





Abstract
Cardiac hypertrophy is a common and often lethal complication of arterial hypertension. Elevation of myocyte cyclic GMP levels by local actions of endogenous atrial natriuretic peptide (ANP) and C-type natriuretic peptide (CNP) or by pharmacological inhibition of phosphodiesterase-5 was shown to counter-regulate pathological hypertrophy. It was suggested that cGMP-dependent protein kinase I (cGKI) mediates this protective effect, although the role in vivo is under debate. Here, we investigated whether cGKI modulates myocyte growth and/or function in the intact organism.
To circumvent the systemic phenotype associated with germline ablation of cGKI, we inactivated the murine cGKI gene selectively in cardiomyocytes by Cre/loxP-mediated recombination. Mice with cardiomyocyte-restricted cGKI deletion exhibited unaltered cardiac morphology and function under resting conditions. Also, cardiac hypertrophic and contractile responses to β-adrenoreceptor stimulation by isoprenaline (at 40 mg/kg/day during 1 week) were unaltered. However, angiotensin II (Ang II, at 1000 ng/kg/min for 2 weeks) or transverse aortic constriction (for 3 weeks) provoked dilated cardiomyopathy with marked deterioration of cardiac function. This was accompanied by diminished expression of the [Ca2+]i-regulating proteins SERCA2a and phospholamban (PLB) and a reduction in PLB phosphorylation at Ser16, the specific target site for cGKI, resulting in altered myocyte Ca2+i homeostasis. In isolated adult myocytes, CNP, but not ANP, stimulated PLB phosphorylation, Ca2+i-handling, and contractility via cGKI.
These results indicate that the loss of cGKI in cardiac myocytes compromises the hypertrophic program to pathological stimulation, rendering the heart more susceptible to dysfunction. In particular, cGKI mediates stimulatory effects of CNP on myocyte Ca2+i handling and contractility.
Introduction
The role of cGMP as a second messenger modulating cardiomyocyte (CM) growth and contractile functions is widely appreciated.1 Elevation of cGMP in CM is associated with attenuation of pathological cardiac hypertrophy, protection against ischaemia/reperfusion injury, and changes of inotropy and lusitropy.1 Three different hormone-receptor systems can elevate cGMP levels in myocytes: atrial (ANP) and B-type natriuretic peptides (BNP) via their shared membrane-bound guanylyl cyclase (GC)-A receptor; C-type natriuretic peptide (CNP) via membrane GC-B; and nitric oxide/NO via the cytosolic, ‘soluble’ GC (sGC).2 Atrial natriuretic peptide and BNP are secreted by CM in response to pressure/volume load.2 C-type natriuretic peptide and NO are primarily released by neighbouring fibroblasts and endothelia.2
The protective role of natriuretic peptides and cGMP in the moderation of cardiac remodelling was emphasized by genetic studies in rodents. Overexpression of BNP3 or CNP4 attenuated hypertensive or ischaemic heart disease. Conversely, CM-specific ablation of GC-A/cGMP or GC-B/cGMP signalling exacerbated hypertensive cardiac hypertrophy.5,6 These observations were supported by pharmacological studies. Inhibition of the cGMP hydrolysing phosphodiesterase (PDE)-5 with sildenafil raised CM cGMP levels and prevented or even reversed cardiac remodelling and dysfunction in mice subjected to cardiac pressure overload or to cardiac ischaemia.1,7 A recent clinical study provided the first human evidence that sildenafil's impact on the failing heart involves improvement of both systolic and diastolic function and reverse remodelling.8 Based on these and other data, the National Institutes of Health initiated a multi-center trial (RELAX) to test the utility of sildenafil for treating diastolic heart failure.
The downstream signalling pathways which transduce the raises in myocyte cGMP levels in changes of myocyte functions are much less clear than that leading to increased cGMP levels. At least three cGMP-modulated third messengers are expressed in CM: cGMP-dependent protein kinase I (cGKI) and cGMP-stimulated PDE-2 as well as cGMP-inhibited PDE-3, which modulate CM cAMP levels.1,9 Many published studies indicated that cGKI is the main third messenger activated by cGMP in CM, mediating antihypertrophic effects of natriuretic peptides or sildenafil. However, conclusive in vivo studies about the cardiac role of cGKI are missing. Studies of cardiac hypertrophy in mice with global deletion of cGKI were hampered by their severe systemic phenotype and early lethality.10 To study the significance of CM cGKI signalling in pathological cardiac hypertrophy, here we generated mice with conditional (αMHC-Cre-mediated) CM-restricted deletion of cGKI (CM cGKI KO mice).
Methods
All mouse experiments included in this manuscript were approved by the local animal care committee and conform with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Detailed methods are given as Supplementary material online.
Genetic mouse model
To achieve a cardiomyocyte (CM)-restricted deletion of cGKI, floxed cGKI mice11 were mated to transgenic mice expressing Cre recombinase under the control of the cardiac αMHC promoter (αMHC-Cretg mice).12 Cardiomyocyte cGKI KO mice and corresponding ‘flox/flox cGKI’ littermates (as controls) on a mixed C57Bl6/129Sv background were studied. Male mice aged 8–10 weeks were studied.
Animal studies
Control and CM cGKI KO littermates were infused subcutaneously with vehicle, isoproterenol (ISO, Sigma, 40 mg//kg BW/day, 7 days; n=8), or angiotensin II (Ang II, Calbiochem, 1000 ng/kg BW/min, 2 weeks; n = 14) via osmotic minipumps (Alzet, Colorado City, CO, USA), or they were subjected to surgical transverse aortic constriction (3 weeks; n = 10), as described in previous studies.5,13 Arterial blood pressure was measured in awake mice by tail cuff.5,13 Echocardiography was performed under light isoflurane anaesthesia before and after ISO or Ang II treatment and after 3-week TAC. In the infusion studies, additional terminal measurements of left ventricular pressure were performed under isoflurane anaesthesia, with a 1.4F micromanometer-tipped catheter. Mice were then sacrificed, the hearts were weighed, and the left ventricles were dissected and frozen in liquid nitrogen (for protein or mRNA extraction) and fixed in 4% buffered formaldehyde (for histology and immunohistochemistry). In a separate series of experiments, left ventricular myocytes were isolated for western blotting and/or functional analyses.
Histology
Cardiomyocyte diameters and the extent of myocardial fibrosis were determined on left ventricular sections stained with periodic acid Schiff or 0.1% picrosirius red.5,13 Connective tissue growth factor (CTGF) expression and localization was examined by immunohistochemistry (see Supplementary material online).
Quantification of apoptosis
Apoptosis was quantified in the myocardium by performing TUNEL staining and using the CaspaTagTMin situ caspase detection kit (Chemicon, Cat. No. APT423) on snap-frozen material. TUNEL-positive nuclei were visualized by FITC-labeled anti-digoxigenin antibody (1:500; Roche) and counterstained with 4,6-diamidino-2-phenylindole (DAPI).
Protein and gene expression
Protein and mRNA were prepared from LV tissue flash frozen in liquid nitrogen, and expression was assessed using standard techniques (see Supplementary material online). Total protein [SERCA, PLB, troponin I (TnI), ERK, AKT, and GAPDH, for normalization] was assessed after stripping the same membrane which was used to detect phospho-proteins. Blots were quantified using the ImageQuant software, and ratios of phosphorylated/total protein calculated and normalized by CTR results.
Isolation of cardiomyocytes for western-blot analysis and for measurement of single cell Ca2+i transients, cell lengths, and contractility
Ventricular myocytes were isolated by liberase/trypsin digestion (for Procedure see protocol PP00000125 from The Alliance for Cellular Signalling (AfCS)) and Cai2+ transients were measured in INDO-1 loaded, electrically paced (0.5 Hz) CM at 27°C as described before.13 Simultaneously, myocyte lengths (in μm) and shortening were visualized by edge detection.13
Statistics
Results are presented as means ± SEM. Group data were compared using two-way ANOVA (with genotype and treatment as categories) followed by the multiple comparison Bonferroni test to evaluate differences between groups. Comparisons between two groups were made using non-paired two-tailed Student's t-test. The paired Student's t-test was applied to evaluate the acute effects of ISO, ANP, and CNP on [Ca2+]i and cell shortening. Statistical analyses were carried out using the StatView statistics program (version 5.01, Abacus Concepts, Inc., Berkley, CA, USA). The significance level was set at P < 0.05. Sample sizes and individual statistical results for all analyses are provided in Figures2–8, and Supplementary material online Figure S1 and Tables.
Results
Generation of mice with cardiomyocyte-specific deletion of the cGKI gene
To generate mice with CM-specific ablation of cGKI, we crossed the αMHC-Cre mouse line12 with mice carrying floxed alleles of the gene coding for cGKI.11 Such CM-specific cGKI-deficient mice (αMHC-Cre+/−cGKIfl/fl, hence termed CM cGKI KOs) were born at expected numbers and were viable and fertile. Their body weight (BW) did not differ from that of respective cGKIfl/fl littermates (controls, CTR) (see Supplementary material online, Table S1). Western blotting of extracts prepared from isolated adult myocytes revealed the ablation of cGKI (Figure 1A), whereas expression levels were fully preserved in non-cardiac tissues, such as the lung, brain and kidney (Figure 1B). In addition, we tested the phosphorylation of PLB at position Ser16, which is mediated both by cGKI and cAMP-dependent protein kinase (PKA). The PLBSer16 phosphorylation-specific antibody detected almost no signal in unstimulated LV myocytes isolated from CTR or CM cGKI KO mice (Figure 1C). Activation of cGKI by the membrane permeant cGMP analogue 8-Br-cGMP (10 μM, 15 min) increased the phosphorylation of PLBSer16 in CTR but not in cGKI KO myocytes (Figure 1C), demonstrating the functional absence of cGKI in the latter. In contrast, the effects of ISO (10 nM) were preserved. Of note, ANP did not stimulate PLB phosphorylation in isolated myocytes (Figure 1C), despite expression of the functional GC-A receptor.5,13 Together, these data demonstrate the efficient and selective deletion of cGKI in CM.
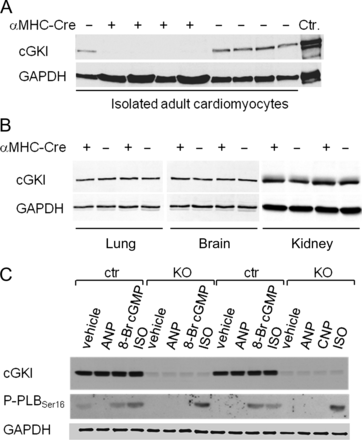
Expression and activity of cGMP-dependent protein kinase I in cardiomyocytes of control mice and mice with cardiac deletion of cGMP-dependent protein kinase I. (A) cGMP-dependent protein kinase I was detected in isolated cardiomyocytes from control mice but was absent in cardiomyocytes from KO mice (+ αMHC-Cre). (B) cGMP-dependent protein kinase I expression levels in the lung, brain and kidney were not different between genotypes. (C) In cardiomyocytes from control mice, 8-Br cGMP (10 μM, 15 min) and isoproterenol (10 nM, 15 min) increased the phosphorylation of phospholamban at Ser16. In cGMP-dependent protein kinase I-deficient cardiomyocyte (KO), the effect of 8-Br cGMP was abolished but the effect of isoproterenol was preserved. Atrial natriuretic peptide (100 nM, 15 min) did not stimulate phospholamban phosphorylation. GAPDH was used as loading control.
Baseline cardiovascular parameters and responses to isoproterenol are unaltered in mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I
Under resting conditions, arterial blood pressure, haematocrit, heart weights (HWs) as well as the ratio to BW (HW/BW) were not different between CM cGKI KO and CTR littermates (Figure 2A and B; see Supplementary material online, Table S1). Morphometric analyses of myocyte diameters and interstitial collagen fractions also did not reveal differences between genotypes (Figure 2C and D). Echocardiography showed normal basal cardiac systolic function, with unaltered heart rate and fractional shortening (FS) (Figure 3A–C; see Supplementary material online, Table S1).
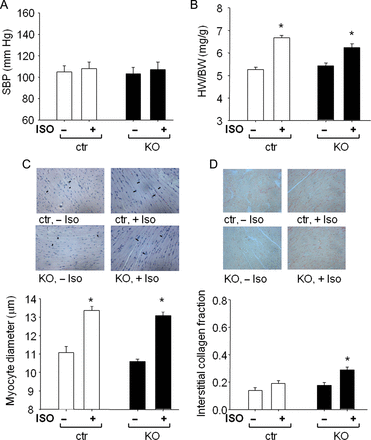
Cardiac remodelling induced by isoproterenol infusion in control mice and mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I. (A) systolic blood pressure (SBP), (B) heart weight (HW) to body weight (BW) ratios, (C) LV myocyte diameters, and (D) LV interstitial collagen fractions of control mice and mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I (n = 8 per group); *P < 0.05 vs. vehicle.
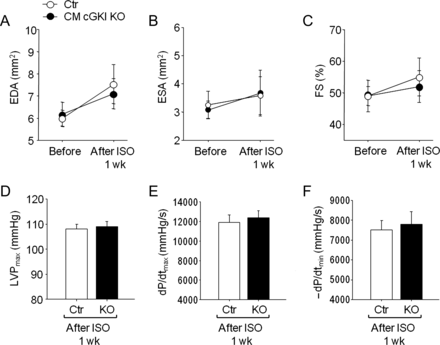
Echocardiography and invasive haemodynamic studies before and after isoproterenol infusion. (A) LV end-diastolic area (EDA), (B) end-systolic area (ESA), and (C) fractional shortening percentage (FS%) from control mice and mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I before and at 6 days of isoproterenol infusion, measured by echocardiography. (D) LV systolic pressure, and (E and F), ±dP/dt by invasive hemodynamics after 1 week of isoproterenol infusion (n = 8 per group).
To study whether cGKI modulates cardiac responses to chronic β-adrenergic stimulation, we subjected CM cGKI KO and CTR littermates to ISO stimulation. Isoproterenol (40 mg/kg/day, 1 week13) had no effect on blood pressure (Figure 2A) but led to significant cardiac hypertrophy at the organ and cellular level (Figure 2B and C). Heart weight, normalized to tibia length (HW/TL), was significantly and similarly increased by ISO in mice from both genotypes (CTR, 88 ± 3; KO, 89 ± 2.6 mg/cm; P < 0.01 vs. vehicle) when compared with vehicle-treated mice (CTR, 68 ± 3; KO, 67 ± 3.3 mg/cm). In CM cGKI KO mice, this hypertrophy was accompanied by a mild increase in interstitial collagen (Figure 2D). Echocardiography demonstrated that heart rate increased in all ISO-treated mice to the same extent (before ISO: CTR, 598 ± 17; KO, 612 ± 18 bpm; after ISO infusion: CTR, 676 ± 10*; KO, 693 ± 12* bpm; n = 8; *P < 0.05 vs. vehicle). Left intraventricular end-systolic area (ESA) and end-diastolic area (EDA), and FS were not significantly altered by ISO infusion (Figure 3A–C). Assessment of contractility by LV catheterization confirmed the positive chronotropic effects of ISO and showed that LV maximal and minimal systolic pressures and rates of LV pressure rise and pressure decline were similar in ISO-treated CM cGKI KO mice when compared with CTR littermates (Figure 3D–F).
Mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I develop dilated cardiomyopathy in response to angiotensin II
We described recently that mice with conditional (αMHC-Cre-mediated) CM-restricted deletion of the GC-A receptor for ANP (CM GC-A KO) show enhanced cardiac hypertrophy in response to Ang II or TAC but not to ISO.5,13 We hypothesized that the ANP/GC-A system, via cGMP/cGKI signalling, might selectively attenuate Ang II- (Gαq-mediated) or pressure-overload-induced but not ISO-stimulated (Gs-dependent) cardiac hypertrophy. To follow this hypothesis, we treated CM cGKI KO and CTR littermates with Ang II (1000 ng/kg BW/min, 2 weeks13). Angiotensin II provoked significant increases in arterial blood pressure (by ∼ 30 mmHg) and cardiac hypertrophy (Figure 4A–C). Heart weight/tibial length was significantly greater in Ang II-treated mice (CTR, 89.5 ± 4.8; KO, 89.1 ± 4.2 mg/cm; P < 0.01 vs. vehicle) when compared with vehicle-treated littermates (CTR, 69.8 ± 3.7; KO, 70 ± 1.3 mg/cm). Hypertrophy was accompanied by increased LV ANP mRNA expression in CTR (by 2.1 ± 0.2-fold*) and even more in KO mice (by 11 ± 3-fold*#; P < 0.05 *vs. vehicle, #vs. CTR). Notably, the LV mRNA expression levels of β-MHC and ratios of β/αMHC were not altered in CTR mice but greatly enhanced in KO mice treated with Ang II (see Supplementary material online, Table S2). Hence, hearts from KO mice showed a greater induction of mRNA for foetal genes associated with heart failure.
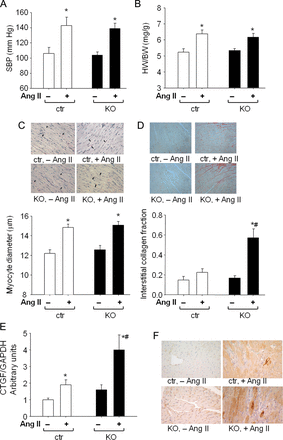
Cardiac remodelling induced by Ang II infusion in control mice and mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I. (A) systolic blood pressure (SBP), (B) heart weight (HW) to body weight (BW) ratios, (C) LV myocyte diameters, (D) LV interstitial collagen fractions of control mice and mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I (n = 16 per group); and (E and F) real time RT–PCR and immunohistochemical analyses of left ventricular CTGF expression (F: representative pictures; original magnification ×400) (n = 8 per group); P < 0.05 *vs. vehicle, #vs. control.
Whereas the hypertensive and hypertrophic effects were not different between CTR and CM cGKI KO mice, the profibrotic actions of Ang II were markedly enhanced in the latter (Figure 4D). This was concomitant to increased LV mRNA expression of CTGF (Figure 4E). In concordance, immunohistochemistry demonstrated strong immunoreactive CTGF in the cytosol of myocytes from Ang II-treated CM cGKI KO mice, especially around areas of focal interstitial fibrosis (Figure 4F).
Unexpectedly, echocardiography showed that CM cGKI KO hearts dilated during Ang II administration, with LV EDA and ESA increasing and FS declining (Figure 5A–C). Such pathological changes were never observed in CTR hearts (Figure 5A–C). Myocyte elongation was documented also by microscopic analysis of single, freshly isolated myocytes using video edge detection. The maximal myocyte lengths were greater in Ang II-treated KO mice (129.7 ± 2.8 μm) when compared with CTR (120 ± 3.1 μm) (P < 0.05; n = 8 mice, four myocytes per mouse). Basal heart rate was not different between genotypes (CTR, 570 ± 11 bpm; KO, 626 ± 10) and was not altered by Ang II (CTR, 568 ± 18; KO, 603 ± 16). These functional echocardiographic data were supported by invasive haemodynamic studies, revealing diminished LV peak systolic pressure and diminished rates of LV pressure rise and pressure decline in Ang II-treated CM cGKI KO mice when compared with CTR littermates (Figure 5D–F).
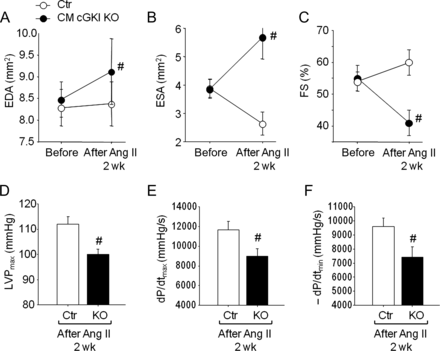
Echocardiography and invasive haemodynamic studies before and after Ang II infusion. (A) LV end-diastolic area (EDA), (B) end-systolic area (ESA), and (C) fractional shortening percentage (FS%) from control mice and mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I before and after 13 days of Ang II infusion, measured by echocardiography. (D) LV systolic pressure, and (E and F) ±dP/dt by invasive haemodynamics after 2 weeks of Ang II infusion (n = 16 per group); #P < 0.05 vs. control.
Despite the onset of cardiac dilatation and failure, no differences in cardiomyocyte death (data not shown) or apoptosis rates were observed between genotypes. Application of the tunnel assay on histological sections detected very few apoptotic cells in hearts from untreated mice (CTR: 41 ± 6; KO: 37 ± 2 tunnel positive nuclei per 100 000 cells) and a small, genotype-independent increase in apoptosis rate after Ang II administration (CTR: 220 ± 10*; KO: 180 ± 25* tunnel positive nuclei per 100 000 cells; *P < 0.05 vs. vehicle; n = 8). Similar results were obtained with caspase stainings (CTR, vehicle: 52 ± 11; KO, vehicle: 60 ± 9; CTR, Ang II: 214 ± 45*; KO, Ang II: 280 ± 70* caspase positive nuclei per 100 000 cells; *P < 0.05 vs. vehicle; n = 8).
Mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I develop dilated cardiomyopathy in response to TAC
As shown in Figure 6A and B, 3 weeks of TAC induced similar cardiac hypertrophy in both genotypes. The HW/TL ratio was significantly increased (CTR, 102 ± 5.6; KO, 97 ± 7 mg/cm; P < 0.01 vs. sham) when compared with sham-operated mice (CTR, 64 ± 3.6; KO, 66 ± 1.8 mg/cm). In both genotypes, this pronounced cardiac hypertrophy was associated with marked interstitial fibrosis (Figure 6C). Left ventricular ANP mRNA expression increased markedly after 3-week TAC in CTR (by 6.3 ± 1.7-fold*) and even more in KO mice (by 31 ± 5-fold*#; P < 0.05 *vs. sham, #vs. CTR). Additionally, LV β-MHC mRNA levels and the ratios β/α-MHC were greatly enhanced in CTR and even more in KO mice subjected to TAC (see Supplementary material online, Table S2). Echocardiography showed that CM cGKI KO hearts dilated in response to TAC, with LV EDA and ESA increasing and FS declining (Figure 6D–F). Such pathological changes were never observed in CTR hearts subjected to TAC or in sham-operated mice (Figure 6D–F). Heart rates in sham-operated CTR and KO mice were similar (567 ± 14 and 603 ± 42 bpm, respectively) and were not altered by TAC (582 ± 20 and 560 ± 14 bpm, respectively).
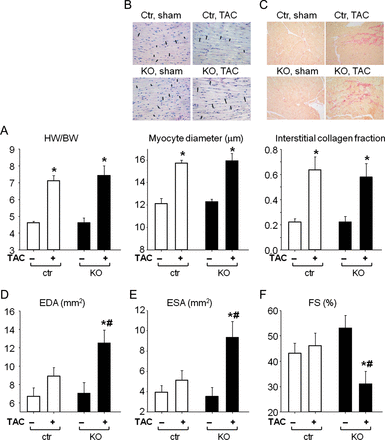
Cardiac remodelling and function in control and cardiomyocyte cGMP-dependent protein kinase I KO mice after TAC or sham operation. (A) Heart weight (HW) to body weight (BW) ratios, (B) LV myocyte diameters, (C) LV interstitial collagen fractions of control mice and mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I. (D) LV end-diastolic area (EDA), (E) end-systolic area (ESA), and (F) fractional shortening percentage (FS%) from control and cardiomyocyte cGMP-dependent protein kinase I KO mice after TAC or sham operation, measured by echocardiography (n = 10 per group); *P < 0.05 vs. sham, #P < 0.05 vs. control.
Taken together, these results suggest that the development of murine heart hypertrophy is not augmented by the absence of endogenous cGKI in cardiac myocytes. However, the loss of cGKI from myocytes compromises the hypertrophic program to pathological pressure load or neurohumoral (Ang II) stimulation, rendering the heart more susceptible to dilated cardiomyopathy (DCM) and dysfunction.
Cardiac dysfunction in mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I is accompanied by diminished expression and phosphorylation of Ca2+i-regulating proteins and altered myocyte Ca2+i handling
Studies in isolated CM and isolated perfused hearts have linked the activation of cGKI to regulation of several target proteins involved in myocyte calcium handling (SERCA, PLB) and calcium sensitivity (TnI).14–16 At baseline, the LV protein expression levels of SERCA2a, total PLB, P-PLBSer16, TnI, and pTnI were not different between genotypes (not shown) and they were not altered by chronic ISO infusion (Figure 7A). However, after Ang II treatment or TAC, LV expression levels of SERCA, total PLB, P-PLBSer16, and Ser23/Ser24-phosphorylated TnI were significantly decreased in CM cGKI KO mice in comparison with CTR littermates (Figure 7B and C).
![Expression of Ca2+i regulating proteins, Ca2+i transients, and cell shortening of myocytes prepared from control mice and from mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I after treatment with isoproterenol or Ang II, or after TAC. (A–C) Western blot analyses. Left ventricular expression levels of SERCA2a, phospholamban, troponin I, and phosphorylated phospholamban and troponin I in control and KO mice. (Left) Representative western blots. (Right) Protein levels of SERCA, phospholamban, and troponin I were normalized to GAPDH; levels of P-phospholamban and P-troponin I were normalized to total phospholamban and total troponin I. Ratios were calculated as x-fold respective control. (D and E) On average, the basal peak amplitude of Ca2+ transients (Indo-1 ratio, 405/495 nm, systolic-diastolic) (D) and basal shortening (E) were diminished in cGMP-dependent protein kinase I-deficient (KO) compared with control myocytes isolated from Ang II-treated mice. Superfusion with isoproterenol (10 nM, 5 min) increased [Ca2+]i and stimulated myocyte shortening, the effects being diminished in cGMP-dependent protein kinase I-deficient cells (n = 8 per group); #P < 0.05 vs. control.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/eurheartj/34/16/10.1093_eurheartj_ehr445/1/m_ehr44507.gif?Expires=1716438656&Signature=Z2OR7LYnXVK04KpfhxPf6el-Xi5zgYFCkNYakj2F8Q2JpEe8E50VKxTuz9-r5uW0Eh~AxJOHlYXLCNkvzzBNbsykGoD0I3s~3AdKQ-98ORT6hzO9MvWQRi-Tukkq5r-~1HCoLOSPszinDvgClMtubNpmuSZ5ovFEBU1n8mdXFH7CEV3q~NI1utqyjvcTINMO7fEKJRBMI~in~BpnbRrF851RrIBCmLKDkBPjhPT5xwPqujJgK7MS9Ax-Vrd7oUq4FMGmFCo80s5xkadjcfMoq80kt6rofFRvhd3yl~nmpbbfggXnoeY9WGcKl3v9beoG6ctPUCIhd8rVW7eUin7Kew__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression of Ca2+i regulating proteins, Ca2+i transients, and cell shortening of myocytes prepared from control mice and from mice with cardiomyocyte-restricted deletion of cGMP-dependent protein kinase I after treatment with isoproterenol or Ang II, or after TAC. (A–C) Western blot analyses. Left ventricular expression levels of SERCA2a, phospholamban, troponin I, and phosphorylated phospholamban and troponin I in control and KO mice. (Left) Representative western blots. (Right) Protein levels of SERCA, phospholamban, and troponin I were normalized to GAPDH; levels of P-phospholamban and P-troponin I were normalized to total phospholamban and total troponin I. Ratios were calculated as x-fold respective control. (D and E) On average, the basal peak amplitude of Ca2+ transients (Indo-1 ratio, 405/495 nm, systolic-diastolic) (D) and basal shortening (E) were diminished in cGMP-dependent protein kinase I-deficient (KO) compared with control myocytes isolated from Ang II-treated mice. Superfusion with isoproterenol (10 nM, 5 min) increased [Ca2+]i and stimulated myocyte shortening, the effects being diminished in cGMP-dependent protein kinase I-deficient cells (n = 8 per group); #P < 0.05 vs. control.
To evaluate the impact of these changes on Ca2+-handling, we compared cytoplasmic Ca2+i transients in LV myocytes isolated from untreated or Ang II-treated CTR and KO mice. Baseline diastolic Ca2+i levels were similar in myocytes isolated from untreated CTR and KO mice (see Supplementary material online, Table S3). Also, the peak amplitude of Ca2+i transients and the time to 50% decay were similar in both genotypes (see Supplementary material online, Table S3). Acute superfusion with ISO (10 nM) provoked a significant, immediate increase in the peak amplitude of the Ca2+i transients, together with an accelerated Ca2+i decay in both CTR and KO myocytes (see Supplementary material online, Table S3). Thus, the β-adrenergic cascade was not affected by genetic deletion of cGKI. However, we observed significant genotype-dependent differences of Ca2+i handling in myocytes prepared from mice previously treated with Ang II. As shown in Figure 7D, the peak Ca2+i-transient amplitudes at baseline and during acute ISO stimulation were significantly diminished in KO myocytes.
We simultaneously measured myocyte shortening using a video edge-detection system. As depicted in Supplementary material online, Table S3, the resting maximal cell shortening and time to relaxation were similar for myocytes isolated from untreated CTR and KO mice. Isoproterenol significantly increased cell shortening and accelerated relaxation, and these responses were similar in both genotypes (see Supplementary material online, Table S3). However, we observed significant genotype-dependent differences in contractility in myocytes prepared from mice previously treated with Ang II. Figure 7E demonstrates that baseline and ISO-stimulated shortening were significantly diminished in cGKI-deficient myocytes.
Collectively, these findings indicate that cGMP/cGKI signalling has an important role in the regulation of SR Ca2+i-regulating proteins and Ca2+i handling during development of pathological cardiac hypertrophy.
C-type natriuretic peptide, via cGMP-dependent protein kinase I, increases phosphorylation of phospholamban, Ca2+i transients, and contractility of isolated ventricular myocytes
As already mentioned at the beginning, ANP did not stimulate myocyte PLBSer16 phosphorylation (Figure 1C). Accordingly, we and others have shown previously that ANP has no direct effects on myocyte Ca2+i handling or contractility.13,17,18 In contrast, CNP was shown to stimulate these parameters.15,17 To investigate the role of cGKI as downstream messenger of the CNP/GC-B/cGMP signalling pathway, we compared the effects of CNP on CTR and cGKI-deficient myocytes. As shown in Figure 8A, CNP (10 nM–1 μM, 15 min) concentration-dependently stimulated PLBSer16 phosphorylation in CTR myocytes. Concomitantly, the peptide increased the peak amplitude of the Ca2+i transients (Figure 8B), accelerated Ca2+i decay, increased cell shortening, and decreased the time to relaxation (see Supplementary material online, Table S3). Remarkably, these effects of CNP were all abolished in cGKI-deficient myocytes (Figure 8A and B and see Supplementary material online, Table S3). As also shown, the effects of ISO (10 nM, acute superfusion) were similar in both genotypes.
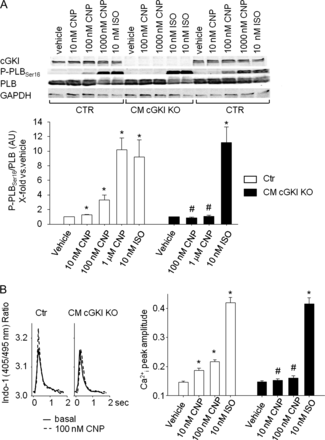
Effect of CNP on Ser16-phosphorylation of phospholamban and Ca2+-transients of isolated myocytes. (A) Effects of CNP (10 nM–1 μM) and isoproterenol (10 nM, all 15 min) on phospholamban phosphorylation at Ser16 in control and cGMP-dependent protein kinase I-deficient myocytes. (Top) Western blots showing the expression of cGMP-dependent protein kinase I, P-PLBSer16, total phospholamban as well as GAPDH in isolated myocytes. (Bottom) Levels of P-phospholamban were normalized to total phospholamban. Ratios were calculated as x-fold respective vehicle-treated control myocytes. (B, left) Representatives examples of effects of CNP (0.1 μM) on Ca2+ transients in control and cGMP-dependent protein kinase I-deficient myocytes. Panels show respective single traces before (basal) and during CNP treatment. (Right) On average, the peak amplitude of Ca2+-transients (Indo-1 ratio, 405/495 nm, systolic–diastolic) was not different in cGMP-dependent protein kinase I-deficient compared with control myocytes. Superfusion with CNP (10 and 100 nM, 5 min) increased Ca2+ transients in control myocytes. These effects were abolished in cGMP-dependent protein kinase I-deficient cells (n = 8); *P < 0.05 *vs. vehicle, #vs. control.
Left ventricular phosphorylation of ERK and Akt
Finally, to investigate cardiac mitogenic pathways that may be influenced by cGKI, we assessed the phosphorylation of members of the mitogen-activated protein kinase (MAPK: extracellular signal-regulated kinase, ERK1/2) and phosphoinositol-3-kinase pathways (AKT) in vehicle vs. Ang II-treated and sham- vs. TAC-operated CTR and KO mice. As shown in Supplementary material online, Figure S1, Ang II infusion or TAC provoked significant increases in cardiac pERK1/2 levels, without differences between genotypes. Also, the phosphorylation status of Akt (phospho-Ser473, active form) increased significantly after Ang II treatment, again without genotype-dependent differences (see Supplementary material online, Figure S1A). In contrast, LV pAKT levels were not different in CTR mice 3 weeks after TAC surgery (when compared with sham), and were even decreased in the KO mice (see Supplementary material online, Figure S1B).
Discussion
Our observations demonstrate that the absence of cGKI in CM does not alter baseline cardiac growth and function, or cardiac hypertrophic and contractile responses of mice to chronic β-adrenergic stimulation. These observations are in line with a recent study by Lukowski et al.19 in mice that express cGKI only in smooth muscle cells (‘cGKI rescue mice’, βRM), but not in other cells such as CM. However, our study adds an important, new piece of information revealing that CM cGKI signalling has a crucial role in modulating cardiac responses to Ang II or pathological pressure load (induced by TAC). Cardiomyocyte cGKI KO mice reacted to chronic Ang II administration or TAC with DCM, which was never observed in respective CTR littermates. Induction of cardiac ANP and β-MHC mRNA and the α- to β-MHC switch was greater in CM cGKI KO mice, indicative of an enhanced cardiomyopathic response. Cardiac dysfunction of mutant mice was accompanied by attenuated expression of the SR [Ca2+]i-regulating proteins SERCA2a and PLB, and diminished phosphorylation of PLB at Ser16, the target site for cGKI. These protein alterations were concomitant to impaired myocyte Ca2+i handling and diminished contractility. In adult myocytes CNP, but not ANP, stimulated PLB-Ser16 phosphorylation, Ca2+i handling, and contractility via cGKI. Overall, these data indicate that cGKI activation is involved in myocyte processes which prevent DCM and cardiac dysfunction in response to sustained pressure-overload and/or neurohumoral imbalance (Ang II).
Myocyte Ca2+i cycling is primarily governed by SERCA2a, which mediates Ca2+ sequestration into the SR, and by PLB, which inhibits SERCA2a activity. Intriguingly, cardiac SERCA2a and (less) PLB expression were down-regulated in CM cGKI KO mice subjected to Ang II infusion or TAC. The ratio SERCA/PLB was diminished by ∼20%. These alterations can account for impaired Ca2+i handling and depressed myocardial performance in the KO mice. The negative impact of the renin–Ang II system or pressure overload (TAC) on expression of SR Ca2+i-regulating proteins, Ca2+i-handling, and contractility was observed in many experimental models.20,21 Similar changes can be observed in human failing hearts.22,23 Corroborating our observations in CM cGKI KO mice, pharmacological enhancement of myocyte cGMP/cGKI activity with the PDE-5 inhibitor, sildenafil, restored SERCA/PLB expression and improved calcium cycling and contractility in mice with heart failure induced by sustained (9 weeks) TAC.21 Together, these data emphasize that cGKI activity plays an important role in the preservation of cardiac contractile functions during hypertrophy. The molecular mechanisms which link myocyte cGMP/cGKI signalling to SERCA/PLB expression remain unknown.
Phosphorylation of PLB by PKA or cGKI at Ser16 relieves the inhibition of SERCA2a and results in increased contractility through enhanced Ca2+i reuptake into the SR. Dilatative cardiomyopathy in CM cGKI KO mice subjected to Ang II or TAC was associated with diminished left ventricular PLBSer16 phosphorylation. Also the ratios P-PLB/PLB were decreased, which may further impair SERCA2a activity. In isolated myocytes, CNP concentration-dependently stimulated PLBSer16 phosphorylation, Ca2+i handling, and contractility, whereas ANP had no effect. These observations are consistent with previous studies.15,17 The effects of CNP were enhanced in cGKI overexpressing15 and were abolished in cGKI-deficient CM (present study), demonstrating unambiguously the mediatory role of the kinase. Notably, the expression levels of CNP and of its cGMP-forming GC-B receptor are upregulated in failing hearts.24,25 Cardiac fibroblasts might be a local source for CNP in this situation.25 Hence, endogenous CNP might participate in intramyocardial fibroblast-myocyte communication during chamber remodelling. C-type natriuretic peptide, via GC-B/cGMP/cGKI signalling and PLB phosphorylation, can improve myocyte Ca2+i handling and contractile functions, effects which are abolished in CM cGKI KO mice.
The lack of direct effects of ANP/GC-A on resting PLB phosphorylation, calcium transients, and myocyte shortening confirms previous studies with adult myocytes and intact hearts.15–18 Together with cell-based studies that employed cGMP reporters,26 these observations emphasize that the cGMP and cGKI pools coupled to ANP/GC-A, CNP/GC-B, or NO-GC stimulation are spatially and functionally compartmentalized in myocytes. Such compartmentation likely contributes to the discrepant phenotypical features of CM GC-A KO (ablation of a subsarcolemmal cGMP signalling: susceptibility to CM hypertrophy) and CM cGKI KO mice (ablation of all cGKI pools: susceptibility to CM contractile dysfunction).
Studies with cGMP analogues such as 8-Br-cGMP, or NO donors, have linked the activation of cGKI to phosphorylation of TnI and subsequent desensitization of myofilaments to calcium.16 In isolated myocytes, this pathway mediates inhibitory effects of NO/cGMP on the acute β-adrenergic activation of inotropy.16 Indeed, the levels of phosphorylated TnI were slightly reduced in CM cGKI KO mice subjected to Ang II or TAC. However, these changes could not ‘rescue’ the deterioration of cardiac contraction–relaxation functions of KO mice, due to the aforementioned alterations in SR protein expression and Ca2+i cycling.
We have also analysed the activation of the MAPK ERK1/2 and of protein kinase B (Akt/PKB) following Ang II infusion or TAC. ERK1/2 was similarly activated in LV of both CTR and KO mice, with greater induction by TAC when compared with Ang II. In contrast, we observed a significant (again genotype-independent) Akt activation after Ang II, but not after TAC. Indeed, published studies27 have shown that pAKT levels are induced at early stages (3 days) after TAC, followed by regression and even decreases at later stages (2 and 3 weeks after TAC, as analysed in our study). According to these studies, the observed decrease in pAKT, together with the marked α- to β-MHC switch, are additional signs of the acceleration of maladaptive LV hypertrophy in CM cGKI KO mice after TAC.
Interestingly, both CM GC-A KO mice13 and CM cGKI KO mice responded to Ang II with very marked cardiac interstitial fibrosis. cGMP-dependent protein kinase I is expressed in fibroblasts and mediates anti-fibrotic effects of cGMP in vitro/in vivo,28 but these should not be affected by our strategy of conditional, CM-restricted gene deletion. Thus, increased fibrosis after deletion of myocyte GC-A or cGKI likely results from increases in the Ang II-stimulated production of pro-fibrotic factors by myocytes. Recent studies showed that Ang II induces myocyte CTGF expression through a PKC activation-mediated pathway.29 In peritoneal fibroblasts and mesothelial cells, ANP/cGMP suppressed Ang II-induced upregulation of CTGF and thereby attenuated inflammatory peritoneal fibrosis.30 As shown by gene expression studies and immunohistochemistry, left ventricular myocyte CTGF levels were significantly increased in Ang II-treated CM cGKI KO mice. Together, these observations suggest that ANP, via GC-A/cGMP/cGKI signalling, inhibits the stimulatory effects of Ang II both on fibroblasts28 and also on myocyte secretion of pro-fibrotic factors, such as CTGF, thereby moderating cardiac fibrosis.
As illustrated, chronic ISO administration, a non-Gαq- (but Gs-) coupled stress, had no adverse effect in CM cGKI KO mice. Together with other studies,14 this observation seems to indicate that cGKI acts as a critical brake specifically against the Gαq/PLCβ pathway in the cardiac stress response to Ang II and pressure load. In particular, the regulator of G-protein signalling (RGS)-2 is phosphorylated and activated by cGKI, and subsequently acts as a critical and specific brake against Gαq/PLCβ signalling.14 However, because we used a relatively mild protocol of chronic ISO infusion, it is also possible that this differential responses of CM cGKI KO mice were just related to a milder cardiac stress, without cardiac pressure-overload, and not to distinct myocyte signalling pathways. We will address this possibility in future studies.
In summary, the present investigation identifies cGKI as an endogenous CM protective signalling molecule preventing cardiac fibrosis and alterations in myocyte Ca2+i handling during excessive neuroendocrine stimulation (e.g. with Ang II) and/or pressure overload. Experimental evidence that PDE-5 inhibition can blunt chronic cardiac stress responses has stimulated ongoing clinical studies on potential cardiac applications of cGMP-enhancing drugs such as PDE-5 inhibitors and therapies that enhance NO signalling or stimulate natriuretic peptide receptors. Our study contributes to the understanding of the interplay between cardiac cGMP and cGKI, which is central to the therapeutic use of agents that modulate them.
Supplementary material
Supplementary material is available at European Heart Journal online.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 688, SFB 487) and Interdisziplinäres Klinisches Forschungszentrum (IZKF) Würzburg (to S.F. and M.K.). Funding to pay the Open Access publication charges for this article was provided by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 688).
Conflict of interest: none declared.
Acknowledgements
Michaela Kuhn would like to thank Dr Stepan Gambaryan (Institute of Clinical Biochemistry, University Hospital Würzburg, Würzburg) for his advice and help for western blot analysis.
References
Author notes
These authors contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}