





Abstract
1,25-Dihydroxyvitamin D3 (1,25-(OH)2D3) is an immune modulator that prevents experimental autoimmune diseases. Receptors for 1,25-(OH)2D3 are present in pancreatic β-cells, the target of an autoimmune assault in nonobese diabetic (NOD) mice. The aim of this study was to investigate the in vivo and in vitro effects of 1,25-(OH)2D3 on β-cell gene expression and death and correlate these findings to in vivo diabetes development in NOD mice. When female NOD mice were treated with 1,25-(OH)2D3 (5 μg/kg per 2 d), there was a decrease in islet cytokine and chemokine expression, which was accompanied by less insulitis. Complementing these findings, we observed that exposure to 1,25-(OH)2D3 in three cell systems INS-1E cell line, fluorescence-activated cell sorting purified rat β-cells, and NOD-severe combined immunodeficient islets) suppressed IP-10 and IL-15 expression in the β-cell itself but did not prevent cytokine-induced β-cell death. This 1,25-(OH)2D3-induced inhibition of chemokine expression in β-cells was associated with a decreased diabetes incidence in some treatment windows targeting early insulitis. Thus, although a short and early intervention with 1,25-(OH)2D3 (3–14 wk of age) reduced diabetes incidence (35 vs. 58%, P ≤ 0.05), a late intervention (from 14 wk of age, when insulitis is present) failed to prevent disease. Of note, only early and long-term treatment (3–28 wk of age) prevented disease to a major extent (more than 30% decrease in diabetes incidence). We conclude that 1,25-(OH)2D3 monotherapy is most effective in preventing diabetes in NOD mice when applied early. This beneficial effect of 1,25-(OH)2D3 is associated with decreased chemokine and cytokine expression by the pancreatic islets.
THE PREVALENCE OF type 1 diabetes (affecting 5.3–41.6 per 100,000 subjects in Europe and the United States) is estimated to rise each year by 3–5%, with a larger increase in some central and eastern European countries (1, 2). Against this background, prediction, prevention, and cure of type 1 diabetes are major health care issues. An increasing amount of data points toward a close relationship between type 1 diabetes and vitamin D. Thus, there is a higher prevalence of type 1 diabetes in regions with low vitamin D supplies, whereas dietary supplementation with vitamin D during infancy reduces risk of developing type 1 diabetes in genetically at-risk subjects later in life (3–5). In nonobese diabetic (NOD) mice, the hormonally active form of vitamin D, 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3] prevents diabetes when given throughout their life span, whereas vitamin D deficiency in early life accelerates the onset and increases the incidence of the disease. This is probably related to deficient macrophage function and/or higher β-cell vulnerability (6–8).
1,25-(OH)2D3 exerts its function by engaging with a specific nuclear receptor [vitamin D receptor (VDR)] present in most tissues and cells, including lymphocytes, macrophages, and pancreatic β-cells (9, 10). Of note, the vitamin D-dependent calcium binding protein (calbindin-D28K), which is thought to serve as a cytosolic calcium buffer, is present in β-cells (11). Besides its well-known effects on calcium and bone metabolism, 1,25-(OH)2D3 inhibits cell growth, induces cell differentiation, and modulates the immune system (12). In NOD mice, 1,25-(OH)2D3 prevents insulitis and diabetes through modulation of the immune system (6, 7). 1,25-(OH)2D3 inhibits antigen-induced T-cell proliferation as well as proinflammation-related cytokine production, enhances T-cell sensitivity to apoptosis-inducing signals, reduces antigen-presenting and costimulating properties of dendritic cells, and induces pancreatic lymph-node CD4+CD25+ T regulator cells (13, 14). Recently several studies suggested that 1,25-(OH)2D3 has a direct protective effect on β-cells, contributing to prevention of diabetes in NOD mice. This issue remains, however, controversial. Whereas normal levels of 1,25-(OH)2D3 are necessary for secretion and de novo biosynthesis of insulin by β-cells (15–17), studies on the effects of 1,25-(OH)2D3 treatment on β-cell vulnerability to immune mediators provided conflicting results. Whereas Mauricio et al. (18) did not observe a protective effect of 1,25-(OH)2D3 against cytokine-induced β-cell death in vitro, Sandler et al. (19) and Riachy et al. (20, 21) described clear protection.
The aim of the present study was to investigate whether in vivo and in vitro effects of 1,25-(OH)2D3 on β-cell gene expression and death were associated with in vivo protection against diabetes in NOD mice.
Materials and Methods
Animals
NOD mice, originally obtained from Professor C. Y. Wu (Department of Endocrinology, Peking Union Medical College Hospital, Beijing, China), were housed and inbred in our animal facility of the Katholieke Universiteit Leuven since 1989. Housing of NOD mice occurred under semibarrier conditions, and animals were fed sterile chow and water ad libitum. NOD mice were screened for the onset of diabetes by evaluating glucose levels in urine (Clinistix; Bayer Diagnostics, Tarrytown, NY) and venous blood (Glucocard; Menarini, Florence, Italy). The cumulative incidence of diabetes of NOD mice in our breeding stock was 85% in females and 61% in males by 28 wk at the time of the present study. Mice were diagnosed as diabetic when having positive glucosuria and two consecutive blood glucose measurements exceeding 200 mg/dl. NOD/LtSz-scid/scid [NOD-severe combined immunodeficient (SCID)] mice were bred from stocks purchased from the Jackson Laboratory (Bar Harbor, ME), whereas BALB/c mice were purchased from Harlan Nederland (Horst, The Netherlands). Wistar male WI rats were purchased from Charles River Belgium (Brussels, Belgium) and maintained in the animal facility of the Université Libre de Bruxelles. Animals were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all experimental procedures were approved and performed in accordance with the Ethics Committees of the Katholieke Universiteit Leuven (Leuven, Belgium) and the Université Libre de Bruxelles (Brussels, Belgium).
Cell culture
The insulin-producing insulinoma (INS)-1E cell clone was kindly provided by Prof. C. Wollheim (Center Medical Universitaire, Geneva, Switzerland) and represents an extremely stable and valuable β-cell model as reflected by a constant doubling time and balanced glucose oxidation/use (Ortis, F., J. Rasschaert, and D. L. Eizirik, unpublished observations) (22). Cells (i.e. passages 60–70) were seeded (250,000 cells in 35 mm dishes) in RPMI 1640 medium (with Glutamax-I) supplemented with 10% heat-inactivated fetal calf serum, 10 mm HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mm sodium pyruvate, and 50 μm 2-mercaptoethanol (23, 24). The next day cells were incubated with vehicle (ethanol) or 10−8m 1,25-(OH)2D3 and 24 h later exposed to IL-1β and/or interferon (IFN)-γ for 8 h for mRNA extraction (see below) or 24 h for assessment of islet viability (see below). Recombinant human IL-1β (10 U/ml; 38 IU/ng) was a kind gift from Dr. C. W. Reynolds (National Cancer Institute, Bethesda, MD) and mouse IFNγ (100 U/ml; 10 IU/ng) was purchased from Invitrogen (Carlsbad, CA). The choice of cytokine concentrations was determined by a dose-response curve as the lowest concentration giving the maximal apoptosis rate after 24 h (25). 1,25-(OH)2D3 was obtained from J. P. van de Velde (Duphar, Weesp, The Netherlands). 1,25-(OH)2D3 was added to the culture medium at a final concentration of 10−8m 24 h before induction of cytokine-mediated cell death and refreshed every other day.
Islet isolation and culture
Pancreatic islets were isolated from Wistar rats (10 wk old) and NOD (4, 8, and 10 wk old) and NOD-SCID mice (10 wk old) by collagenase digestion as previously described (26). Islets were directly frozen in liquid nitrogen and stored at −80 C (for mRNA extraction, see below) or cultured as whole islets (for assessment of islet viability, see below). Unless otherwise indicated, islet cells were cultured free floating in RPMI 1640 medium containing 10% heat-inactivated fetal calf serum, 2 mm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin and maintained at 37 C in a humidified incubator (95% air-5% CO2).
Purification of β-cells by autofluorescence-activated cell sorting (FACS)
Rat islet β-cells were purified by FACS as described (27). These preparations contain single β-cells that are 90–95% pure (Rasschaert, J., L. Ladrière and D. L. Eizirik, unpublished data) (28). Purified β-cells were cultured single and attached to polylysine-coated microtiter dishes at 37 C in Ham’s F-10 medium (Invitrogen) as previously described (28). After overnight culture, primary β-cells were incubated for 24 h with vehicle (ethanol) or 10−8m 1,25-(OH)2D3 and thereafter exposed for 48 h to IL-1β (50 U/ml) and/or IFNγ (1000 U/ml) for assessment of cell viability (see below). The choice of cytokine concentrations was based on previous studies by our group (29, 30).
Islet viability
Percentages of viable, apoptotic, and necrotic cells were assessed in INS-1E cells [in vitro treated with or without 1,25-(OH)2D3 at concentration of 10−8m, 6000 cells/condition], rat FACS-purified β-cells [in vitro treated with or without 1,25-(OH)2D3 at concentration of 10−8m, 10,000 cells/condition], and NOD-SCID islets [in vivo treated with or without 1,25-(OH)2D3 at a dose of 5 μg/kg per 2 d from weaning till 10 wk of age, 15 islets/condition]. Briefly, single cells or whole islets were incubated for 15 min with the DNA binding dyes Hoechst (HO) 342 (20 μg/ml) and propidium iodide (10 μg/ml) and the cells examined on an inverted fluorescence microscopy as previously described for rodent and human β-cells (31–33). In each well a minimum of 500 cells were counted by two independent individuals, one of them unaware of sample identity. For whole islets, discrimination between necrosis and apoptosis is difficult due to superposition between cells; therefore, an approximate estimate of the percentage of living/dead cells was performed by two observers, one of them unaware of sample identity, as described (33).
RNA isolation, reverse transcription, and quantitative PCR
Total RNA was extracted from INS-1E cells and whole islets using the High pure RNA extraction kit (Roche Diagnostics Corp., Indianapolis, IN). Either 0.5 or 1 μg RNA was reverse transcribed using 100 U Superscript II reverse transcriptase (Invitrogen) at 42 C for 80 min in the presence of 5 μm oligodT16. Real-time PCR was performed using the 7700 SDS (Applied Biosystems, Foster City, CA) and the TaqMan system, as described previously (34–36). Primers and probes for calbindin-D28K, IL-1, IL-15, IFNγ, monocyte chemoattractant protein (MCP)-1 (CCL-2), macrophage inflammatory protein (MIP)-3α (CCL-20), interferon inducible protein (IP)-10 (CXCL-10), VDR, and β-actin as housekeeping gene were as described (34–37). Cytokines as well as 1,25-(OH)2D3 did not modify β-actin expression (data not shown).
In vivo experimental design
For in vivo administration, 1,25-(OH)2D3 was suspended in peanut oil and administered at a dose of 5 μg/kg ip every other day. Control mice received the treatment vehicle only (50 μl). For in vivo experiments dealing with insulitis development, islet viability and mRNA extraction studies, NOD and NOD-SCID mice were treated from weaning until the end of the follow-up period (10 wk of age).
For in vivo experiments dealing with prevention of diabetes, NOD mice were randomly assigned to the control group or three treatment groups: treatment from 3 to 14 wk of age, 14–28 wk of age, or 3–28 wk of age. All mice were followed up until 28 wk of age.
Calcium and bone parameters
At the end of the in vivo experiments, blood was collected by heart puncture, and a femur was removed. Serum and femur were stored at −20 C until determinations were performed. Calcium content of the femur, measured on HCl dissolved bone ashes, and the serum was measured using a photocolorimetric method (Sigma-Aldrich, St. Louis, MO). Osteocalcin was measured by an in-house RIA as described previously (38).
Histology and insulin determination
Percentage of insulitis and insulin positivity were assessed by histological screening of pancreatic glands of at least four animals per group, snap frozen in 2-methyl-butane 99+% (ACROS Organics, Geel, Belgium), and chilled in liquid nitrogen, taken from experimental NOD mice at 4, 8, and 10 wk of age. Two independent experiments were performed to establish data. Five-micrometer cryostat sections were stained with standard Mayers’ hematoxylin and eosin and scored for immune cell infiltration by light microscopy (6). The examiner was unaware of the origin of the samples. A mean of 10 or more islets per pancreatic sample were scored as follows for islet infiltration: 0, no infiltration; 1, periinsulitis; 2, islets with lymphocyte infiltration in less than 50% of the area; 3, islets with lymphocyte infiltration in more than 50% of the area; and 4, islets completely destroyed. The remaining half of each pancreas was used for insulin content determination as described (6).
Statistics
Values are expressed as means ± sem. Statistical analysis of the in vitro results was performed using ANOVA and post hoc Student’s t test. For comparing the in vivo insulitis and diabetes incidence, Kaplan-Meier survival curves, log-rank test, and χ2 test were used. Statistical analysis was performed with NCSS 2000 software (Kaysville, UT).
Results
Effects of in vivo treatment with 1,25-(OH)2D3 in NOD mice on insulitis and islet gene expression during insulitis development
To evaluate possible linkage between evolution of insulitis and gene expression in the islets of NOD mice, female NOD mice were treated with 1,25-(OH)2D3 (5 μg/kg per 2 d) as described above. Islets from these mice were studied before the onset of diabetes at 4, 8, and 10 wk of age. As shown in Fig. 1, administration of 1,25-(OH)2D3 had a clear reducing effect on overall islet infiltration as well as the severity of insulitis, as determined by the insulitis scores at 10 wk of age (mean score of 0.55 ± 0.42 vs. 2.06 ± 0.54 in pancreases of vehicle-treated NOD mice, P ≤ 0.01) (Table 1). This preservation of islet function was translated in a higher insulin content in the pancreases of 1,25-(OH)2D3-treated NOD mice at 10 wk of age (17.45 ± 19.17 vs. 4.17 ± 3.06 pmol/mg tissue in vehicle-treated NOD mice, P ≤ 0.001) (Table 1).
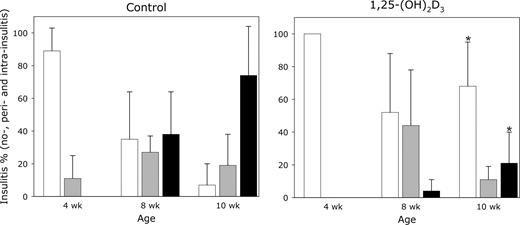
Effect of in vivo treatment with 1,25-(OH)2D3 on severity of islet infiltration in pancreases of diabetes-prone NOD mice. NOD mice were treated with either vehicle or 1,25-(OH)2D3 by ip injection every other day from weaning until 4, 8, and 10 wk of age (n = 8 animals). Each pancreas was subjected to a blinded analysis for all islets (mean of 10 or more islets per pancreatic sample). Bars indicated the percentage of islet infiltration: white bars, no insulitis; gray bars, periinsulitis; black bars, intrainsulitis. *, P ≤ 0.05 vs. age-related vehicle-treated NOD mice.
Effect of in vivo treatment with 1,25-(OH)2D3 on insulitis score and insulin content in pancreases of diabetes-prone NOD mice
| Mean insulitis score | P value | Insulin content (pmol/mg pancreas) | P value | |
|---|---|---|---|---|
| Control (wk) | ||||
| 4 | 0.11 ± 0.14 | 28.14 ± 10.13 | ||
| 8 | 1.18 ± 0.48 | 7.22 ± 7.84 | ||
| 10 | 2.06 ± 0.54 | 4.17 ± 3.06 | ||
| 1,25-(OH)2D3 (wk) | ||||
| 4 | 0.00 ± 0.00 | NS | 30.41 ± 28.99 | NS |
| 8 | 0.53 ± 0.39 | NS | 35.53 ± 22.23 | ≤0.05 |
| 10 | 0.55 ± 0.42 | ≤0.01 | 17.45 ± 19.17 | ≤0.001 |
| Mean insulitis score | P value | Insulin content (pmol/mg pancreas) | P value | |
|---|---|---|---|---|
| Control (wk) | ||||
| 4 | 0.11 ± 0.14 | 28.14 ± 10.13 | ||
| 8 | 1.18 ± 0.48 | 7.22 ± 7.84 | ||
| 10 | 2.06 ± 0.54 | 4.17 ± 3.06 | ||
| 1,25-(OH)2D3 (wk) | ||||
| 4 | 0.00 ± 0.00 | NS | 30.41 ± 28.99 | NS |
| 8 | 0.53 ± 0.39 | NS | 35.53 ± 22.23 | ≤0.05 |
| 10 | 0.55 ± 0.42 | ≤0.01 | 17.45 ± 19.17 | ≤0.001 |
NOD mice were treated either with vehicle or 1,25-(OH)2D3 by ip injection every other day from weaning until 4, 8, and 10 wk of age (n = 8 animals). Each pancreas was subjected to a blinded analysis for all islets (a mean of ≥10 islets per pancreatic sample). Scoring was done as described in Materials and Methods. Pancreatic insulin content was determined by ELISA.
NS, Not significant.
Effect of in vivo treatment with 1,25-(OH)2D3 on insulitis score and insulin content in pancreases of diabetes-prone NOD mice
| Mean insulitis score | P value | Insulin content (pmol/mg pancreas) | P value | |
|---|---|---|---|---|
| Control (wk) | ||||
| 4 | 0.11 ± 0.14 | 28.14 ± 10.13 | ||
| 8 | 1.18 ± 0.48 | 7.22 ± 7.84 | ||
| 10 | 2.06 ± 0.54 | 4.17 ± 3.06 | ||
| 1,25-(OH)2D3 (wk) | ||||
| 4 | 0.00 ± 0.00 | NS | 30.41 ± 28.99 | NS |
| 8 | 0.53 ± 0.39 | NS | 35.53 ± 22.23 | ≤0.05 |
| 10 | 0.55 ± 0.42 | ≤0.01 | 17.45 ± 19.17 | ≤0.001 |
| Mean insulitis score | P value | Insulin content (pmol/mg pancreas) | P value | |
|---|---|---|---|---|
| Control (wk) | ||||
| 4 | 0.11 ± 0.14 | 28.14 ± 10.13 | ||
| 8 | 1.18 ± 0.48 | 7.22 ± 7.84 | ||
| 10 | 2.06 ± 0.54 | 4.17 ± 3.06 | ||
| 1,25-(OH)2D3 (wk) | ||||
| 4 | 0.00 ± 0.00 | NS | 30.41 ± 28.99 | NS |
| 8 | 0.53 ± 0.39 | NS | 35.53 ± 22.23 | ≤0.05 |
| 10 | 0.55 ± 0.42 | ≤0.01 | 17.45 ± 19.17 | ≤0.001 |
NOD mice were treated either with vehicle or 1,25-(OH)2D3 by ip injection every other day from weaning until 4, 8, and 10 wk of age (n = 8 animals). Each pancreas was subjected to a blinded analysis for all islets (a mean of ≥10 islets per pancreatic sample). Scoring was done as described in Materials and Methods. Pancreatic insulin content was determined by ELISA.
NS, Not significant.
To test whether in vivo treatment with 1,25-(OH)2D3 could alter the inflammatory and chemoattractive status of the pancreatic islets, we performed real-time PCR on isolated islets from prediabetic female NOD mice (4–8 and 10 wk of age). As an external control, islets from BALB/c mice were analyzed. As shown in Fig. 2, mRNA levels of all genes tested were clearly linked to the course of insulitis in the NOD mouse. Note that for some chemokines, expression was found only from 10 wk onward, when insulitis was already present in most mice. Treatment with 1,25-(OH)2D3 decreased more than 2-fold the expression of IL-1β locally in the islets of NOD mice at 8 and 10 wk of age (P ≤ 0.05 vs. expression in age-related control NOD islets) (Fig. 2A). Moreover, treatment with 1,25-(OH)2D3 clearly reduced the local islet IP-10 and IL-15 gene expression, compared with levels of vehicle-treated controls at 8 and 10 wk of age (Fig. 2, D–F). MCP-1 and MIP-3α were inhibited only at either 8 or 10 wk of age in the islets of prediabetic NOD mice after in vivo treatment with 1,25-(OH)2D3 (Fig. 2, B and C). Finally, we observed that in vivo treatment with 1,25-(OH)2D3 did not influence the intraislet expression levels of calbindin-D28k and VDR during the course of insulitis (Fig. 2, G and H).
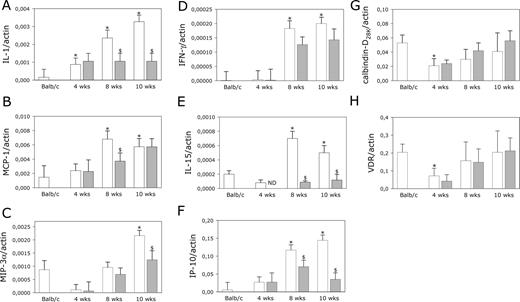
Effect of in vivo treatment with 1,25-(OH)2D3 on time course of IL-1β (A), MCP-1 (B), MIP-3α (C), IFNγ (D), IL-15 (E), IP-10 (F), calbindin-D28K (G), and VDR (H) mRNA expression in islets of diabetes-prone NOD mice. mRNA was isolated from islets of female NOD mice; treated with either vehicle (peanut oil, white bars) or 1,25-(OH)2D3 (5 μg/kg body weight, gray bars) by ip injection every other day from weaning until 4, 8, and 10 wk of age; and analyzed by real-time PCR. Four-week-old female BALB/c islets were used as nondiabetes-prone controls. The results obtained are means ± sem for three similar experiments and are expressed as cytokine or chemokine copies per β-actin copies. Significance (symbol) is expressed compared with mRNA expression levels in islets of control mice. *, P ≤ 0.05 vs. 4-wk-old BALB/c islets; $, P ≤ 0.05 vs. age-related vehicle-treated NOD mice.
Effects of in vitro treatment with 1,25-(OH)2D3 on islet cell death and gene expression after combined cytokine treatment
To assess whether the observed in vivo effects of 1,25-(OH)2D3 on the β-cell phenotype of NOD mice was associated with direct effects on the β-cell, we tested in a first set of experiments whether 1,25-(OH)2D3 was able to preserve viability of the clonal rat insulinoma cell line (INS-1E cells) when exposed to IL-1β and IFNγ. The cells were pretreated with vehicle or 10−8m 1,25-(OH)2D3 for 24 h. Neither IL-1β nor IFNγ alone induced cell death in INS-1E cells (data not shown), whereas a combined cytokine treatment led to extensive cell death already at 24 h of cytokine exposure (67.0 ± 1.9 vs. 94.0 ± 0.7% living cells in medium control, P ≤ 0.05). 1,25-(OH)2D3 (10−8m) per se was not toxic toward islet cells (90.0 ± 1.2% living cells, P = NS vs. medium control) but did not protect the INS-1E cells against cytokine-induced β-cell death (63.0 ± 1.2% living cells) (Fig. 3A). Discrimination between apoptosis and necrosis rates revealed no differences between vehicle and 1,25-(OH)2D3 conditions, and cytokine-induced accumulation of medium nitrite [an indicator of nitric oxide production] was also unaffected by 1,25-(OH)2D3 (data not shown).
![Effect of in vitro treatment with 1,25-(OH)2D3 on cytokine-induced cell death in INS-1E cells (A) and primary rat β-cells (B). INS-1E cells (6,000 cells/condition) and FACS-purified rat β-cells (10,000 cells/condition) were exposed for 24 and 48 h, respectively, to IL-1β (10 U/ml) plus IFNγ (100 U/ml) (CYTK, dashed bars) as indicated in the figure. Cultures were either untreated (white bars) or pretreated with 10−8m 1,25-(OH)2D3 for 24 h (gray bars). Cell viability was determined with the nuclear dyes HO 342 and PI. Nuclei of viable cells are stained in blue, whereas the nuclei of dead cells are stained in red. Islet cell dead is expressed as means ± sem (percentage of living cells) from three independent experiments. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/146/4/10.1210_en.2004-1322/1/m_zee0040507600003.jpeg?Expires=1716485480&Signature=baRxK7kwo7UkSsXDaMT2FfiyrfEsMVY07nJsIy-zV2ma~t09d8vzcoD8HGBGgk37fsEij0Z-tNw3iN82Oew6DApLsoyOcyAJsA4mqLuWUSUQcOeBQgNzSar7xP3O06yfdrB4t7fEGGVCVSX2KQGmykoN-QO9czS~JvVJHbsysE0fsGcU04sPEhdmKZu1PX6Y~eWO13Z0obVEkg9LgFsBzoxl3wUeMqGPE6DvsVgU5OMsttoJqIblaxzl7ZE47iYsDADxxL8GbS7WfPfDFA-cPx6xIrZIk3f~dTc7IUg~IC6Au3pFI-R2QRA3B0KHZlKLQwbMGuvBiva~uQL-LXsUsw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of in vitro treatment with 1,25-(OH)2D3 on cytokine-induced cell death in INS-1E cells (A) and primary rat β-cells (B). INS-1E cells (6,000 cells/condition) and FACS-purified rat β-cells (10,000 cells/condition) were exposed for 24 and 48 h, respectively, to IL-1β (10 U/ml) plus IFNγ (100 U/ml) (CYTK, dashed bars) as indicated in the figure. Cultures were either untreated (white bars) or pretreated with 10−8m 1,25-(OH)2D3 for 24 h (gray bars). Cell viability was determined with the nuclear dyes HO 342 and PI. Nuclei of viable cells are stained in blue, whereas the nuclei of dead cells are stained in red. Islet cell dead is expressed as means ± sem (percentage of living cells) from three independent experiments. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].
Cytokine-induced cell death of FACS-purified rat β-cells was studied after a 48-h instead of a 24-h cytokine culture because cytokine-induced cell death was shown to appear later in primary β-cells (25). Again no difference in the total number of viable islet β-cells was observed after cytokine treatment on addition of 1,25-(OH)2D3 (68.0 ± 2.0, compared with 61.0 ± 4.6%, living cells in cytokine control, P = NS) (Fig. 3B).
Although treatment with 1,25-(OH)2D3 was unable to protect against cytokine-induced islet damage in both experimental models, 1,25-(OH)2D3 was able to alter the gene expression profile of islets (INS-1E cells) after combined cytokine treatment. In line with previous observations from gene array analyses (25, 30), mRNA levels of IL-1β, MCP-1, IP-10, and the cytokine IL-15 were significantly up-regulated in INS-1E cells after exposure to inflammatory cytokines (Fig. 4). No MIP-3α and IFNγ mRNA expression could be observed in the INS-1E cells under basal as well as cytokine conditions (data not shown). 1,25-(OH)2D3 reduced cytokine-induced INS-1E IL-1β expression (Fig. 4A) and clearly decreased cytokine-triggered MCP-1, IP-10, and IL-15 gene expression during the period of incubation (Fig. 4, B–D). Of note, calbindin-D28K and VDR mRNA levels in INS-1E cells were slightly up-regulated by 1,25-(OH)2D3 under both basal and cytokine stimulated conditions (Fig. 4, E and F).
![Effect of in vitro treatment with 1,25-(OH)2D3 on differential expression of cytokine-induced inflammation-related molecule IL-1β (A), MCP-1 (B), IL-15 (C), IP-10 (D), calbindin-D28K (E), and VDR (F) mRNA in INS-1E cells. INS-1E cells were exposed for 8 h to IL-1β (10 U/ml) plus IFNγ (100 U/ml) (CYTK, dashed bars) with (gray bars) or without (white bars) pretreatment of 1,25-(OH)2D3 (10−8m) for 24 h, as indicated in the figure. The cells were then harvested, mRNA extracted, and measured by real-time PCR with the use of gene-specific oligonucleotide primers and fluorogenic probes. mRNA levels (the ratio between the gene of interest and β-actin) are expressed as means ± sem from three independent experiments. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/146/4/10.1210_en.2004-1322/1/m_zee0040507600004.jpeg?Expires=1716485480&Signature=zT~m1K7WfUhIY-f4dPNj5wePLkitpzGo6Okm0sTYF7YcEA~6sPqpxpc0k6d-UYkA64g7gNvofGcDvepjliIchbZ7X90E1L3LAcuUf0A7IYjikgac26lXNtuucioF-6J~xBFSE6N-3hHx1hzmHmly01Pd09e2KhDALiX5l2VkF0LkntKDsYZmjV7uiwHHW6gDjx03CDxXwyQf5pghyvpAgTRpimZJlEK7J7GF31ClTdMKyjEVJ0Tb3QLi-AlVfnhbfPqS2OriwcWDZ32To6VN1z1BbwEnG5fDGdq7mqw8dSmv5ZZ2WkRc3G88ZzXbCnvK5Nlpkzoea5a7M-1u16xioA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of in vitro treatment with 1,25-(OH)2D3 on differential expression of cytokine-induced inflammation-related molecule IL-1β (A), MCP-1 (B), IL-15 (C), IP-10 (D), calbindin-D28K (E), and VDR (F) mRNA in INS-1E cells. INS-1E cells were exposed for 8 h to IL-1β (10 U/ml) plus IFNγ (100 U/ml) (CYTK, dashed bars) with (gray bars) or without (white bars) pretreatment of 1,25-(OH)2D3 (10−8m) for 24 h, as indicated in the figure. The cells were then harvested, mRNA extracted, and measured by real-time PCR with the use of gene-specific oligonucleotide primers and fluorogenic probes. mRNA levels (the ratio between the gene of interest and β-actin) are expressed as means ± sem from three independent experiments. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].
Effects of in vivo treatment with 1,25-(OH)2D3 in NOD-SCID mice on islet cell death and gene expression after combined cytokine treatment
To evaluate whether in vivo treatment with 1,25-(OH)2D3 also resulted in less chemokine expression in the islet cells, we treated NOD-SCID mice, lacking T and B lymphocytes and natural killer cells, with 1,25-(OH)2D3 (5 μg/kg per 2 d) until 10 wk of age. Exposing control islets to the combined cytokines (IL-1β + IFNγ) killed already 26.8% of islet cells after 48 h of culture (islet viability 73.2 ± 14.8 vs. 95.8 ± 3.1% living cells in medium control, P ≤ 0.05). A prolonged exposure of 5 d further increased cell death as reflected by decreased general islet viability (36.0 ± 16.0 vs. 97.4 ± 1.9% living cells in medium control, P ≤ 0.05) (Fig. 5). After treatment with 1,25-(OH)2D3in vivo, 97.4% of cells remained viable in cytokine-free medium (P = NS vs. medium control), but again no protection against cytokine-induced cell death was observed (30.0 ± 7.6% living cells, P = NS vs. cytokine control) (Fig. 5).
![Effect of in vivo treatment with 1,25-(OH)2D3 on cytokine-induced cell death in NOD-SCID islets. Islets (15 per condition) isolated from NOD-SCID mice were exposed for 5 d to IL-1β (50 U/ml) plus IFNγ (1000 IU/ml) (CYTK, dashed bars), as indicated in the figure. Cultures were from either 10-wk-old vehicle- (white bars) or 1,25-(OH)2D3-treated NOD-SCID mice (5 μg/kg body weight ip from weaning per 2 d, gray bars). Cell viability was determined with the nuclear dyes HO 342 and PI. Nuclei of viable cells are stained in blue, whereas the nuclei of dead cells are stained in red. Islet cell dead is an estimation of the percentage of living vs. dead cells. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/146/4/10.1210_en.2004-1322/1/m_zee0040507600005.jpeg?Expires=1716485480&Signature=jeHIUtrYIxAohqBt1TbkyQCXUw51YIgXy8zEhKn5EOs0gbPl7ylfZiGe~go26JA8WPTEeARwrC8Jbj8M8ZalYNSwrGVB-OtgaWZH0Sq44p2kY7pDFvlwahIvX3qrUhN4t9HJpTWbamH8u16LezxheKD9EXyQukxkuu5HY0f-CYJepfxXWiOwkazjMmGmZfm9OWPPiqAml-wKG1ICoqMYzQ6j6X79gxuCrLpAfRujICaaVQmokcfeauTRIP14S0yKlJJIx0W0Ryp9ixOuACZrp~Zo4e5zPpctLI50bJ125hv0GalaIMCQ3MsluDu6r0z648GwanDFOCvATIog3k8xkg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of in vivo treatment with 1,25-(OH)2D3 on cytokine-induced cell death in NOD-SCID islets. Islets (15 per condition) isolated from NOD-SCID mice were exposed for 5 d to IL-1β (50 U/ml) plus IFNγ (1000 IU/ml) (CYTK, dashed bars), as indicated in the figure. Cultures were from either 10-wk-old vehicle- (white bars) or 1,25-(OH)2D3-treated NOD-SCID mice (5 μg/kg body weight ip from weaning per 2 d, gray bars). Cell viability was determined with the nuclear dyes HO 342 and PI. Nuclei of viable cells are stained in blue, whereas the nuclei of dead cells are stained in red. Islet cell dead is an estimation of the percentage of living vs. dead cells. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].
IL-1β + IFNγ up-regulated levels of IL-1β, MCP-1, MIP-3α, IP-10, and IL-15 in islet cells from control mice (Fig. 6). No IFNγ mRNA expression could be observed in the NOD-SCID islets under basal as well as cytokine conditions (data not shown). In vivo treatment of NOD-SCID mice with 1,25-(OH)2D3 led to a small, but significant, decrease in IP-10 and especially IL-15 levels in islet cells after cytokine exposure (P ≤ 0.05 vs. cytokine control) (Fig. 6E).
![Effect of in vivo treatment with 1,25-(OH)2D3 on differential expression of cytokine-induced inflammation-related molecule IL-1β (A), MCP-1 (B), MIP-3α (C), IL-15 (D), and IP-10 (E) mRNA in NOD-SCID islet cells. NOD-SCID islets were exposed for 24 h to IL-1β (50 U/ml) plus IFNγ (1000 U/ml), as indicated in the figure. mRNA was isolated from islets of female NOD-SCID mice; treated with either vehicle (peanut oil, white bars) or 1,25-(OH)2D3 (5 μg/kg body weight, gray bars) by ip injection every other day from weaning until 10 wk of age; and analyzed by real-time PCR. mRNA levels (the ratio between the gene of interest and β-actin) are expressed as means ± sem from three independent experiments. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/endo/146/4/10.1210_en.2004-1322/1/m_zee0040507600006.jpeg?Expires=1716485480&Signature=wlgtALO-WzwV3GmkzOwyh-EqEotgNFe5hz~uyyCgjPqX6O6e~sUMpo~pQWrEf~dT-yz4o88EM0AsHLi1zpkOP6q0pypa7mHU90nVJ8Tg-zya5fLFXRzGlUbzXiNvX9SIb9sUc3W1bAnIjWeVhLjyaLhqV6hqNgR~ueXo8-EsDX-pieo3iGjIhDLJOcJFbZZmWl6RIQ3YyOY9rMmC4hpstI89gJyOSqkpORAUcItPjSS5h2iQk2-nYiRj~RaxoJlm4ua2T~2ILNO-k5SJ2c0ebzqeqPpayUdCIcxHbhZB~9ZxKHSLQk-eITp0IqF1dS-lEZAhPPrqavGxVGqc-C76nA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of in vivo treatment with 1,25-(OH)2D3 on differential expression of cytokine-induced inflammation-related molecule IL-1β (A), MCP-1 (B), MIP-3α (C), IL-15 (D), and IP-10 (E) mRNA in NOD-SCID islet cells. NOD-SCID islets were exposed for 24 h to IL-1β (50 U/ml) plus IFNγ (1000 U/ml), as indicated in the figure. mRNA was isolated from islets of female NOD-SCID mice; treated with either vehicle (peanut oil, white bars) or 1,25-(OH)2D3 (5 μg/kg body weight, gray bars) by ip injection every other day from weaning until 10 wk of age; and analyzed by real-time PCR. mRNA levels (the ratio between the gene of interest and β-actin) are expressed as means ± sem from three independent experiments. *, P ≤ 0.05 vs. medium control (control cells, without cytokines); $, P ≤ 0.05 vs. CYTK control (control cells, with cytokines); §, P ≤ 0.05 vs. medium 1,25-(OH)2D3 [cells treated with 1,25-(OH)2D3, without cytokines].
Early and long-term treatment with 1,25-(OH)2D3 prevents diabetes in NOD mice
Different treatment windows with 1,25-(OH)2D3 were tested in female NOD mice to study the impact of 1,25-(OH)2D3 on diabetes development when administered during early insulitis development (3–14 wk of age), after insulitis development (14–28 wk of age), and throughout the whole process (3–28 wk of age). As anticipated, at 28 wk of age, diabetes was clearly prevented [20% (18 of 90)] in the long-term-treated group, compared with 58% (40 of 69) in the control group (P ≤ 0.0001) (Fig. 7). This was accompanied by a delay in diabetes onset (mean age 147 ± 17 vs. 139 ± 18 d in controls, P ≤ 0.0005). When therapy was initiated only at 14 wk of age (when more than 75% of control NOD mice already have islet infiltration) 40% (eight of 20) of these mice became diabetic by wk 28 (P = NS vs. controls). The mean age at onset was again delayed, compared with controls (153 ± 24 d vs. controls, P ≤ 0.0005). When mice were treated from 3 until 14 wk of age, a small delay of diabetes onset was seen (142 ± 23 d, P ≤ 0.0005 vs. controls), and a reduction in incidence by 28 wk was observed (35 vs. 58% in controls, P ≤ 0.05).
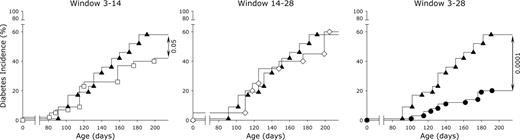
Effect of in vivo treatment with 1,25-(OH)2D3, applied in different time windows, on diabetes incidence in diabetes-prone NOD mice. Female NOD mice were treated with either vehicle (peanut oil) until 28 wk of age (▴, n = 90) or 1,25-(OH)2D3 (5 μg/kg body weight) by ip injection every other day from weaning until 28 wk of age (•, n = 69) or 14 wk of age (□, n = 43) or from 14 until 28 wk of age (⋄, n = 20). Mice with blood glucose levels of more than 200 mg/dl at 28 wk of age were scored as having diabetes. Significance is expressed compared with vehicle-treated NOD mice.
None of the different treatment regimens had an effect on body weight at the end of the experiment, and total serum calcium levels remained in the normal range [9.1 ± 1.7 (3–14), 9.4 ± 0.6 (14, 15–28), 9.8 ± 2.4 (3–28) vs. 9.6 ± 2.0 mg/dl in vehicle-treated control, P = NS, respectively]. Effects of 1,25-(OH)2D3 on bone metabolism appeared from serum concentrations of osteocalcin, a sensitive marker of osteoblast activity and bone turnover, and bone calcium content measured at the end of the experiment. Osteocalcin levels in the group treated during their entire life span was increased (102.1 ± 11.2 vs. 37.8 ± 3.9 μg/liter in vehicle-treated control, P ≤ 0.001), whereas the bone turnover in the mice from the shorter treatment duration was in the normal range (62.6 ± 14.7 μg/liter, P ≤ 0.05 vs. vehicle-treated control). Bone calcium content as indicated by the calcium content of the right femur reflected again the impact of 1,25-(OH)2D3 treatment during the whole life span because the bone calcium content of these animals was decreased (3.9 ± 0.4 vs. 7.9 ± 1.1 mg/femur in vehicle-treated control, P ≤ 0.001). On the other hand, when treatment was either ended or initiated at 14 wk of age, no bone loss under 1,25-(OH)2D3 treatment was seen (6.7 ± 0.3 and 5.1 ± 0.4 mg/femur, P = NS, respectively, vs. vehicle-treated control).
Discussion
The search for innovative therapies to prevent type 1 diabetes in genetically at-risk children and adolescents and/or arrest further loss of islet β-cells in recent-onset diabetics stands high on the priority list of researchers in the diabetes field. At present no treatment can prevent type 1 diabetes in humans, although many interventions are successful in the NOD mouse (reviewed in Ref. 39). Keeping this argument in mind, we point out that this animal model is not a perfect surrogate for the human disease but provides us with important lessons and insights into the possible heterogeneity underlying the complex pathogenesis of human diabetes. Based on data in the NOD mouse and also recent epidemiological data of the European Community Concerted Action Programme in Diabetes study and observations made in a Finnish study in humans, treatment with analogs of 1,25-(OH)2D3, the active form of vitamin D, or interventions with supplements of regular vitamin D are presently being considered in patients at risk for type 1 diabetes (5, 7, 8, 40). Many studies have demonstrated that inflammation is orchestrated by cytokines and chemokines secreted by activated T cells and macrophages during the autoimmune response (reviewed in Ref. 41). Recently we observed that expression of these cytokines and chemokines were also detectable in β-cells on exposure to combinations of IL-1β and IFNγ in vitro and in pancreatic islet cells during insulitis progression in the NOD mice in vivo (29, 42), suggesting an active role of the target organ it its own destruction.
In the present study, we combined in vivo and in vitro approaches to evaluate whether 1,25-(OH)2D3 inhibits insulitis and autoimmune diabetes in NOD mice and the mechanisms involved in these putative beneficial effects at the islet level.
We observed that in vivo treatment of diabetes-prone NOD mice with 1,25-(OH)2D3, initiated before insulitis onset, not only diminished the severity of islet infiltration and preserved β-cell functionality during the course of insulitis, as was previously observed by our group (6), but also decreased the levels of IL-1β, IL-15, and IP-10 mRNA levels in the islet cells from NOD mice at 8 and 10 wk of age. Complementing these findings, we found that in vivo and in vitro treatment with 1,25-(OH)2D3 decreased the cytokine-induced expression of IL-1β and IL-15 as well as the chemokine IP-10 in islet β-cells during cell culture. Our data in INS-1E cells and islet cells of immune-deficient NOD-SCID mice confirm that those cytokines and chemokines are expressed by the β-cells themselves. However, in contrast to NOD-SCID mice, in the case of NOD mice, these cytokines and chemokines may be produced by not only β-cells but also the infiltrating cells. We hypothesize that the reduced expression of inflammatory cytokines, as well as inhibition of chemotaxis in the islet cells by 1,25-(OH)2D3, might limit in vivo the development of insulitis by affecting the migration and recruitment of effector T cells and macrophages to the islets. While this manuscript was in preparation, Giarratana et al. (43) showed that in vivo treatment with an analog of 1,25-(OH)2D3, given from 8 until 16 wk of age to NOD-SCID mice, inhibited basal islet expression of the chemokines [MCP-1, IP-10, and RANTES (regulated upon activation, normal T cell expressed, and secreted)] and delayed diabetes development after transfer experiments with diabetogenic T cells. Finally, although others have reported protective effects of calbindin-D28K-overexpression in βTC-3 cells incubated with cytokines (44), we did not find any clear involvement of calbindin-D28K and VDR in the 1,25-(OH)2D3-mediated disease prevention.
On the other hand, in vitro and in vivo treatment with 1,25-(OH)2D3 alone (prior to islet isolation) did not protect rodent insulinoma INS-1E cells, rat FACS-purified single β-cells, and whole NOD-SCID mouse islets against cytokine-induced cell death in vitro. This confirms observations of Mauricio et al. (18) but contrasts with the findings of Riachy et al. (20, 21), who demonstrated that in vitro treatment with 1,25-(OH)2D3 protects rat insulin-producing RINm5F cells and human islets against cytokine-induced cell death. Differences between these studies and our own experiments include different exposure periods to 1,25-(OH)2D3 (ranging from 48 h to 6 d) and different islet cell systems used, which might have modified sensitivity to inflammatory cytokines and/or response to treatment with 1,25-(OH)2D3. Of note, in our hands, 1,25-(OH)2D3 does not provide protection against in vitro cytokine-induced β-cell death even when using variable incubation times and multiple cellular islet systems.
Taking our previous and present data together, these observations suggest that 1,25-(OH)2D3 acts on both immune cells and β-cells, affecting the expression of key proinflammatory genes expressed in the islet cells and thus reshaping them into less inflammation-prone and chemoattractive targets. Indeed, we previously observed that 1,25-(OH)2D3 is a strong immune modulator, inhibiting phytohemagglutinin A-induced lymphocyte proliferation (45), stimulating macrophage phagocytosis of bacteria and suppressing the antigen-presenting capacity of these cells and dendritic cells in vitro (46). In vivo, we observed that administration of 1,25-(OH)2D3 to NOD mice induced suppressor cells (7), mainly CD4+CD25+ cells (14), and eliminated effector cells (47), inducing an immune shift with lower Th1 cytokine levels locally in the inflamed islets of the native pancreas (48). Therefore, protection against insulitis and diabetes in NOD mice by 1,25-(OH)2D3 may be due to a combination of effects on both effector (e.g. immune cells) and victim (e.g. β-cells) of the autoimmune process culminating in type 1 diabetes mellitus.
Departing from this hypothesis, we tested different treatment windows for 1,25-(OH)2D3 as preventive agent against diabetes in NOD mice. We confirmed that protection against development of autoimmune diabetes was observed when mice were treated from weaning until the end of life. However, also, a small but consistent protection was seen when treatment was initiated early and terminated at 14 wk of age. On the other hand, no protection was observed when therapy was initiated at 14 wk of age, a time point when most of the islets of the subjects were already heavily infiltrated and there remained few insulin-producing β-cells. From these data it is clear that early-life treatment (before the onset of insulitis) had long-lasting effects on the development of diabetes and that interfering with an already ongoing immune attack against the β-cells is more difficult than interfering at the start of autoimmunity. These observations are in line with previous papers (49), in which we have shown that long-term treatment with analogs of 1,25-(OH)2D3, initiated early in life, was able to prevent disease. However, when treatment was initiated in mice with already ongoing insulitis, the analogs could not prevent progression to overt disease, unless they were combined with a short course of an anti-T-cell agent (e.g. cyclosporine A) (50). Thus, inhibiting homing of mononuclear cells into the islets, by decreasing chemokine expression, might be an attractive approach to prevent insulitis and diabetes. In line with this hypothesis, Christen et al. (51) demonstrated that blocking of IP-10 by monoclonal antibody therapy significantly reduced the incidence and onset of diabetes by decreasing clonal expansion and migration into the pancreas of CD8+ T cells.
In conclusion, this study showed that in vitro and in vivo treatment with 1,25-(OH)2D3 did not protect β-cells against cell death by direct immune attack but was able to modify the inflammation-induced gene expression in islet cells, shaping them into less inflamed and less chemoattractive targets. This β-cell effect, together with the previously described immune effects of the treatment, leads to reduced severity of islet infiltration and preserved β-cell functionality during the course of insulitis, eventually inhibiting the development of type 1 diabetes, especially when therapy is initiated early and given during the entire life span. These observations thus delimit the place for 1,25-(OH)2D3 and its analogs and provide a rationale for its use as potential tools in the prevention of type 1 diabetes in humans.
Acknowledgments
The authors thank W. Cockx, J. Depovere, J. Laureys, D. Valckx, M. Neef, J. Schoonheydt, and M. Urbain for their excellent technical assistance.
This work was supported by the Juvenile Diabetes Research Foundation (JDRF) Center for Prevention of β-Cell Destruction in Europe Grant 4-2002-457 and the European Community concerted Sixth Framework Program Grant TONECA (LSHM-CT-2004-503245). Additional grants were from the Belgium Program on Interuniversity Poles of Attraction initiated by the Belgian State (IUAP P5/17), the Katholieke Universiteit Leuven (KUL) (Geconcerteerde Onderzoeksacties, 2004/10), the Flemish Research Foundation [Fonds Voor Wetenschappelijk Onderzoek (FWO)—Vlaanderen, G.0084.02 and G.0233.04], and the National Research Foundation (Fonds National de la Recherche Scientifique). C.M. has a clinical research fellowship (FWO); C.A.G. has a postdoctoral FWO fellowship; A.K.C. has a postdoctoral JDRF fellowship; and A.G. has a doctoral scholarship from the KUL (Interfaculty Council for Development Co-operation).
Abbreviations
- FACS
Fluorescence-activated cell sorting
- IFN
interferon
- HO
Hoechst 342
- IP
inducible protein
- MCP
monocyte chemoattractant protein
- MIP
macrophage inflammatory protein
- NOD
nonobese diabetic
- 1
25-(OH)2D3, 1,25-dihydroxyvitamin D3
- PI
propidium iodide
- VDR
vitamin D receptor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}