





Abstract
Gene knockout and transgenic mice are important tools that are widely used to dissect the mammalian hosts' responses to microbial invasion. A novel alternative is to engineer the pathogen itself to secrete host factors that stimulate or suppress specific immune defense mechanisms. Herein, we have described and validated an approach to facilitate the production and export of ectopic host proteins, from the most prevalent human fungal pathogen, Candida albicans. Our strategy utilized a prepropeptide from the C. albicans secreted aspartic proteinase, Sap2p. The prepeptide facilitates entry of Sap2p into the secretory pathway, while the propeptide maintains the protease as an inactive precursor, until proteolytic cleavage in the Golgi apparatus releases the mature protein. The Sap2p prepropeptide coding sequence was linked to that of two mammalian calcium-binding proteins, S100A8 and S100A9, which are associated with symptomatic vaginal candidiasis. The resulting expression constructs were then introduced into C. albicans. While the S100A8 protein is secreted into the growth medium intact, the S100A9 protein is apparently degraded during transit. Nonetheless, culture supernatants from both S100A8 and S100A9 expressing C. albicans strains acted as potent chemoattractants for a macrophage-like cell line and polymorphonuclear leukocytes. Thus, the pathogen-derived mammalian proteins possessed the expected biological activity.
Introduction
The molecular era has provided a diverse set of tools, which can be applied to dissect the pathogenesis of infectious disease. This has included genetic manipulation of many pathogenic microorganisms to establish pathogen requirements for colonization and infection of the host, as well as a variety of approaches to modulate the host response to microbial invasion. A key approach to subvert the mammalian immune response is to use gene ‘knockout’ mice. However, this can have significant complications. For example, many gene knockouts result in lethality, or have a multitude of indirect pleiotropic defects unrelated to the process under investigation, which can confound the interpretation of results. This is especially the case where the protein in question functions in multiple physiological processes or has distinct functions in different tissues. Another approach is to utilize recombinant mammalian proteins produced in various microbial expression systems. Unfortunately, many of those produced in bacteria do not possess biological activity due to inappropriate or insufficient molecular processing and/or glycosylation in prokaryotic cells. In addition, the purification steps necessary to isolate recombinant proteins and eliminate contamination with microbial byproducts can be challenging.
When investigating the pathogenesis of diseases caused by a eukaryotic microbe, an interesting alternative is to engineer the pathogen itself to synthesize and secrete the desired host modulatory factor in situ. Locksley and colleagues applied this approach, constructing a Leishmania strain that secretes IFN-γ (Tobin et al., 1993). More recently, Wormley and colleagues have constructed a Cryptococcus neoformans strain that also secretes murine IFN-γ (Wormley et al., 2007). Infection of mice with the C. neoformans IFN-γ strain results in a skewed immune response, that protects against lethality, and offers immunity against subsequent challenge with a ‘wild-type’ strain. Such an approach can be applied to test the effect of a single host protein upon the outcome of infection using either in vitro or in vivo models. One advantage is that the host protein of choice is produced in a eukaryotic system, which is more likely to yield a correctly processed, biologically active macromolecule than recombinant protein produced in bacteria. This approach also enables modulation of the hosts' normal physiological responses to the infectious microbe, in otherwise healthy ‘wild-type’ animals. Finally, production of the host factor is restricted to the site of infection, potentially limiting unintended physiological effects in distal tissues, or the host organism as a whole.
Recent advances in our capability to introduce ectopic DNA into several important fungal pathogens make this an attractive approach to manipulate host physiology during the course of infection. The relative ease of pathogen manipulation may yield significant benefits with respect to cost and time-saving vs. genetic manipulation of the mammalian host, or production of bacterial recombinant proteins, and thus provide a useful and complimentary alternative. Accepting these arguments, the challenge lies in developing pathogen-based expression system that facilitate the biosynthesis and secretion of biologically active host proteins. The goal of this study was to devise an expression system to facilitate the secretion of mammalian proteins by the most prevalent human fungal pathogen, Candida albicans.
The Sap2p secreted aspartic proteinase of C. albicans is synthesized as an inactive precursor, with a 56 amino acid N-terminal prepropeptide (Wright et al., 1992; Naglik et al., 2003). The prepeptide targets Sap2p into the endoplasmic reticulum and is subsequently removed by signal peptidase, leaving the propeptide that is thought to keep Sap2p inactive. Following transit through the secretory pathway, the propeptide is cleaved in the trans-Golgi by the Kex2p protease following a LYS-ARG sequence, to liberate the mature, active protease, prior to exocytosis (Brenner & Fuller, 1992; Togni et al., 1996; Newport & Agabian, 1997). We therefore sought to exploit this export mechanism, using the Sap2p prepropeptide to facilitate secretion of a mammalian protein from C. albicans.
To test the validity of this approach, we selected two small calcium-binding proteins, the S100A8 and S100A9 alarmins. These are cytosolic and secretory proteins expressed by a wide variety of cells including polymorphonuclear neutrophils (PMNs) and epithelial cells (Kumar et al., 2001; Ross & Herzberg, 2001; Ryckman et al., 2003; Gebhardt et al., 2006; Foell et al., 2008). In murine models, secreted S100A8 and S100A9 exert both antimicrobial and potent PMN chemotactic properties and are produced abundantly during protective and pathological inflammatory conditions, including vaginal candidiasis (Devery et al., 1994; Cornish et al., 1996; Kocher et al., 1996; Sohnle et al., 1996; Ryckman et al., 2003; Vandal et al., 2003; Yano et al., 2010,). Thus, these proteins are potentially of great importance as novel therapeutic targets. While investigating the role of these proteins in vaginal candidiasis, it was determined that commercially available recombinant S100A8 and S100A9 produced in bacteria are not biologically active with respect to chemotactic activity (J. Yano, G.E. Palmer, K.E. Eberle, T. Vogl, A.N. McKenzie & P.L. Fidel, unpublished data). Thus, our interest in studying these alarmins role in candidiasis provided an ideal opportunity to establish and validate the aforementioned C. albicans ectopic protein expression/export strategy.
Materials and methods
Candida albicans strains
CAF2-1 and CAI4 (Fonzi & Irwin, 1993) were kindly provided by Dr William Fonzi (Georgetown University). SC5314 has been described (Gillum et al., 1984).
Growth conditions
Candida albicans was routinely grown in YPD (1% yeast extract, 2% Bacto peptone, 2% dextrose) at 30 °C, supplemented with uridine (50 μg mL−1) when necessary (Guthrie & Fink, 1991). For growth curves, overnight cultures were subcultured to 20 mL fresh YPD medium to OD600 nm = 0.2 and incubated at 30 or 37 °C with shaking. OD600 nm was determined from samples taken hourly. Transformants were selected on minimal media (6.75 g L−1 yeast nitrogen base plus ammonium sulfate and without amino acids, 2% dextrose, 2% Bacto agar; YNB) supplemented with the appropriate auxotrophic requirements, as described for Saccharomyces cerevisiae (Burke et al., 2000) except for uridine, which was added at 50 μg mL−1.
Synthetic gene fusions
SAP21-56-S100A8 or SAP21-56-S100A9 fusions (Table 1) were synthesized by IDTDNA technologies. The sequence of either fusion was derived by combining the first 56 codons of the C. albicans SAP2 ORF, in frame with C. albicans optimized coding sequence of either mouse S100A8 (89 codons) or S100A9 (113 codons), followed by two TAA stop codons. Codon optimization was achieved by reverse translation of either S100 protein using the most frequently occurring C. albicans codons for each amino acid (http://www.candidagenome.org/). SalI and ClaI sequences were engineered at the 5′ and 3′ ends, respectively, of either fusion ORF.
Synthetic genes used in this study
Restriction enzyme sites used for cloning are underlined. Start and stop codons are shown in bold type, and the sequence encoding for the Sap2p prepropeptide is italicized.
Synthetic genes used in this study
Restriction enzyme sites used for cloning are underlined. Start and stop codons are shown in bold type, and the sequence encoding for the Sap2p prepropeptide is italicized.
Plasmids
Plasmid pKE1 was previously described (Johnston et al., 2013). The SAP21-56-S100A8 or SAP21-56-S100A9 fusions were cloned downstream of the ACT1 promoter between SalI and ClaI sites of pKE1 to yield the S100A8 and S100A9 expression constructs, respectively.
Candida albicans transformation
Candida albicans was transformed using the lithium acetate method (Gietz et al., 1992). pKE1 or the derived expression vectors were linearized with NheI, prior to transformation into the recipient strain CAI4 (Fonzi & Irwin, 1993), and Ura+ transformants selected. Correct integration of the pLUX-based vectors and thus full restoration of the URA3 locus in each transformant was confirmed by PCR using primers LUXDETF + LUXDETR primers (Table 2). Integration of each construct was also confirmed by Southern analysis using standard protocols. Genomic DNA from each strain was digested with EcoRI prior to separation on an agarose gel. A URA3-specific probe was amplified using primer pair URA3PBF1 + URA3PBR1, labeled using the NEBlot® Phototope® kit (New England Biolabs), and detected using the Phototope®-Star chemiluminescence detection kit, in accordance with the manufacturer's directions. Volumetric analysis was performed using Quantity One software (Bio-Rad).
Oligonucleotides used in this study
| Oligonucleotide | Sequence 5′→3′ |
| LUXDETF | CTGACCTTTAGTCTTTCCTGC |
| LUXDETR | CAGTAGTACTTGTTGTTGTATCG |
| URA3PBF1 | CCACCGTCGATAGTTTTACGG |
| URA3PBR1 | TGGATACTATCAAACAAGAGG |
| Oligonucleotide | Sequence 5′→3′ |
| LUXDETF | CTGACCTTTAGTCTTTCCTGC |
| LUXDETR | CAGTAGTACTTGTTGTTGTATCG |
| URA3PBF1 | CCACCGTCGATAGTTTTACGG |
| URA3PBR1 | TGGATACTATCAAACAAGAGG |
Oligonucleotides used in this study
| Oligonucleotide | Sequence 5′→3′ |
| LUXDETF | CTGACCTTTAGTCTTTCCTGC |
| LUXDETR | CAGTAGTACTTGTTGTTGTATCG |
| URA3PBF1 | CCACCGTCGATAGTTTTACGG |
| URA3PBR1 | TGGATACTATCAAACAAGAGG |
| Oligonucleotide | Sequence 5′→3′ |
| LUXDETF | CTGACCTTTAGTCTTTCCTGC |
| LUXDETR | CAGTAGTACTTGTTGTTGTATCG |
| URA3PBF1 | CCACCGTCGATAGTTTTACGG |
| URA3PBR1 | TGGATACTATCAAACAAGAGG |
Candida albicans phenotypic assays
For agar plate growth assays, C. albicans was grown overnight in YPD at 30 °C, cells washed in sterile distilled water, cell density adjusted to 107 mL−1, and serial 1 : 5 dilutions performed in a 96-well plate. Cells were then applied to agar using a sterile multipronged applicator. Resistance to temperature stress was determined on YPD agar at 37 and 42 °C, osmotic and ionic stresses on YPD agar + 2.5 M glycerol, 1.5 M NaCl, or 500 mM CaCl2. We also tested each strains tolerance of cell surface disrupting agents on YPD agar + 25 μg mL−1 Congo Red, 25 μg mL−1 Calcofluor white, or 0.02% SDS. Secreted protease activity was examined on bovine serum albumin (BSA) + YE agar at 37 °C (Crandall & Edwards, 1987).
Morphogenesis assays
Cells from overnight cultures were washed twice in distilled water, resuspended at 107 cells mL−1 and 2.5 μL spotted to either M199 (pH 7.5) or 10% FBS agar (Palmer & Sturtevant, 2004). Hyphal growth was also induced in liquid M199 or 10% FBS media at 37 °C, following inoculation of yeast at 106 cells mL−1.
Western blot analyses
S100 and vector only control transformants were grown overnight in 5 mL YPD broth at 30 °C. Cells were pelleted in a benchtop centrifuge, and the culture supernatant passed through a 0.22-μM filter. The cell-free culture supernatant (10 μL) was then separated on a 12% SDS-PAGE gel, transferred to a nitrocellulose membrane, and immunoblotted with either polyclonal goat anti-mouse S100A8 or goat anti-mouse S100A9 (R&D systems). Recombinant S100A8 or S100A9 produced in a bacterial expression system was loaded as a positive control (R&D systems). An HRP-conjugated rabbit anti-goat secondary antibody (sc-2768; Santa Cruz) was used for detection by enhanced chemiluminescence (Thermo Scientific).
ELISA
Concentrations of S100A8 and S100A9 in C. albicans culture supernatants were determined by enzyme-linked immunosorbent assay (ELISA) using mouse S100A8 and S100A9 DuoSet ELISAs, according to manufacturer's instructions (R&D Systems). Briefly, 96-well EIA/RIA plates (Costar, Corning, NY) were prepared by coating with monoclonal rat anti-mouse S100A8 (4.0 μg mL−1) or polyclonal goat anti-mouse S100A9 antibodies (0.82 μg mL−1) overnight at room temperature and blocked with 1% BSA for 1 h. The plates were washed with ELISA wash buffer (0.05% Tween 20 in PBS), and supernatants from overnight culture of the transformed C. albicans described above were added in triplicate. Serial dilutions of recombinant mouse S100A8 and S100A9 were used as standards. Following incubation for 2 h, the plates were washed and incubated with detection antibodies (biotinylated polyclonal goat anti-mouse S100A8 at 0.4 μg mL−1 or S100A9 at 0.1 μg mL−1) for 1 h. After washing, the plates were incubated with streptavidin-horseradish peroxidase for 20 min, washed and reacted with tetramethylbenzidine (Thermo). The reaction was stopped with sulfuric acid (2 N), and the optical density was determined at 450 nm on a microplate photometer.
Cell culture
Murine monocyte/macrophage J774A.1 cells were maintained as adherent cultures in DMEM (Life Technologies, Grand Island, NY) with 10% FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin in a humidified incubator at 37 °C and 5% CO2.
Mice
Female CBA/J mice, 6–10 weeks of age, were purchased from Charles River and used as a source for mouse PMNs. All animals were housed and handled according to institutionally recommended guidelines. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the LSU Health Sciences Center, New Orleans.
Chemotaxis assay
(1) Macrophage chemotaxis: Chemotaxis assays were performed using a modified Boyden chamber protocol (Boyden, 1962). Subconfluent J774A.1 cells were harvested by gentle scraping, washed twice in PBS, and resuspended at 4 × 106 cells mL−1 in DMEM with 1% FBS. 100 μL of the cell suspension was added to the upper inserts of Transwell Supports (6.5-mm-diameter wells, 8-μm pores) (Corning Life Sciences, Lowell, MA) and allowed to settle at 37 °C for 10 min followed by the addition of 600 μL of preconditioned test media into corresponding lower chambers. Test media were prepared as cell-free culture supernatants from overnight YPD cultures as described above, then diluted 100-fold in fresh DMEM medium. Cells were allowed to migrate for 4 h at 37 °C in 5% CO2. 1 μg mL−1Escherichia coli LPS (Sigma, St. Louis, MO) was used as the positive control for chemotaxis (Tajima et al., 2008; Kleveta et al., 2012). Inserts were aspirated, fixed in 4% paraformaldehyde for 10 min, stained with 0.3% crystal violet, and rinsed in PBS. Nonmigrating cells on the upper surface of each membrane were removed with a cotton swab, and the cells that had migrated to the lower side were counted in four random fields per well by light microscopy (100 ×). Each assay was performed in triplicate wells, and the assay was repeated three times. (2) PMN chemotaxis: Mouse PMNs were obtained from peritoneal exudate cells harvested 16 h after intraperitoneal injection of 10% casein and isolated by density gradient centrifugation using Ficoll-Paque (GE Healthcare, Uppsala, Sweden). PMN chemotaxis levels of C. albicans overnight culture supernatants were determined using a transwell system (Corning). Chemotaxis buffer (110 μL) consisting of RPMI 1640, 2 nM l-glutamine, 25 mM HEPES, and 1% BSA was added to the upper chambers of the transwell supports. Recombinant mouse MIP-2 (100 ng mL−1, a PMN chemoattractant) and RANTES (100 ng mL−1, a monocyte chemoattractant) (R&D Systems) were used as positive and negative controls, respectively. Culture supernatant samples were diluted at 1 : 10 with the chemotaxis buffer and added to the bottom chambers in parallel with the assay controls. After 5 min of equilibration, 5 × 104 PMNs in a volume of 50 μL were transferred to the upper chambers, and the plate was incubated for 1 h at 37 °C in 5% CO2. After incubation, PMNs in the bottom chambers were harvested and enumerated microscopically using a hemocytometer. All controls and samples were tested in duplicate.
Results and discussion
Candida albicans can be engineered to export ectopic proteins
We synthesized two gene fusions consisting of the first 56 codons of the C. albicans Sap2p ORF, with either C. albicans optimized S100A8 or S100A9 coding sequences (Table 1). Each fusion was cloned downstream of the ACT1 promoter in pKE1. Vector pKE1 is derived from pLUX (Ramon & Fonzi, 2003) and can be targeted for integration at, and fully reconstitute the URA3 locus in the most widely used C. albicans strains CAI4 and BWP17 (Fonzi & Irwin, 1993; Wilson et al., 1999). This negates the well-documented URA3 positional effect, and also fully restores the adjacent IRO1 gene deleted in strain CAI4 and its derivatives (Staab & Sundstrom, 2003; Chibana et al., 2005). The resulting expression constructs, or pKE1 vector alone, were introduced into C. albicans strain CAI4, and correct restoration of the URA3 locus confirmed by PCR and Southern blot analysis. Correct insertion and restoration of one URA3 allele occurred in all transformants examined, with about half having tandems insertions of usually one extra copy (Fig. 1a).
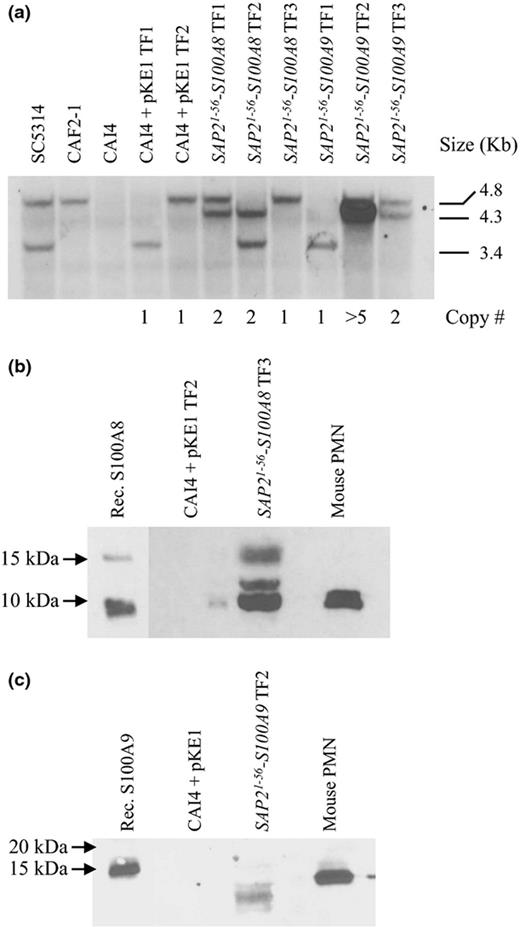
Construction of Candida albicans transgenic strains for the production and export of mammalian S100 proteins. (a) Southern blot analysis of SAP21-56-S100A8, SAP21-56-S100A9, and expression vector alone (pKE1) transformants. Genomic DNA was digested with EcoR1, and hybridized with a probe specific to the URA3 locus, that is internal to the vector. SC5314 = URA3/URA3; CAF2-1 = URA3/ura3Δ; CAI4 = ura3Δ/ura3Δ. This approach detected two distinct EcoRI fragments, presumably representative of two distinct chromosomal alleles, as only the larger 4.8-kb band was identified in the CAF2-1 strain. Tandem insertions are indicated by the 4.3-kb EcoRI fragment. The number of tandem insertions was determined by volumetric analysis of the 4.3-kb band intensity, and values normalized to the single restored 4.8- or 3.4-kb band. Total number of vector insertions for each transformant is indicated below the blot. (b, c) Culture supernatants from S100-producing strains or the isogenic control strain (CAI4 + pKE1) were immunoblotted with either anti-S100A8 (b) or anti-S100A9 (c). Recombinant S100A8 and S100A9 proteins or mouse PMN cell extracts were used as controls. Data are representative of triplicate experiments.
To detect secretion of S100 proteins, cell-free culture supernatants were immunoblotted with anti-S100A8 or anti-S100A9 antibodies. A protein of approximately 10 kDa was detected in all three SAP21-56-S100A8 transformants examined, that was absent from the vector alone control strains, and abundant in mouse PMN extracts (Fig. 1b). This is close to the expected molecular mass of the S100A8 protein (10.3 kDa), indicating the correct transport of the fusion molecule and removal of the Sap2p prepropeptide. Two additional species of around 12 and 15 kDa were also detected in the S100A8 expressing transformants that may represent a small fraction of unprocessed fusion protein retaining the Sap2p prepropeptide (predicted mass 16.3 kDa), and partially processed cleaved after an alternate LYS-ARG dipeptide at residue 25 within the Sap2p prepropeptide (predicted mass 13.5 kDa). Only 1 of 10 SAP21-56-S100A9 transformants yielded detectable anti-S100A9 reactive species from culture supernatants, and this transformant had an abnormally high number of tandemly inserted expression constructs (estimated to be > 5 by volumetric analysis; Fig. 1a). Furthermore, this was detected as a series of anti-S100A9 reactive species (6–15 kDa), possibly indicating degradation (Fig. 1c). This may be due to secreted aspartic proteinase or Kex2p-mediated proteolysis of S100A9, which contains an internal LYS-ARG sequence, and two LYS-LYS sequences that are potential Kex2p cleavage sites (Naglik et al., 2003). While S100A8 possesses an internal LYS-LYS sequence, there is no obvious proteolysis of the secreted protein.
S100 protein concentrations in overnight culture supernatants were determined by ELISA, using either a ‘single-copy’ S100A8 strain, or the ‘multicopy’ S100A9-producing strain. Data indicated that the amounts of secreted S100A8 and S100A9 in the culture supernatants were approximately 400 and 200 ng mL−1 (Fig. 2), respectively. Although a small proportion of the secreted S100A8 fusion protein exhibited incomplete maturation/cleavage, it is exported at potentially physiologically relevant concentrations (Yano et al., 2010).
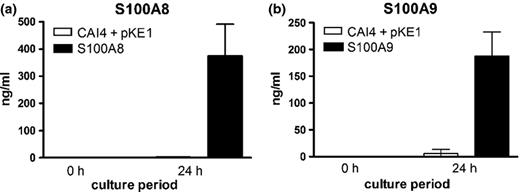
S100 proteins are present in culture supernatants of S100A8- and S100A9-expressing Candida albicans. Quantification of (a) S100A8 and (b) S100A9 by ELISA. Supernatants from overnight cultures of S100-producing strains (24 h) or YPD growth medium alone (0 h) were evaluated for concentrations of the respective proteins. The results are cumulative of three repeat experiments.
Production of some ectopic proteins may adversely affect C. albicans physiology
A crucial step in demonstrating the validity of our approach is to ensure that expression of the ectopic protein does not have deleterious consequences upon the pathogens ‘fitness’, or upon virulence-related attributes. Forced expression of the S100A8 protein (single or double insertion strains) did not cause any detectable deficiencies with respect to growth kinetics at 30 or 37 °C, ionic (NaCl or CaCl2) or osmotic stresses, sensitivity to the cell wall/surface disrupting agents Congo Red, Calcofluor white or SDS, hyphal growth, or levels of secreted aspartyl protease activity (Fig. 3a–c, and data not shown). Thus, our strategy for S100A8 protein secretion does not have any obvious effects upon C. albicans ‘fitness’ or virulence-related attributes. However, the S100A9-producing strain exhibited a slight reduction in hyphal growth compared with the isogenic control strain on M199 and FBS agar plates (Fig. 3c), but not in M199 or 10% FBS liquid medium. The S100A9 transformant also exhibited sensitivity to Calcofluor white (Fig. 3b). Together with the expression data described above, this suggests that S100A9 production may have some adverse effect on fungal fitness. These data are consistent with a recent study which found that while S100A8 has no antifungal activity, S100A9 has moderate antifungal activity (Sroussi et al., 2009).

S100A8 production does not have deleterious consequences upon Candida albicans. (a) Growth of each strain was compared as OD600 nm in YPD broth at 30 °C. (b) Cell suspensions of each strain were prepared by serial dilution, applied to YPD agar, and incubated at 30 or 42 °C, or to YPD agar supplemented with 1.5 M NaCl, 500 mM CaCl2, 0.02% SDS, 25 μg mL−1 Congo red, 25 μg mL−1 Calcofluor white, or 5 nM rapamycin and incubated at 30 °C for 1 or 2 days. (c) Hyphal growth was compared on 10% FBS (shown) or M199 agar (not shown).
Fungal-derived alarmins retain biological activity
Finally, we wanted to determine whether the C. albicans-derived S100 proteins were biologically active, and produced at sufficient levels to induce a response from mammalian leukocytes. Culture supernatants from the S100-producing strains were diluted 100-fold and tested for chemotactic activity upon the mouse macrophage-like, J774A.1 cell line. This revealed that supernatants from both S100A8- and S100A9-producing strains possessed chemotactic activity, compared with supernatants from the isogenic control (Fig. 4a). Combining S100A8 and S100A9 culture supernatants neither enhanced nor diminished this chemotactic activity. Culture supernatants were further evaluated for chemotactic activity upon primary mouse PMNs isolated from peritoneal lavage. The supernatants from the S100A8 strain showed a significant increase in PMN migration compared with those from the isogenic control (P = 0.034), while the activity of S100A9 culture supernatant was similar to those from the isogenic control (P = 0.20). This clearly established that the C. albicans-derived S100 proteins possess the expected biological activity and are exported in sufficient quantity to induce a physiological response from mammalian cells. It also demonstrates that, despite aberrant proteolytic cleavage, the C. albicans-derived S100A9 retained its capacity to induce a chemotactic response. The S100A8 secreted by our C. albicans strain has also been shown to possess chemotactic activity upon PMNs in vivo using a mouse model of vaginal candidiasis (J. Yano, G.E. Palmer, K.E. Eberle, T. Vogl, A.N. McKenzie & P.L. Fidel, unpublished data).
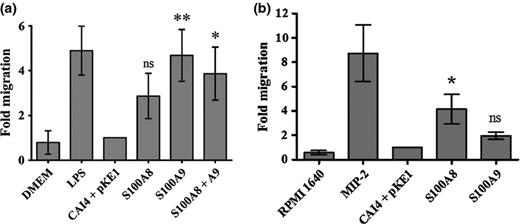
Candida albicans-derived S100A8 and S100A9 proteins possess chemotactic activity. (a) Culture supernatants from S100-producing or isogenic control strain were diluted 100-fold in DMEM medium, and chemotactic activity determined using a transwell assay with J774A.1 cells. Cell migration across the membrane was enumerated after 4 h, and scaled to express results as ‘fold migration’ relative to the isogenic control strain (CAI4 + pKE1). Results represent the mean and standard deviation of three separate experiments. DMEM alone provided our negative control, while LPS-spiked medium was a positive control. P values were calculated vs. the isogenic control using Tukey's honestly significant differences test n *P = 0.0254; **P = 0.0043; ns = not statistically significant. (b) PMN chemotaxis. Supernatants from overnight cultures of S100-producing or isogenic control strains were diluted 10-fold in chemotaxis buffer and tested in a transwell assay with primary mouse PMNs isolated from peritoneal lavage. The numbers of PMNs that migrated to the bottom chamber were quantified and expressed as the fold migration relative to the isogenic control strain. Results represent cumulative data from three separate experiments. Medium alone served as a negative control, while MIP-2 (100 ng mL−1) containing medium was tested in parallel as a positive control. P values were calculated vs. the isogenic control using Tukey's honestly significant differences test, *P = 0.0473; ns = not statistically significant.
The previous studies of Locksley and Wormley (Tobin et al., 1993; Wormley et al., 2007) established a precedent for engineering an invasive eukaryotic microbe to produce host proteins, capable of modulating the mammalian hosts' immune responses. We argue that the application of pathogen-based expression systems to produce host proteins can provide a novel and powerful approach to dissect mammalian defenses against invasive eukaryotic microorganisms. In this study, we have defined and validated a protein expression and export system for the most prevalent human fungal pathogen, C. albicans. Important caveats that need to be considered for such an approach include possible fungal-derived proteolysis of the said factor (which does not necessarily preclude biological function, as for S100A9 in this study). Specifically, it may be important that the ectopic protein does not contain any potential Kex2p cleavage sites (LYS-ARG). Expression of a foreign protein in any fungal pathogen is also likely to require optimization of the coding sequences. This is especially important in several Candida species due to the alternative translation of CTG codons as serine instead of leucine (Santos et al., 1993) and AT-rich genomes. In each case, it will be crucial to ensure that the pathogen-derived protein retains the expected biological activity and is secreted in sufficient quantities to induce a biological response, without impairing the viability or inherent pathogenicity of the microbe itself. The expression level and activity of the host protein is likely to vary with the nature and properties of the host protein itself, as well as the conditions under which C. albicans is grown. Careful selection of transcriptional promoters, as well as codon usage during ‘optimization’ may enable some control over the amount of protein produced and exported from the pathogen.
In this study, we have exploited the C. albicans Sap2p prepropetide to support the production and export of a host immunomodulatory factor by the most prevalent human fungal pathogen, C. albicans. This method is likely to have broad application, potentially enabling the fungal-derived production of a variety host immune factors at the site of infection in vivo.
Acknowledgements
The authors would like to thank Dr William Fonzi (Georgetown University) for providing strains and plasmids.
Authors' contributions
D.A.J. and J.Y. contributed equally to this work.
References
Author notes
Editor: Stefanie Poeggeler

{kind=link}
{kind=link}
{kind=link}
{kind=link}