





Abstract
This study examines whether the serine/threonine protein kinase, Akt, is involved in the cross-talk between epidermal growth factor (EGF) and insulin-related growth factor I (IGF-I) receptors and ER-α. Treatment of MCF-7 cells with either EGF or IGF-I resulted in a rapid phosphorylation of Akt and a 14- to 16-fold increase in Akt activity, respectively. Akt activation was blocked by inhibitors of phosphatidylinositol 3-kinase, but not by an inhibitor of the ribosomal protein kinase p70S6K. Stable transfection of cells with a dominant negative Akt mutant blocked the effects of EGF and IGF-I on ER-α expression and activity, whereas stable transfection of cells with a constitutively active Akt mutant mimicked the effects of EGF and IGF-I. In the latter cells, there was a decrease in the amount of ER-α protein and messenger RNA (70–80%) and an increase in the amount of progesterone receptor protein, messenger RNA (4- to 9- and by 3.5- to 7-fold, respectively) and pS2 (3- to 5-fold). Coexpression of wild-type ER-α and the dominant negative Akt mutant in COS-1 cells also blocked the growth factor-stimulated activation of ER-α, but coexpression of the wild-type receptor with the constitutively active Akt mutant increased ER-α activity. Receptor activation was blocked by an antiestrogen. Studies using mutants of ER-α demonstrated that Akt increased estrogen receptor activity through the amino-terminal activation function-1 (AF-1). Serines S104 S106, S118, and S167 appear to play a role in the activation of ER-α by Akt.
BREAST CANCER is frequently characterized by hormonal control of its growth. Because the estrogen receptor (ER) plays a central role in growth control, the presence of ER is employed to predict the hormone dependency of a tumor. However, the correlation between expression of ER and response to endocrine therapy is not perfect. Although significant amounts of ER are detected in more than 60% of human breast cancers, at best only two-thirds of these ER positive tumors respond to hormone therapy. In addition, 5–10% of the ER negative patients also respond to endocrine therapy. To improve the predictability of the hormone dependency of a tumor, the presence of the progesterone receptor is also measured, but this relationship is not perfect either. To fully appreciate the prognostic and therapeutic value of ER in breast cancer, a clearer understanding of the mechanisms that regulate expression and activity of the receptor, as well as the cross-talk between ER-α and growth factors is important.
The estrogen receptor belongs to a superfamily of ligand-inducible transcription factors (1). Two distinct regions within the ER contribute to its transcriptional activity; the AF-1 domain, located in the amino-terminus and the ligand-dependent AF-2 domain, located in the carboxy-terminal hormone binding domain. The AF-1 and AF-2 domains regulate transcription both independently and synergistically, depending on the promoter and cell type (2). In the absence of hormone, the inactive receptor is complexed with a host of proteins, including heat shock proteins, which prevent it from interacting with the cellular transcription apparatus. Upon binding estradiol, the receptor undergoes a conformational change that permits it to bind to coactivators and initiate the transcription of target genes. Activation of the ER is also associated with an increase in phosphorylation (3) on serines S104, S106, S118, and S167 (4) located in the amino-terminal A/B domain (5) and on tyrosine T537, located in the ligand binding domain (6).
A great body of evidence has accumulated demonstrating that growth factors also activate the ER in the absence of estradiol. However, the signal transduction pathways involved in its activation are poorly understood. The MAPK cascade may mediate steroid-independent activation of ER by epidermal growth factor (EGF), whereas insulin-like growth factor I (IGF-I) and transforming growth factor-α act through pathways independent of MAPK and protein kinase C (7). Recent data from this laboratory have demonstrated that EGF and IGF-I regulate the expression and activity of ER-α by activating PI 3-K (8, 9). One of the downstream targets of PI 3-K is the serine/threonine protein kinase, Akt, also called protein kinase B or the related protein kinase A and C (RAC) (10). Akt is a member of a conserved family of kinases that includes AKT1/RAC-α, AKT2/RAC β, and AKT3/RAC γ in humans (11). AKT1 is amplified in gastric adenocarcinoma (12) and is overexpressed in cancer cell lines including MCF-7 cells (13). AKT2 is amplified in 12% of ovarian cancers (14), 10% of pancreatic cancers (15, 16), and 3% of breast carcinomas (17). AKT3 expression and activity is elevated in ER negative breast cancer cells and is associated with more aggressive forms of breast tumors (18). Akt mediates the physiological effects of several peptide growth factors, including platelet-derived growth factor (10), EGF, basic fibroblast growth factor (19), insulin, and IGF-I (20). Cellular responses to serum, phosphatase inhibitors (21), and stress, such as heat shock and hyperosmolarity (22) are also mediated by Akt. Akt contains a pleckstrin homology (PH) domain in its amino-terminal region, a kinase domain in the middle, and a regulatory domain in the carboxy-terminal region (23). The binding of phosphoinositides to the PH domain of Akt recruits Akt to the plasma membrane where it is phosphorylated on threonine T308 and on serine S473 (24). Activation of the Akt pathway results in cellular proliferative, as well as antiapoptotic cell responses (25–27).
In this paper, we provide evidence that the cross-talk between EGF and IGF-I with ER-α in MCF-7 breast cancer cells involves Akt. Treatment of cells with EGF and IGF-I activates Akt through PI 3-K. Inhibitors of PI 3-K, as well as a dominant negative Akt mutant, block the effects of EGF and IGF-I on ER-α expression and activity, whereas a constitutively active Akt mutant mimics the effects of the growth factors. Estrogen receptor phosphorylation on either serine S118 and S167, and on serines S104 or S106 appears to play a role in receptor activation by Akt.
Materials and Methods
Cell culture
Monolayer cultures of MCF-7 cells were grown in improved MEM (IMEM) supplemented with 5% FCS. At 80% confluence, the medium was replaced with phenol red-free IMEM containing 5% charcoal-treated calf serum (CCS) (28). The calf serum was pretreated with sulfatase and dextran-coated charcoal to remove endogenous steroids. After 2 days, the medium was changed into serum-free, phenol red-free IMEM supplemented with fibronectin, glutamine, HEPES, trace elements, and transferrin, after which 100 ng/ml epidermal growth factor (EGF), 40 ng/ml IGF-I or 1 nm estradiol was added. Cells were harvested at the times indicated.
COS-1 cells were cultured in IMEM supplemented with 10% CCS, penicillin (10 IU/ml), and streptomycin (10 μg/ml). Serum starvation was performed in phenol red-free IMEM for 24 h. The next day, cells were stimulated with growth factors at the concentrations and times indicated. EGF was purchased from Upstate Biotechnology, Inc. (Lake Placid, NY); IGF-I was obtained from Biosource International (Camarillo, CA); estradiol and wortmannin were purchased from Sigma (St. Louis, MO). Rapamycin and LY294002 were purchased from Calbiochem (San Diego, CA).
Plasmids
The probe for the ER, pOR-300, was constructed by subcloning a 300-bp restriction fragment of pOR3 into the pGem4 polylinker regions using the restriction enzymes PstI and EcoRI (29). The clone 36B4 was constructed by subcloning a 220-bp fragment of 36B4 into the PstI restriction site of the pGem polylinker (29). In addition, the clones for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (30), pS2, and progesterone receptor (PR) (31) are described elsewhere.
The expression vector for wild-type Akt (32), the kinase defective Akt mutant, K179M, and the constitutively active Akt mutant, myrAkt, were generated as HindIII-BamHI inserts in pCMV-6 (10). Expression vectors for the human wild-type ER-α, HEG0, and the deletion mutants HE15, HE19, as well as the amino acid mutants S104A S106A, S118A, S153A, and S167A are described elsewhere (4, 33, 34). The estrogen-responsive reporter construct, pbCAT-(S)MERE was employed in the transient transfection assays. The estrogen-responsive reporter construct contains two consensus estrogen response elements separated by 20 bp inserted into the MMTV promoter in place of the glucocorticoid response element (35). The chimeric receptors GAL-ER and A/B-GAL contain the DNA binding domain of the yeast transcription factor GAL-4 fused to either the hormone binding domain or the A/B region (amino acids 1–184) of ER-α, respectively. The chimeric receptors and the reporter plasmid 17 m2GCAT are described elsewhere (34, 36).
Estrogen receptor-α and progesterone receptor protein assays
For analysis of ER-α and progesterone receptor protein concentration, MCF-7 cells were cultured and treated as described above. The concentration of receptor protein was determined using enzyme immunoassay kits from Abbott Laboratories (North Chicago, IL) containing the two rat monoclonal antibodies H222 and D547 to ER or rat monoclonal anti-progesterone receptor antibodies. To obtain total receptor protein, the cells were homogenized by sonication in a high salt buffer (10 mm Tris, 1.5 mm EDTA, 5 mm Na2 Mo O4, 0.4 m KCl, and 1 mm monothioglycerol with 2 mm leupeptin) (29). The homogenate was incubated on ice for 30 min and centrifuged at 100,000 × g for 1 h at 4 C. Aliquots of the total extracts were then analyzed according to the manufacturer’s instructions.
Measurement of estrogen receptor-α messenger RNA (mRNA), progesterone receptor mRNA, and pS2 mRNA
Total cellular RNA was extracted from MCF-7 cells by the RNazol method. The amounts of ER-α, 36B4, progesterone receptor, pS2, and GAPDH were determined by an RNase protection assay (29). Briefly, homogeneously 32P-labeled antisense complementary RNA (cRNA) were synthesized in vitro from pOR-300, 36B4, pS2, and pGAPDH using T7 polymerase and from the progesterone receptor probe using SP6 polymerase. Sixty micrograms of total RNA were hybridized for 12–16 h to the radiolabeled cRNA. After a 30-min digestion at 25 C with RNase A, 32P-labeled cRNA probes protected by total RNA were separated by electrophoresis on 6% polyacrylamide gels. The bands were visualized by autoradiography and quantified using the phospho imager. The amounts of ER-α mRNA, pS2 mRNA and progesterone receptor mRNA were normalized to the internal control 36B4 and GAPDH, respectively.
Immunoprecipitation and in vitro Akt kinase assay
To assay for Akt kinase activity, MCF-7 cells were serum starved, treated with growth factor, and lysed in Nonidet P-40 (NP-40) lysis buffer (1% NP-40, 10% glycerol, 137 mm NaCl, 20 mm Tris-HCl, pH 7.4) containing 2 μg/ml aprotinin, 2μ g/ml leupeptin, 1 mm pefabloc, 20 mm NaF, 1 mm sodium phosphate, and 1 mm Na3VO4. Equal amounts of lysates (300 μg) were precleared by centrifugation and preabsorbed with protein A-protein G (1:1) agarose slurry. Immunoprecipitation was carried out for 16–18 h using anti-Akt antibody (1:500 dilution) (Transduction Laboratories, Inc., Lexington, KY). Immunoprecipitates were washed three times with lysis buffer, once with water, and once with the Akt kinase buffer (20 nm HEPES-NaOH, 10 mm MgCl2, 10 mm MnCl2, pH 7.4). Kinase assays were carried out in Akt kinase buffer containing 10 μCi[γ -32P]ATP (3000 Ci/mmol), 5 μm ATP, and 1 mm dithiothreitol. Histone H2B (Roche Molecular Biochemicals, Indianapolis, IN) was added as exogenous substrate at a final concentration of 0.05 mg/ml. After 20 min at room temperature, kinase assays were stopped by the addition of loading buffer and separated on 12.5% SDS polyacrylamide gels. Detection was performed by autoradiography and phospho imaging.
Western blot analysis
MCF-7 cells were preincubated for 20 min with the inhibitors of PI 3-K, wortmannin (100 nm), and LY294002 (10μ m), or with an inhibitor of p70S6K, rapamycin (NOREF>20 ng/ml). Cells were then treated with EGF (NOREF>100 ng/ml) or IGF-I (NOREF>40 ng/ml) for 10 min. The cells were lysed in NP-40 lysis buffer and the lysates were heated to 95–100 C for 5 min. Equal amounts of protein (100 μg) were loaded onto SDS polyacrylamide gels. The samples were electrotransferred onto nitrocellulose membranes and the membranes were washed in PBS five times at room temperature. Membranes were kept in blocking buffer overnight at 4 C and incubated with either an anti phospho-Akt antibody (NOREF>S473) or an anti-Akt antibody (New England Biolabs, Inc., Beverly, MA) for 1 h at room temperature. After three additional washes in PBS, membranes were incubated with the horseradish peroxidase-conjugated secondary antibody (1:2000) in blocking buffer for 1 h at room temperature. Detection was performed by chemiluminescence, using the Super Signal chemiluminescent substrate (Pierce Chemical Co., Rockford, IL).
Transfections and CAT assays
COS-1 cells were employed in the transient transfection assays. Cells were maintained at 37 C in 5% CO2 in IMEM phenol red-free medium with 10% CCS. COS-1 cells were plated at 3× 106 cells per 150-mm plate. After 24 h, the cells were transfected using the low temperature, low pH calcium phosphate precipitation technique (37). Cells were transfected with 3 ml of DNA precipitate containing 5 μg of either the wild-type or mutant ER-α expression vectors, 25 μg of the estrogen responsive CAT reporter construct, and 2 μg of aβ -galactosidase vector in the presence or absence of 5 μg of the wild-type or mutant Akt expression vectors. Salmon sperm DNA was added to a total of 30 μg of DNA. For transfection assays employing the chimeric receptors, COS-1 cells were transfected with 5 μg of either GAL-ER or A/B-GAL and 25 μg of the GAL-4-CAT reporter vector. Eighteen hours after transfection, the cells were washed, the medium was changed to serum-free, and 10−9m estradiol, 100 ng/ml EGF, or 40 ng/ml IGF-I, in the presence or absence of 5 × 10−7m ICI 182,780 were added for 6 h. The cells were harvested and cell lysates were assayed for CAT activity. The conversion of [14C] chloramphenicol to its acetylated forms was determined by TLC. The reaction products were measured with a phospho imager. β-Galactosidase activity was determined as a measure of the transfection efficiency. CAT activity was expressed as the percent conversion of chloramphenicol to its acetylated forms and was normalized to the β-galactosidase activity.
Stable transfection of MCF-7 cells with wild-type and mutant Akt was performed with Lipofectamine Plus (Life Technologies, Inc., Rockville, MD), according to the manufacturer’s instructions. The amount of complementary DNA (cDNA) was 5 μg in the presence of 0.2 μg of the neomycin resistant gene, pcDNA 3.1 (−) (Invitrogen, Carlsbad, CA) per 106 cells in 100 mm dishes. Stably transfected cells were selected in IMEM supplemented with 10% FCS and 500 μg/ml G418 for about 1 month. Four to six clones of each mutant, as well as pooled clones were selected and characterized. Clones containing the kinase defective mutant, K179M, were designated K1 through K4 and the pooled clone was designated Kp. For the constitutively active mutant, myr-Akt, the clones were designated m1 to m6 and the pooled clone was identified as mp. Statistical analysis was performed using the Student’s t test, 1P < 0.05; ** P < 0.02; *** P < 0.0005.
Results
EGF and IGF-I signal transduction is mediated by Akt
Previous results from this laboratory demonstrate that PI 3-K mediates the effects of EGF and IGF-I on the amount and activity of ER-α in the breast cancer cell line, MCF-7 (8, 9). To determine whether Akt is also a mediator of this regulatory pathway, the ability of EGF and IGF-I to activate Akt through PI 3-K was tested. In this study, MCF-7 cells were serum starved and treated with either 100 ng/ml EGF or 40 ng/ml IGF-I for 10 min in the presence or absence of the PI 3-K inhibitors wortmannin (100 nm) or LY 294002 (10 μm). The cells were lysed and Akt was immunoprecipitated. The amount of kinase activity was measured in the presence and absence of the exogenous substrate, histone 2B (H2B). Autophosphorylation of Akt is shown in Fig. 1A. In serum-starved MCF-7 cells, Akt is catalytically inactive, as noted by the absence of autophosphorylation. Treatment with either EGF or IGF-I induced a significant increase in autophosphorylation of Akt. The ability of EGF and IGF-I to activate Akt was inhibited by wortmannin or LY 294002 (data not shown), suggesting that PI 3-K mediates the growth factor effects on Akt. The phosphorylated kinase migrated at approximately 60 kDa as a tightly spaced triplet, consistent with the proposed multiple phosphorylation sites on the enzyme (21, 38, 39). Similar results were obtained when H2B was employed as a substrate (Fig. 1, B and C). In the absence of growth factor treatment, the amount of phosphorylated H2B was low. However, there was a 14-fold increase in H2B phosphorylation following treatment with EGF and a 16-fold increase following treatment with IGF-I. Wortmannin and LY 294002 (data not shown) blocked the effects of the growth factors providing additional evidence that activation of Akt is mediated by PI 3-K. A time course of the effects of EGF and IGF-I on activation of Akt is presented in Fig. 1D. Growth factor treatment resulted in a rapid activation of Akt. EGF induced a 14-fold increase in activity by 10 min, which remained elevated for at least 1 h, whereas IGF-I induced a maximum increase of 16-fold by 20 min, which declined to an 8-fold increase by 1 h.
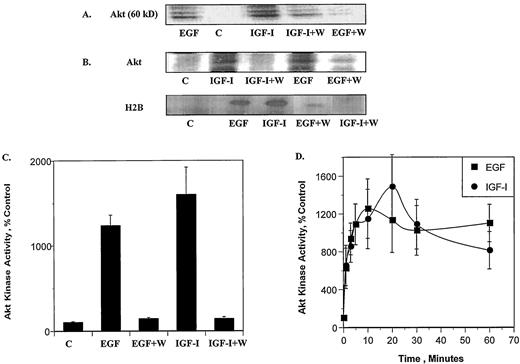
EGF and IGF-I induce Akt kinase activity. MCF-7 cells were serum starved for 24 h, preincubated for 20 min with the PI 3-K inhibitor wortmannin (100 nm) and treated with EGF (100 ng/ml) or IGF-I (40 ng/ml) for 10 min. Akt was immunoprecipitated with an anti-Akt antibody from cell lysates, and an in vitro kinase assay was performed in the presence or absence of the histone protein H2B. A, In vitro kinase assay of Akt in the absence of H2B. Representative immunoblot. B, In vitro kinase assay of Akt in the presence of H2B. Representative immunoblots. C, H2B was quantitated by autoradiography using a phospho imager and results are expressed as percent of control cells (n = 3 ± sd). D, Time course of the effects of EGF and IGF-I on Akt activity as measured by H2B phosphorylation (n = 3 ± sd). The above experiments were repeated three times.
Activation of Akt has been shown to result in the phosphorylation of serine S473 (24). To determine whether S473 is phosphorylated following activation with either EGF or IGF-I, Western blot analysis was performed using an antibody that specifically recognizes Akt, phosphorylated on S473. Serum starved MCF-7 cells were treated with either 100 ng/ml EGF or 40 ng/ml IGF-I in the presence or absence of 100 nm wortmannin, 10 μm LY 294002, or 20 ng/ml of rapamycin, the ribosomal protein kinase p70S6K inhibitor. The cells were lysed, and the amount of total and phosphorylated Akt was measured on immunoblots. The results in Fig. 2 demonstrate that in the absence of EGF and IGF-I the amount of phosphorylation on S473 was minimal. Treatment with growth factor induced a significant increase in the phosphorylation of Akt on S473. The effects of EGF and IGF-I were inhibited by wortmannin and LY 294002, but not by rapamycin, suggesting that Akt is a downstream target of PI 3-K, but not of p70S6K. Taken together, these data demonstrate that the rapid activation of Akt by EGF and IGF-I is mediated by PI 3-K.
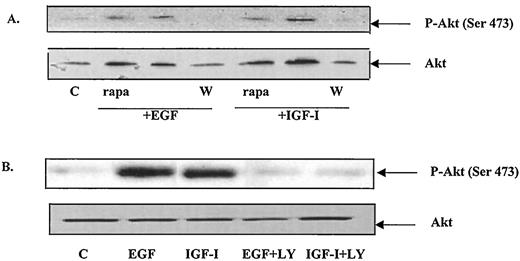
EGF and IGF-I induce the phosphorylation of S473. Cell lysates were prepared as described in the legend to Fig. 1. Western blot analysis was performed with equal amounts of protein (100μ g) using either anti phospho-Akt or anti Akt antibodies. A, Western blot of serum-starved, growth factor-treated cell extracts in the presence and absence of 100 nm wortmannin or 20 ng/ml rapamycin. A representative immunoblot of three independent experiments. B, Western blot of serum-starved, growth factor-treated cell extracts in the presence or absence of 10 μm LY 294002. A representative immunoblot of three independent experiments.
Regulation of ER-α gene expression by growth factors is mediated by Akt
To determine whether Akt regulates ER-α gene expression, MCF-7 cells were stably transfected with either a dominant negative or a constitutively active mutant of Akt. The expression vector alone was employed as a negative control. Four to six clones, as well as a pool of stably transfected clones, were selected and Akt activity was characterized (Fig. 3). Clones containing the kinase defective mutant, K179M, were designated as K1 to K4 and the pooled clone was identified as Kp. For the constitutively active mutant, myr-Akt, the clones were designated m1 through m6 and the pooled clone was designated as mp. In cells stably transfected with the dominant negative mutant, Akt was inactive, and the ability of EGF and IGF-I to activate Akt was inhibited. In cells transfected with the constitutively active mutant, the kinase was active as measured by the amount of autophosphorylation of Akt on S473. MCF-7 cells transfected with the expression vector alone behaved similarly to the parental MCF-7 cell line.
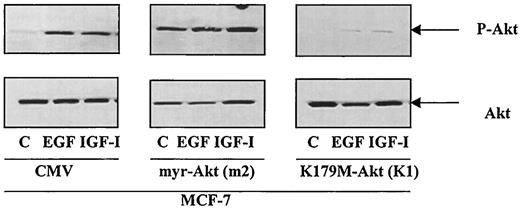
Characterization of MCF-7 cells stably transfected with constitutively active and dominant negative Akt. MCF-7 cells were stably transfected with the expression vector alone, the constitutively active mutant, myr-Akt, or the dominant negative mutant, K179M-Akt, as described in Materials and Methods. Clones were selected, serum-starved, and treated with either EGF or IGF-I. Western blot analysis was performed using an anti phospho-Akt (S473) antibody. One representative clone from each transfection is shown. The m2 clone contains the constitutively active Akt mutant, whereas the K1 clone expresses the dominant negative Akt mutant. Experiments were repeated twice for each clone, as well as for pooled clones.
To demonstrate that the effects of EGF and IGF-I on ER-α expression were mediated by Akt, cells that were stably transfected with the Akt mutants were treated with growth factor and the effects on ER-α protein and mRNA were measured. Before treatment with 100 ng/ml EGF or 40 ng/ml IGF-I, the cells were serum starved for 24 h. The amounts of ER-α protein and mRNA were measured by an enzyme immunoassay and an RNase protection assay, respectively. Estradiol was used as a positive control. In the parental MCF-7 cells and in MCF-7 cells stably transfected with the empty vector, growth factor treatment resulted in a 60% decrease in total receptor protein and in a 60–80% decrease in ER-α mRNA (Fig. 4). Expression of the kinase inactive Akt mutant in clones K1, K2, and Kp did not alter ER-α expression and blocked the effects of EGF and IGF-I on receptor protein and mRNA by 80–90%. In contrast, the overexpression of the constitutively active Akt mutant in clones m2, m4, m5, and mp mimicked the effects of the growth factors on receptor expression. There was a 70–80% decrease in ER-α protein and mRNA. Treatment of the constitutively active mutants with either EGF or IGF-I did not result in a further decrease in ER-α. These results suggest that the regulation of ER-α gene expression by EGF and IGF-I is mediated by Akt. In addition, Akt activates ER-α resulting in receptor down-regulation, similar to the down-regulation observed following receptor activation by estradiol (29, 31, 33, 34, 40).
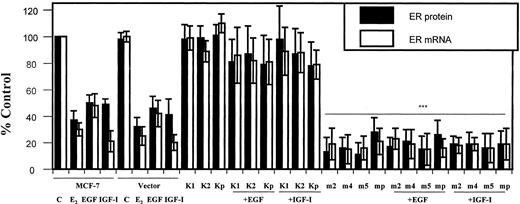
Akt modulates ER-α gene expression. Parental and stably transfected MCF-7 cells with myr-Akt (clones m2, m4, m5, and mp) or K179M-Akt (clones K1, K2, and Kp) were grown in improved MEM (IMEM) supplemented with 5% charcoal-stripped serum (CCS 5%). At 80% confluence, the medium was changed to phenol red-free IMEM with 5% CCS. After 2 days, the medium was replaced with serum-free medium and the cells were treated for 6 h with 100 ng/ml EGF and 40 ng/ml IGF-I. ER-α protein was measured using an enzyme immunoassay and ER-α mRNA was determined by an RNase protection as described in Materials and Methods. Results are expressed as percent of control cells and represent the mean value of three independent experiments performed in duplicate ± sd. Statistical differences between cells in the presence of constitutively active Akt vs. in the absence of myr-Akt were determined using the Student’s t test, * P < 0.05; ** P < 0.02; and *** P < 0.0005.
Growth factor activation of ER-α can be modulated by Akt
EGF and IGF-I have been shown to activate the estrogen receptor independent of hormone (8, 9). To determine whether Akt is involved in the steroid-independent activation of ER-α, the ability of the dominant negative and constitutively active Akt mutants to either block or mimic the effects of EGF and IGF-I was investigated. The ability of the dominant negative mutant to block the effects of EGF and IGF-I was tested in the K1, K2, and Kp clones, whereas the ability of the constitutively active Akt mutant to induce the estrogen regulated genes progesterone receptor and pS2 was determined in the m2, m4, m5, and mp clones. The amounts of progesterone receptor protein and mRNA, as well as pS2 mRNA were measured by an enzyme immunoassay and an RNase protection assay, respectively. The results were compared with the parental MCF-7 cells (Fig. 5). Estradiol was used as a positive control.
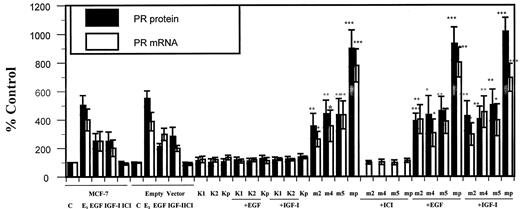
Effects of Akt on progesterone receptor expression. MCF-7 cells were grown and treated as described in the legend of Fig. 4. Progesterone receptor protein was measured using an enzyme immunoassay and mRNA was measured by an RNase protection assay. Results are expressed as percent of control cells and represent the mean value of three independent experiments ± sd. Statistical differences between cells in the presence of constitutively active Akt (treated or nontreated with EGF or IGF-I) vs. in the absence of myr-Akt were determined using the Student’s t test, * P < 0.05; ** P < 0.02; and *** P < 0.0005.
In the parental MCF-7 cells, EGF and IGF-I induced progesterone receptor protein and mRNA by 2- to 3-fold, P < 0.009 and pS2 mRNA by 2- to 3-fold, P < 0.05 (data not shown). In MCF-7 cells stably transfected with the dominant negative mutant, the effects of EGF and IGF-I were significantly blocked. In MCF-7 cells stably transfected with the constitutively active mutant, a 4-fold and a 3.5-fold increase in progesterone receptor protein and mRNA, respectively, was observed in the absence of treatment with growth factor. In the pooled clone, a 7- to 9-fold induction in progesterone receptor mRNA and protein, respectively, was detected. Treatment of the constitutively active Akt mutants with either EGF or IGF-I did not result in further increase in progesterone receptor. Similar increases (3.33 ± 0.98 in m2, P = 0.0147; 3.54 ± 1.28 in m4, P = 0.0264; 3.48 ± 1.37 in m5, P = 0.035; and 4.66 ± 0.64 in mp, P = 0.0006) were observed in the amount of pS2 mRNA (data not shown). The increases in progesterone and pS2 mRNA (data not shown) were blocked by the antiestrogen ICI 182,780, suggesting that these effects were mediated by ER-α. The expression vector alone did not alter the effects of growth factors on ER-α activity, suggesting that growth factor activation of ER-α is mediated by Akt.
To identify the region of ER-α activated by Akt, transient cotransfection assays of Akt and ER-α mutants (Fig. 6) were performed in COS-1 cells. The ability of Akt to activate ER-α through either the A/B domain or the hormone binding domain was tested using the deletion mutants HE15 and HEG19 and the chimeric receptors A/B-GAL and GAL-ER. The deletion mutant HE15 contains the amino-terminal and DNA binding domains of ER-α whereas HEG19 contains the DNA and hormone binding domains of the receptor. The chimeric receptors A/B-GAL and GAL-ER contain the DNA binding domain of the yeast transcription factor GAL-4 and the A/B and hormone binding domains of ER-α, respectively. In the first study, COS-1 cells were transiently transfected with the ER-α mutants and a reporter construct and the ability of either 100 ng/ml EGF or 40 ng/ml IGF-I to activate the mutant receptors was assayed (Fig. 7A). Treatment with growth factor activated the ER-α mutants containing the A/B domain, resulting in an approximately 2.5-fold increase in CAT activity, but failed to activate mutants containing the hormone binding domain. Although EGF and IGF-I treatment failed to activate receptor mutants containing the hormone binding domain, these mutants were activated by estradiol.
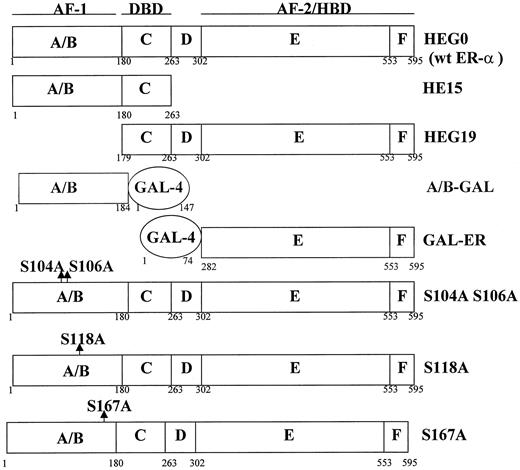
Schematic representation of the human wild-type ER-α and ER-α mutants studied. Structural and functional domains of ER-α are indicated. Transcription activation functions AF-1 and AF-2 map to the amino-terminal (A/B) and carboxy-terminal (E/F) domains, respectively. The DNA binding domain (DBD) consists of the C domain and the hormone binding domain (HBD) consists of domains E/F. In chimeras GAL-ER and A/B-GAL, human estrogen receptor-α (ER) domains A/B, C, D and C, D, E/F, respectively, were replaced by the yeast transcription factor GAL-4. Amino acid positions are numbered from the amino-terminal of the ER and indicate the boundaries of the functional domains of the receptor and the truncated or chimeric receptors. Positions of amino acid substitutions are indicated with arrows in the A/B domain. The names on the right refer to the proteins, which were expressed from the respective expression vectors.
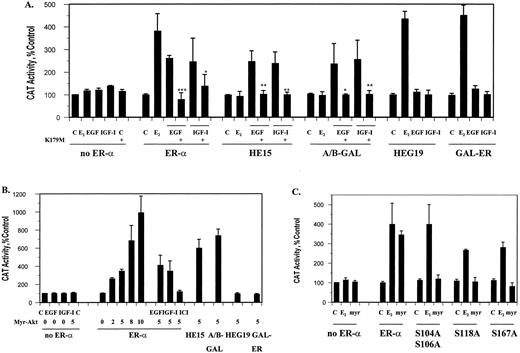
Akt activation of ER-α mutants. Transient cotransfections were performed in COS-1 cells with the expression vector, wild-type or mutant ER-α (HE15, HEG19, GAL-ER, A/B-GAL, S104A S106A, S118A, or S167A) and an estrogen-responsive CAT construct in the presence or absence of either the kinase defective mutant of Akt (K179M) or the constitutively active Akt mutant (myr-Akt). Transfected cells were treated for 6 h with 10−9m estradiol, 100 ng/ml EGF, or 40 ng/ml IGF-I in the presence or absence of the antiestrogen ICI 182,780. CAT activity was measured as described in Materials and Methods and normalized to the amount of β-galactosidase activity. The control was arbitrarily assigned the value of 100. Results represent the mean value of three independent experiments ± sd. Statistical differences between treatment with growth factors in the absence or presence of the dominant inactive mutant were determined using the Student’s t test, * P < 0.05, ** P < 0.02; *** P < 0.0005. A, Activation of chimeric and deletion mutants of ER-α by EGF and IGF-I. B, Activation of chimeric and deletion mutants of ER-α by the constitutively active Akt mutant. C, Activation of ER-α serine mutants by the constitutively active Akt mutant.
To determine whether the growth factor effects on the A/B domain were mediated by Akt, the ability of the dominant negative mutant to block the effects of EGF and IGF-I was tested (Fig. 7A). Expression of the Akt kinase defective mutant (K179M-Akt) inhibited the growth factor-stimulated activation of the chimera and deletion mutant containing the A/B domain and the wild-type receptor. When the constitutively active Akt mutant (myr-Akt) was cotransfected with ER-α, it produced a concentration-dependent increase in CAT activity, which was blocked by the antiestrogen, ICI-182,780 (Fig. 7B). Treatment of the cells with growth factor had no additional effect on CAT activity. Similar to the results with EGF and IGF-I, constitutively active Akt stimulated ER-α mutants containing the A/B domain of ER-α but not the mutants containing the hormone binding domain of the receptor. To determine whether serines S104, S106, S118, and S167 in ER-α played a role in Akt activation of the receptor, the ability of constitutively active Akt to stimulate ER-α mutants, containing alanines instead of serines, was tested (Fig. 7C). Although the serine mutants were activated by estradiol, the mutants were not activated by Akt, suggesting a role for serine phosphorylation in receptor activation. Taken together, the results of this study suggest that Akt activates ER-α through the A/B domain.
Discussion
Evidence for distinct signaling pathways that are coupled is accumulating. However, the pathways are not clearly defined. This paper provides evidence that the cross-talk between the EGF and IGF-I signal transduction pathways and ER-α is mediated by Akt in ER-α positive human breast cancer cells. Previous studies demonstrated that treatment of MCF-7 cells with EGF and IGF-I decreases ER-α protein and mRNA and increases ER-α activity as measured by an induction of estrogen regulated genes such as the progesterone receptor and pS2 (8, 9). Similar to EGF and IGF-I treatment, expression of constitutively active Akt decreased expression of ER-α protein and increased expression of progesterone receptor protein and pS2. In contrast, expression of kinase defective Akt inhibited the effects of the growth factors on ER-α, providing additional evidence that Akt is a mediator of the EGF and IGF-I pathways. Studies using ER-α mutants also suggest that Akt activates ER-α through particular serines in the AF-1 domain of the receptor.
Activation of the PI 3-K pathway appears to be an essential step in the estrogenic action of EGF and IGF-I in MCF-7 cells (8, 9). The EGF and IGF-I receptors may activate PI 3-K by binding directly to the p85 subunit (41). Although evidence exists for the activation of PI 3-K (41) and Akt (42) by IGF-I in MCF-7 cells, this is the first report that links activation of Akt by either growth factor to the activation of ER-α. Akt has several downstream targets (reviewed in Ref. 43), but it is not known whether the estrogen receptor is immediately downstream of Akt. It is possible that the effects of Akt on ER-α activity are mediated by one of its downstream targets, but it is equally possible that Akt directly phosphorylates ER-α, and, thereby, alters its transcriptional activity. Steroid receptors, such as ER, progesterone receptor, glucocorticoid receptor, androgen receptor, and vitamin D receptor, are constitutively phosphorylated and phosphorylation increases upon activation with estradiol, EGF, and IGF-I (2–6, 44). Tyrosine 537, located in the ligand binding domain, is thought to be involved in basal phosphorylation and may be a prerequisite for hormone binding (6), whereas serines S104, S106, S118, and S167 (4), located in the amino-terminal domain (5), are ligand-dependent phosphorylation sites. Experiments in COS-1 cells suggest that, although serines S104 and S106 are estradiol-inducible phosphorylation sites, serine S118 is the major phosphorylation site following ligand binding (4, 7, 45–47). In MCF-7 cells and insect cells, the major estradiol-inducible site is serine S167 (5, 47). The explanation for multiple hormone-inducible sites is not clear but may be dictated by cell- and/or promoter-specific factors including coactivators, corepressors, and heat shock proteins. Serines S104, S106, S118, and S167 are also phosphorylation sites for signal transduction pathways. Serines S104, S106, and S118 are targets for a proline-directed protein kinase (4). Serine S118 is also phosphorylated by MAPK in cells treated with EGF (7, 46) and serine S167 is phosphorylated by casein kinase II in vitro (6). Activation of pp90 rsk1 by EGF or phorbol myristate acetate specifically phosphorylates serine S167 both in vivo and in vitro (48). Although activation of the estrogen receptor has been shown to be associated with phosphorylation of the AF-1 domain, the precise role of individual serines is not known. Mutation of one phosphorylation site appears to result in a compensatory phosphorylation at another site, leading to the suggestion that the clustering of phosphorylation sites within a region of the receptor affects a large conformational change and/or increases the area of negative charge (1, 48). However, the results of this study suggest a more specific role for individual serines in receptor phosphorylation because mutation of either serine S118 or serine S167 to alanine resulted in a complete loss of ER-α activation by Akt. It is not clear at the present time whether Akt phosphorylates ER-α directly or indirectly through a downstream kinase or cross-talk with other signaling pathways. Serine S167 is a good candidate phosphorylation site for Akt; it is contained within the sequence RERLAS, which corresponds to the consensus phosphorylation site RXRXXS (43), suggesting a direct interaction of Akt with ER-α and phosphorylation of at least one of the serines. The ability of Akt to directly phosphorylate ER-α on serine S167, as well as on the other serines, remains to be tested.
The MAPK pathway is also thought to play an important role in mediating the effects of growth factors on cell proliferation and activation of ER-α. Although a role for the MAPK pathway in the growth response of MCF-7 cells to EGF has not been clearly demonstrated, the MAPK pathway and the PI 3-K pathway have been shown to mediate the proliferative response of these cells to IGF-I (41, 49). In addition, activation of MAPK potentiates the transactivation function of ER-α by phosphorylating serines in the AF-1 domain of the receptor (7, 46, 50). Similar to Akt, activation of MAPK induces phosphorylation of serine S118 but, in contrast to Akt, does not appear to phosphorylate serine S167. The ability of these pathways to affect ER-α function suggests that the estrogen receptor is a point of convergence of the Akt and MAPK pathways. However, it is unclear whether the Akt and MAPK pathways are alternate pathways or whether the Akt and MAPK pathways cross-talk with each other, resulting in receptor activation. In the former scenario, the Akt and MAPK pathways would function as separate and alternate pathways that activate ER-α by similar, but different, mechanisms; whereas, in the latter scenario, Akt would be expected to phosphorylate serine S167 and to activate the MAPK pathway that would subsequently phosphorylate serine S118. Although the pathways that mediate the effects of growth factors on ER-α activity remains to be defined, these data suggest that the mechanism of regulation is complex, involving multiple signaling pathways.
In this paper, we report that activation of Akt by EGF and IGF-I potentiates the AF-1 function of ER-α, possibly through the phosphorylation of serines. There are a limited number of studies on the regulatory function of Akt and this is the first study to suggest a role for Akt in the regulation of ER-α expression and activity. Although the precise mechanism by which Akt activates ER-α remains to be defined, these results offer a novel role for Akt in the cross-talk between growth factor signaling cascades and steroid receptors.
We thank Drs. S. Spiegel and A. Wellstein for helpful discussions, and Drs. M. E. Lippman and R. B. Dickson for critical reading of the manuscript.
Author notes
This work was supported by grants from the Milheim Foundation and Georgetown University (Dean of Research) (to A.S.); NIH Grants CA-50445 and CA-70708 (to M.B.M.); NIH Grant CA-18119 (to B.S.K); and, in part, by DAMD-17–99-1–9153 (to T.F.F). The Tissue Culture Shared Resource was supported by P-30-CA-51008 and P-50-CA-58185.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}