour (39).
Statistical analysis
All data were subjected to a one-way ANOVA analysis using GraphPad Prism software (GraphPad Inc., San Diego, CA), and treatment differences were subjected to a Duncan’s multiple comparison test at the 5% probability. Data in each study were derived from at least three independent experiments and expressed as mean ± se.
Results
Genistein stimulates cAMP in vascular ECs
We first examined the effect of genistein on intracellular cAMP in BAECs. Cells were incubated with 5 μm genistein for 15 min to 6 h. Genistein caused a significant and prolonged elevation of intracellular cAMP in BAECs (Fig. 1A). The genistein-stimulated intracellular cAMP accumulation was maximal at 30 min after exposure to genistein and then decreased but was still significantly greater than control after 6 h of genistein incubation. Dose-response studies demonstrated that exposure of the cells to genistein for 30 min elevated intracellular cAMP levels by 105.9 ± 12.9 to 199.9 ± 24.1% with a maximal increase at 10 μm genistein (Fig. 1B). Genistein (5 μm) also increased intracellular cAMP by 129.8 ± 8.7% in HUVECs, a similar response as observed in BAECs (data not shown), suggesting that the effect of genistein on the cAMP signaling pathway is not species specific.
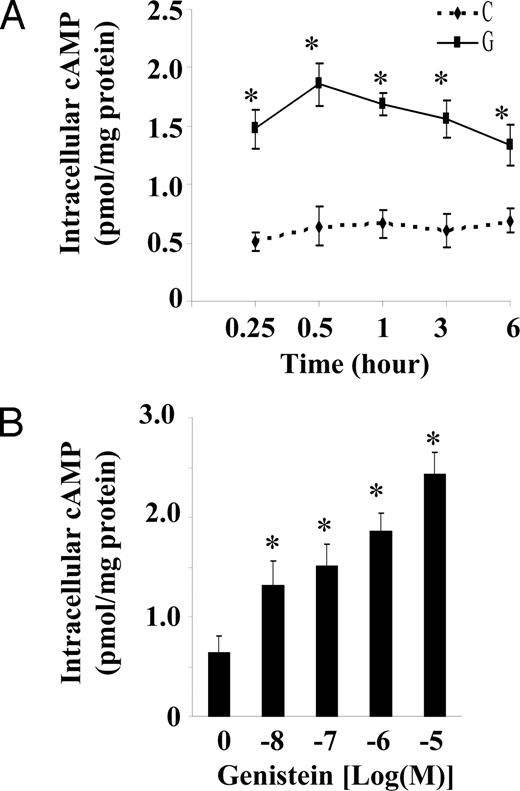
Genistein stimulates intracellular cAMP accumulation. Serum starved BAECs were incubated in HBSS buffer with 5 μm genistein (G) or vehicle (C) for various time periods (A) or with various concentrations of genistein (10 nm to 10 μm) for 30 min (B) at 37 C. Intracellular cAMP was extracted and measured by EIA and normalized to cellular protein. Data were expressed as mean ± se of four separate experiments determined in duplicate. *, P < 0.05 vs. vehicle-treated control.
ER- and transcription-independent effect of genistein on cAMP
Because genistein has weak estrogenic effects in some tissues by binding to ERs (1), we examined whether the genistein effect was mediated through ERs. The ER antagonist ICI caused no change in basal (data not shown) or genistein-induced cAMP (Fig. 2). The activity of ICI used in this study was validated through blocking the 17β-estradiol-induced eNOS activity (29). In addition, 10 nm 17β-estradiol, a concentration that has been shown to induce the maximal cellular response in vascular ECs (40) and further validated in our previous study (29), failed to stimulate cAMP from BAECs after 30 min of treatment (Fig. 2). These results suggest that the effect of genistein on cAMP in ECs is independent of estrogen signaling machinery. Next, we examined whether RNA or protein synthesis was required for the stimulatory effect of genistein on intracellular cAMP accumulation. Pretreatment of the cells with the mRNA transcription inhibitor actinomycin D or the protein synthesis inhibitor cycloheximide had no effect on basal (data not shown) or genistein-stimulated accumulation of cAMP (Fig. 2), but they inhibited RNA or protein synthesis by 93 and 82%, respectively (29, 41), using the same inhibitor concentration and duration of incubation as in our experimental studies, based on uridine and proline incorporation assay (41). These data suggest that genistein stimulates cAMP through nontranscriptional mechanisms, which is consistent with a very early time course of the genistein-stimulated cAMP increase (Fig. 1A).
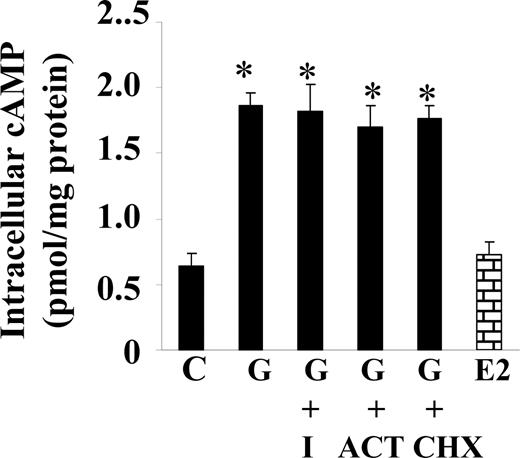
ER- and transcription-independent effect of genistein on cAMP. Serum-starved BAECs were incubated with or without the specific ER antagonist ICI 182,780 (I, 10 μm), the RNA synthesis inhibitor actinomycin D (ACT, 10 μm), or the protein synthesis inhibitor cycloheximide (CHX, 10 μm) in HBSS buffer for 30 min. Cells were then stimulated with genistein (G, 5 μm), 17β-estradiol (E2, 10 nm), or vehicle (C) in the continued presence or absence of the inhibitors for 30 min at 37 C. cAMP was extracted and measured as indicated in Fig. 1. The experiment was repeated three times in triplicate, and data were expressed as mean ± se. *, P < 0.05 vs. vehicle-treated control.
AC and cAMP-specific PDE activity
To explore whether genistein induces accumulation of cAMP through stimulation of cAMP production and/or inhibition of cAMP hydrolysis, the effect of genistein on AC and cAMP-specific PDE activity in BAECs was evaluated. Dose-response studies revealed that a concentration of genistein as low as 10 nm was able to stimulate substantial increases in AC activity in BAECs (Fig. 3). Full stimulation was achieved at 1 μm genistein, a change that was about half that achieved by exposure to forskolin, a powerful agonist of AC activity. Genistein also suppressed PDE activity of BAECs, but a significant inhibitory effect was achieved only at 10 μm genistein (51% decrease) (Fig. 4). As expected, the inhibitor IBMX was very potent in suppressing PDE activity (Fig. 4).
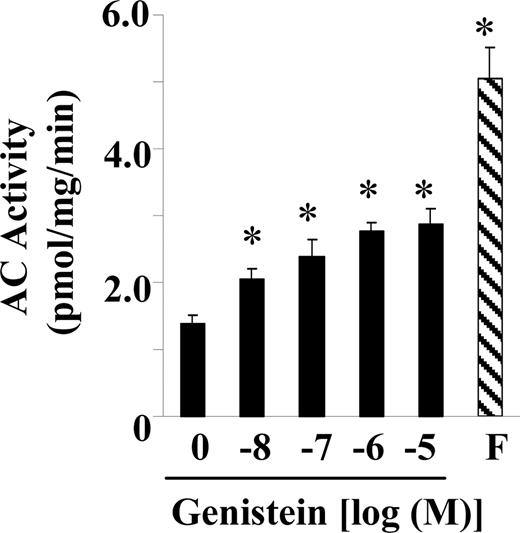
Genistein stimulates AC activity. Serum-starved BAECs were exposed to increasing concentrations of genistein, vehicle (C), or forskolin (F, 20 μm) for 15 min. Membrane preparations were isolated by differential centrifugation. The AC activity was measured as described in Materials and Methods. The experiment was repeated four times in triplicate, and data were expressed as mean ± se. *, P < 0.05 vs. vehicle-treated control.
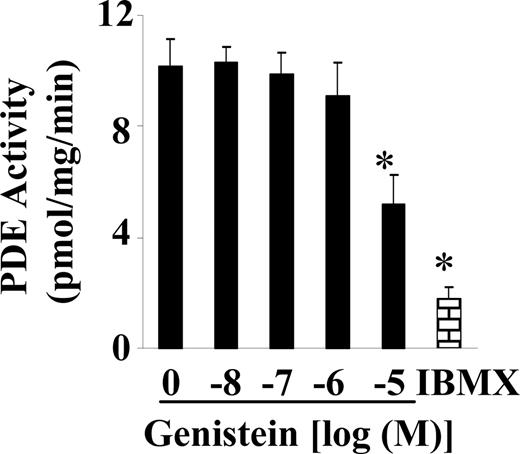
Effect of genistein on cAMP-specific PDE activity. Cell extracts of serum-starved BAECs were exposed to increasing concentrations of genistein or IBMX (0.2 mm) at 30 C for 30 min. cAMP in cell extracts was determined. PDE activity was expressed as the rate of cAMP hydrolyzation. The data were obtained from four separate experiments determined in triplicate and expressed as mean ± se. *, P < 0.05 vs. vehicle-treated control.
Activation of PKA
To investigate whether the elevation of cAMP by genistein activates downstream signals, we first assessed the activity of PKA in BAECs as measured by a fluorescent kemptide kinase assay (37). As shown in Fig. 5, genistein significantly stimulated PKA activity. Forskolin evoked a larger response, which is consistent with its greater stimulation of intracellular cAMP than genistein as observed in the present study.
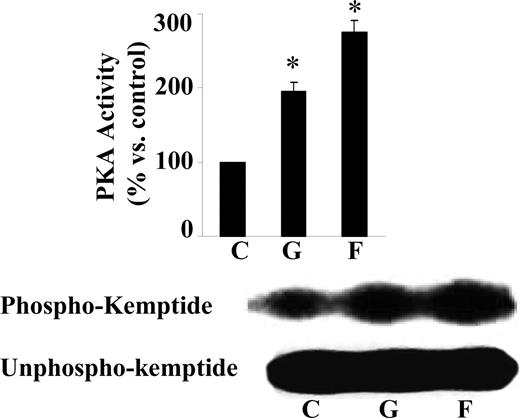
Genistein stimulates PKA activity. Serum-starved BAECs were stimulated with genistein (G, 5 μm), forskolin (F, 20 μm), or vehicle (C) in HBSS buffer for 15 min at 37 C. Cell extracts were incubated with the fluorescent-labeled PKA substrate kemptide in a kinase buffer for 15 min. Phosphorylated kemptide (upper panel) was separated from unphosphorylated kemptide (lower panel) by agarose gel electrophoresis and visualized under UV light. A representative photograph of the agarose gel used for the kemptide assay is shown on the left, and the quantification of the assay is on the right. The experiment was repeated three times with similar results, and data were expressed as mean ± se. *, P < 0.05 vs. vehicle-treated control.
CREB phosphorylation
To investigate whether activation of PKA by genistein induces CREB phosphorylation, we performed Western blot analysis using a specific phospho-CREB antibody that recognizes CREB only when phosphorylated at serine 133. As shown in Fig. 6A (upper panel and bar graph), incubation of BAECs with genistein (5 μm) for 30 min caused about a 2-fold increase in CREB phosphorylation. Pretreatment of cells with PKA inhibitors completely abolished the CREB phosphorylation induced by genistein (Fig. 6B, upper panel and bar graph), suggesting that PKA mediates the genistein-induced phosphorylation of CREB. The total level of CREB protein expression as detected with an antibody against CREB was not changed in this study (Fig. 6, A and B, lower panels), indicating the nontranscriptional mechanisms of genistein-induced CREB activation.
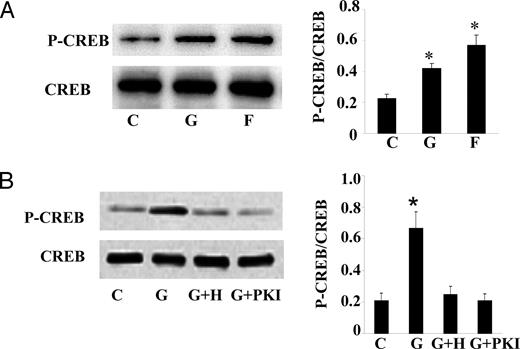
Genistein induces PKA-mediated CREB phosphorylation. A, Serum-starved BAECs were treated with genistein (G, 5 μm), forskolin (F, 20 μm), or vehicle (C) in HBSS buffer for 15 min at 37 C. B, BAECs were preincubated with H89 (H, 10 μm), PKI (2 μm), or vehicle (C) for 30 min before the addition of genistein (G, 5 μm) for 15 min. Phosphorylation of CREB (P-CREB) was detected by Western blot analysis using a phospho-specific CREB antibody (upper panel). The membrane was stripped and reprobed with a CREB antibody (lower panel). The specific bands were scanned and quantified, and the ratio of phosphorylated CREB to total CREB is shown on the right (bar graphs). The bar graphs represent three independent experiments. *, P < 0.05 vs. vehicle-treated control.
Activation of CRE-luciferase activity
Because elevation of cAMP and activation of CREB mediates cAMP-regulated genes, we then tested whether this change in cAMP and the activation of CREB by genistein is sufficient to affect cAMP-regulated gene transcription. BAECs were transfected with a cAMP-responsive gene construct (pCRE-Luc) containing a TATA-like promoter linked to a luciferase reporter gene. The transfection efficiency is 34.8 ± 7.7% based on β-galactidose cotransfection assay. The transfected cells were incubated with 5 μm genistein for 1–24 h. The basal luciferase activity was 4560 ± 513 relative luciferase units/mg protein. As indicated in Fig. 7, genistein significantly increased luciferase activity as early as 2 h of incubation, which is consistent with an early time course of the genistein-stimulated cAMP accumulation. Maximal increase (∼2.5-fold) in luciferase activity was observed between 8 and 16 h after exposure to genistein in BAECs. In contrast, 17β-estradiol that did not stimulate the elevation of intracellular cAMP also failed to increase CRE-driven luciferase expression. This result further demonstrated that that genistein, but not estrogen, targets the cAMP/PKA pathway.
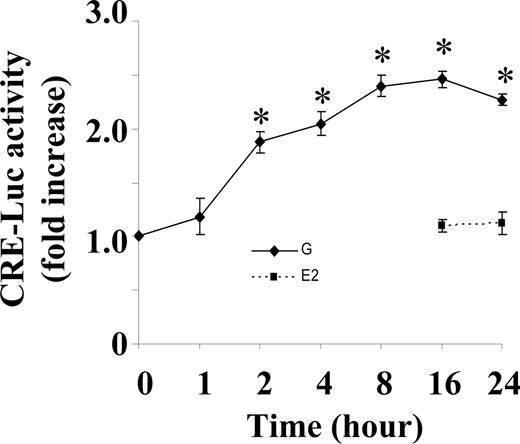
Genistein induces CRE-driven luciferase activity. BAECs were cotransfected with reporter plasmid pCRE-Luc and PRL-CMV plasmid. After serum starvation for 12 h, cells were incubated with genistein (G, 5 μm), 17β-estradiol (E2, 10 nm), or vehicle for time periods as indicated on graph. Luciferase activity was detected with the dual luciferase assay system and normalized to the control plasmids in the cell extracts. Values are expressed as fold increase over vehicle derived from three separate experiments performed in triplicate. *, P < 0.05 vs. vehicle-treated control.
Effect of genistein on in vitro endothelial barrier function
Because elevation of intracellular cAMP level improves endothelial barrier function (42–47), we tested whether the elevation of cAMP by genistein is sufficient to play such a role by measuring the effect of genistein on endothelial permeability, a hallmark of atherosclerotic development. To do this, BAECs were grown to confluence on fibronectin-coated porous filters, and the passage of avidin-FITC (∼64 kDa) through the endothelial monolayer was measured as an index of barrier function. Pretreatment of BAECs with 5 μm genistein for 30 min significantly inhibited the thrombin-induced increase in avidin-FITC passage (Fig. 8A). This genistein pretreatment inhibited thrombin-induced avidin-FITC passage during a 1-h incubation period by 59.3 ± 1.7 to 76.0 ± 1.3%, depending on the genistein concentrations used (Fig. 8B). Preincubation of the cells with Rp-cAMP, a cAMP antagonist, or PKI, the cell-permeable specific PKA inhibitor peptide, abolished the inhibitory effect of genistein on thrombin-induced avidin-FITC passage (Fig. 8C), whereas the baseline was not altered by these agents, indicating that genistein reduced thrombin-induced avidin-FITC passage by cAMP-dependent activation of PKA.
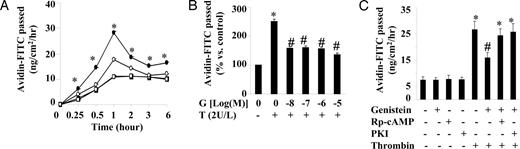
Effect of genistein on barrier function of BAECs. A, BAEC monolayers were preincubated with 5 μm genistein (○, □) or vehicle (•, ▪) for 30 min at 37 C. Cells were then treated with (○, •) or without (□, ▪) 2 U/liter thrombin for various times in the continued presence of genistein. B, BAEC monolayers were preincubated with various concentrations of genistein (G) or vehicle for 30 min. Cells were then treated with or without 2 U/liter thrombin (T) for 1 h in the continued presence of genistein. C, BAEC monolayers were preincubated with Rp-cAMP (0.5 mm), PKI (2 μm), or vehicle for 30 min before the addition of genistein (5 μm) for another 30 min. Cells were then treated with or without 2 U/liter thrombin for 1 h in the continued presence of genistein and the inhibitors. Avidin-FITC passage was measured as described in Materials and Methods. Data were expressed as mean ± se of four different experiments in duplicate. *, P < 0.05 vs. cells treated with vehicle; #, P < 0.05 vs. cells treated with thrombin alone.
Discussion
Genistein has various biological functions including beneficial effects on the vascular system. However, the cellular and molecular mechanisms of these effects are still unclear. In the present study, we demonstrated that genistein targets the cAMP signaling pathway in vascular ECs. Genistein directly stimulates AC through mechanisms that are independent of mRNA or protein synthesis, leading to a marked increase in intracellular cAMP levels. The induced intracellular accumulation of cAMP by genistein is not mediated by ERs. Furthermore, the elevation of cAMP stimulates PKA activity, which subsequently activates CREB by phosphorylation and stimulates the cAMP-regulated gene transcription. As a result, increases in intracellular cAMP by genistein protect the thrombin-induced endothelial barrier dysfunction, an effect mediated by PKA. Our findings define a novel signaling pathway involved in genistein action in vascular ECs and provide an explanation for these previously reported actions of genistein in vascular tissues.
The effect of genistein was rapid, with maximal cAMP accumulation in BAECs at 30 min incubation. A similar rapid increase in cAMP accumulation was seen in HUVECs, showing that the effect was not species specific. The genistein-stimulated cAMP is significantly decreased from 0.5 to 6 h of incubation, which was completely blocked by inhibition of PDE activity (data not shown), suggesting that genistein may not enhance cAMP metabolism. Such a time-course response pattern is also observed in steroid-induced cAMP production in breast cancer and uterine cells (35). Concentration as low as 10 nm significantly elevated intracellular cAMP, although the maximal effect was achieved with 10 μm genistein. The serum concentrations of genistein are reported to be 0.3–0.6 μm in Japanese men (48). However, serum genistein levels in humans consuming three meals per day containing soy milk or a single soy meal can reach 4.6 and 4.1 μm, respectively (49, 50). Our results therefore have relevance for humans because the concentrations of genistein used in the present study (10 nm-10 μm) are overlapped with those physiologically achieved in the plasma of individuals consuming soy products.
AC and cAMP-specific PDE are the primary enzymes responsible for regulation of the intracellular level of cAMP. Genistein at lower concentrations (10 nm to 1 μm) had no significant effect on PDE activity in cell extracts, suggesting that elevated cAMP by genistein at these doses may be primarily attributable to activated AC. Our recent preliminary study demonstrated that genistein directly acts on plasma membranes to activate AC in vascular ECs (data not shown). Therefore, genistein may require an intact cell membrane for its action on the cAMP system including PDE. In the present study, cell extracts instead of intact cells were treated with genistein for performing PDE activity assay as described (27). Therefore, the disruption of plasma membranes in our assay system could contribute to the lack of significant effect of genistein at lower doses on PDE activity, an aspect that remains to be determined. The elevated cAMP by genistein at a higher concentration (10 μm) may be partially due to suppression of PDE activity, as observed in this study.
The mechanism by which genistein inhibited PDE is unclear. Genistein is an inhibitor of protein tyrosine kinase (TRK) (51) and is often used to study tyrosine kinase-mediated intracellular signaling events. Recent studies have shown that PDE activity may be directly or indirectly regulated by TRK (52). However, genistein at 10 μm had no effect on basal or agonist-stimulated TRK activity in our previous study (29). TRK inhibition was obtained only at a higher concentration (100 μm), consistent with previous findings (53, 54). Therefore, we believe that TRK does not play a role in a genistein effect on PDE activity. Although our studies observed an increase in AC activity stimulated by genistein, leading to cAMP production, the mechanism by which genistein enhances AC is unclear. AC is positively regulated by plasma membrane-associated Gs-GTP binding proteins (Gαs) to increase intracellular cAMP production (55). Therefore, genistein may, at least in part, directly act on the cell surface to facilitate cAMP production involving a receptor, Gαs, and AC (56). In this aspect, genistein may act through a nongenomic mechanism. Indeed, the rapidity of onset of the genistein action is against a possible genomic effect. In addition, inhibition of gene transcription and protein translation has no effect on genistein-stimulated cAMP, strongly supporting the concept of nongenomic mechanisms underlying this activation. However, whether genistein activates AC through a plasma membrane receptor-mediated mechanism involving Gαs protein requires further investigation.
Recent studies provide evidence that the nongenomic effects of estrogen in ECs are mediated by cell surface ERs (57–59). Genistein has weak estrogenic effects by binding to ERs (60). However, estrogen had no significant effect on endothelial cAMP production. In addition, blocking ERs with ICI did not inhibit the cellular response to genistein. It is unlikely that the inability of this agent to block the effect of genistein on endothelial cAMP production is due to a lack of efficacy because we previously reported that, at the same concentration used, ICI completely abolished the 17β-estradiol-induced eNOS activity (29). These results suggest that the known ERs are not involved in this genistein action in vascular ECs. Recently it has been shown that genistein may enhance cAMP accumulation by modifying α-adrenoceptors in rat brain (61). In addition, there is evidence indicating that some xenoestrogens act by binding at the plasma membrane catecholamine receptors (62, 63). Furthermore, a recent report suggests the presence of membrane binding sites for genistein in human vascular system (64). It is therefore tempting to speculate that genistein may activate the cAMP signaling by interaction with membrane binding sites, possibly the catecholamine receptors that can modulate intracellular cAMP in ECs (65). Characterizing the possible plasma membrane-initiated, Gs-protein mediated effect of genistein is an ongoing area of investigation in the laboratory.
One important downstream target of cAMP signaling is CREB, a nuclear transcription factor that binds to the CRE to activate the transcription of target genes. Phosphorylation of CREB at serine 133 residue is necessary for transcriptional activation of this protein. Our findings indicate that genistein has a significant stimulatory effect on CREB, mediated by PKA in response to elevated cAMP. These observations are important because they suggest that genistein, by increasing cAMP and subsequent activating CREB within target cells, may modulate expression of cAMP-regulated genes and thereby possibly influence other cAMP-mediated biological activities. In the present study, by using a CRE-regulated luciferase reporter construct, we demonstrated for the first time that genistein evoked cAMP-regulated gene expression in ECs. Functional CRE has been found in a few endothelial genes, including thrombomodulin, tissue-type plasminogen activator, and eNOS (66–68), which have important vasoprotective effects by inhibiting coagulation and enhancing fibrinolysis and vasorelaxation, respectively. Given the present results and those of previous studies, we speculate that genistein can act via a cAMP cascade to regulate expression of these cAMP-regulated genes and thereby likely play a wide range of vasoprotective roles. Indeed, a recent study suggests that genistein, independent of ERs, enhances eNOS expression (Liu, D., unpublished observation). However, whether the up-regulation of eNOS by genistein is mediated through the cAMP/PKA pathway remains to be determined.
cAMP is a central signaling molecule in a variety of cellular systems and plays an important role in maintaining normal vascular function. Activation of the cAMP/PKA pathway directly phosphorylates multiple residues of eNOS, leading to the activation of eNOS and nitric oxide production (36, 69). In addition, the presence of functional CRE sites within the human eNOS promoter (68) suggests that the eNOS expression may be directly regulated by CREB. cAMP also inhibits injury-induced cell growth of the arterial wall thereby reducing the formation of the neointima (31). In addition, activation of cAMP/PKA signaling inhibits vascular inflammation by depressing the adhesion of leukocytes to ECs (32), possibly through PKA-mediated CREB phosphorylation (70). Furthermore, elevation of intracellular cAMP concentration improves barrier function by decreasing intercellular gap formation and endothelial permeability that result from various inflammatory mediators such as thrombin (42–47). This effect is likely mediated through a PKA-dependent signaling mechanism (34, 46). All these events are implicated in various pathological conditions such as the development of arteriosclerosis, suggesting that the cAMP elevating agent genistein may retard the process of some chronic vascular diseases by targeting the cellular cAMP/PKA pathway.
Indeed, it has been shown that genistein potently inhibits platelet-derived growth factor-induced proliferation, migration, and extracellular matrix synthesis in human aortic smooth muscle cells (4), suggesting a cardioprotective effect on vascular dysfunction. A recent study (26) also suggests that cAMP-dependent mechanisms may be involved in genistein-induced vascular relaxation. Genistein also inhibits the interaction of leukocytes and vascular ECs (71, 72). We recently demonstrated that genistein activates eNOS and NO production through the cAMP/PKA-mediated, nongenomic mechanisms (29). In the present study, by using ECs grown on porous filters, an established in vitro model for study of endothelial permeability, we found that genistein reduced endothelial barrier dysfunction induced by thrombin. This genistein effect was associated with an elevation of intracellular cAMP concentration and blocked by the inhibition of PKA. Because the development of atherosclerosis is always associated with increased permeability of vascular endothelial monolayers for macromolecules such as low-density lipoprotein (73). This result suggests a novel cAMP/PKA-mediated protective effect of genistein on vascular function. Collectively, many of these genistein effects are either mediated through cAMP signaling or are compatible with the declared action of cAMP, suggesting that the effect of genistein on the cellular cAMP/PKA cascade may represent a central mechanism and play a key role in a wide range of vascular protective effects. Our findings therefore may provide an explanation for these previously reported versatile actions of genistein observed in animal and human studies. In the meantime, on the basis of the results from this study, it is tempting to speculate that genistein may be a novel vasoactive agent for the prevention of cardiovascular diseases, an aspect that needs further investigation.
Acknowledgments
The authors thank Kathy Reynolds for excellent technical assistance. We are grateful to Dr. Joseph Dillon (Department of Internal Medicine, The University of Iowa) for helpful discussions.
This work was supported by A Support Program Innovative Research Strategies Seed Grant from Virginia Polytechnic Institute and State University (to D.L.).
Abbreviations
- AC,
Adenylate cyclase;
- BAEC,
bovine aortic endothelial cell;
- CRE,
cAMP response element;
- CREB,
cAMP response element-binding protein;
- EC,
endothelial cell;
- EIA,
enzyme immunoassay;
- ER,
estrogen receptor;
- eNOS,
endothelial nitric oxide synthase;
- FBS,
fetal bovine serum;
- FITC,
fluorescein;
- Gαs,
Gs-GTP binding protein;
- HBSS,
Hank’s balanced salt solution;
- HUVEC,
human umbilical vein endothelial cell;
- IBMX,
isobutylmethylxanthine;
- ICI,
ICI 182,780;
- PDE,
phosphodiesterase;
- PKA,
protein kinase A;
- PKI,
PKA inhibitory peptide;
- TRK,
tyrosine kinase.
References
van der
van Nieuw

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}