





Abstract
The evolutionary theories of mutation accumulation (MA) and disposable soma (DS) provide possible explanations for the existence of human aging. To better understand the relative importance of these theories, we devised a test to identify MA- and DS-consistent sites across the genome using familial DNA methylation data. Two key characteristics of DNA methylation allowed us to do so. First, DNA methylation exhibits distinct and widespread changes with age, with numerous age-differentially-methylated sites observed across the genome. Second, many sites show heritable DNA methylation patterns within families. We extended heritability predictions of MA and DS to DNA methylation, predicting that MA-consistent age-differentially-methylated sites will show increasing heritability with age, while DS-consistent sites will show the opposite. Variance components models were used to test for changing heritability of methylation with age at 48,601 age-differentially-methylated sites across the genome in 610 individuals from 176 families. Of these, 102 sites showed significant MA-consistent increases in heritability with age, while 2266 showed significant DS-consistent decreases in heritability. These results suggest that both MA and DS play a role in explaining aging and aging-related changes, and that while the majority of DNA methylation changes observed in aging are consistent with epigenetic drift, targeted changes exist and may mediate effects of aging-related genes.
Evolutionary theories of aging
Aging is the progressive and general deterioration of an organism, defined by postmaturation declines in survival and fertility (Medawar 1952; Williams 1957; Hamilton 1966). Basic evolutionary theory suggests that such a trait should be selected against given its associated reductions in Darwinian fitness. However, seemingly paradoxically, aging is a universal feature of human life. The existence of aging remains an evolutionary puzzle.
Evolutionary theories provide possible explanations for the existence of human aging. Two major theories include: (1) mutation accumulation (MA) (Medawar 1952) and (2) antagonistic pleiotropy (AP) (Williams 1957), with disposable soma (DS) (Kirkwood 1977; Kirkwood and Holliday 1979; Kirkwood and Rose 1991) as a special case. Very generally, these theories suggest aging to occur because of a decline in the strength of selection with age. Even in the absence of aging, extrinsic mortality, or death due to external factors such as accident or starvation, causes fewer people to survive to higher ages (Medawar 1952). As a result, older ages matter increasingly less to lifetime reproductive success, and selection becomes increasingly ineffective (Hamilton 1966).
More specifically, MA suggests aging to be a nonadaptive consequence of the decline in the strength of selection with age (Medawar 1952). Mutations with deleterious effects confined to late in life have only small impacts on fitness, as carriers likely reproduce before the onset of mutation action. Such mutations are hidden from the full force of selection and are essentially neutral. As a result, these mutations can grow to high frequency and accumulate within a population’s germline over many generations (Hughes and Reynolds 2005). The resulting burden of late-acting deleterious mutations is suggested to cause aging under MA.
In contrast to MA, AP suggests aging to be an adaptive consequence of an evolutionary trade-off between survival and reproduction (Williams 1957). AP proposes the existence of a specific type of pleiotropic gene that has opposite effects on fitness at different ages. These genes are said to be antagonistically pleiotropic, and present a potential trade-off between early and late life, or survival and reproduction. Selection of these antagonistically pleiotropic genes depends both on the magnitude and timing of the opposing effects. Fitness advantages conferred early in life can easily selectively outweigh accompanying late-life costs due to the weakening strength of selection with age. AP suggests aging to be caused by the deleterious late-life effects of antagonistically pleiotropic genes that have accumulated in the population germline through active selection of their early-life benefits.
A special case of AP is DS (Kirkwood and Holliday 1979). Similar to AP, DS considers aging to be an adaptive consequence of the evolutionary optimization of a general trade-off between survival and reproduction (Kirkwood 1977; Kirkwood and Holliday 1979; Kirkwood and Rose 1991). In particular, DS suggests aging to result from trade-offs in the allocation of finite energy resources between biological functions such as growth, reproduction, and maintenance. Selection works to optimize energy allocation strategies to maximize fitness, and evolves energetic limits for each function. Given the limited amount of energy that can be allocated toward maintenance, mechanisms for somatic maintenance and repair mechanisms cannot be perfect. Imperfect maintenance and repair mechanisms cause unrepaired cellular and molecular damage to accumulate over the lifetime of an individual. DS suggests aging to be caused by this accumulation of somatic damage throughout life.
DS is often considered to be a special case of AP due to their shared general trade-off framework. However, since AP and DS differ in their suggested trade-off mechanisms and underlying causes of aging (Kirkwood and Rose 1991; Kirkwood and Austad 2000), we consider them to be related but separate theories and investigate their predictions independently.
The theories of MA, AP, and DS are not mutually exclusive and large bodies of literature provide support for aspects of each individual theory (Gavrilov and Gavrilova 2002). It is possible that all three theories play some role in explaining the features and existence of human aging, but the relative importance of each theory has not yet been well established (Partridge and Barton 1993). A better understanding of each theory’s contribution will help to clarify the roles of the environment and different types of genes in the aging process. Many of the methods previously used to test these theories have been able to provide support for one theory over another, but have not been able to speak to the size of the contribution each theory makes in explaining aging (Robins and Conneely 2014). Here, we have devised a unique test using DNA methylation data that will allow us to better understand the relative importance of MA and DS evolutionary models of aging.
DNA methylation and aging
DNA methylation is an epigenetic modification that is dynamic with age (Fraga and Esteller 2007; Bocklandt et al. 2011; Koch and Wagner 2011; Alisch et al. 2012; Hannum et al. 2013; Horvath 2013; Xu and Taylor 2014) and has been shown to be heritable in cross-sectional family studies (McRae et al. 2014; Day et al. 2016). It involves the addition of a methyl group to the 5-position of a cytosine base, and typically occurs at a CpG site, where a cytosine base is directly followed by a guanine base. Functionally, DNA methylation of gene promoter regions is often associated with gene expression silencing (Razin and Riggs 1980; Jaenisch and Bird 2003; Bell et al. 2012).
Robust age-associated changes in DNA methylation occur throughout the genome (Fraga and Esteller 2007; Bocklandt et al. 2011; Koch and Wagner 2011; Alisch et al. 2012; Hannum et al. 2013; Horvath 2013; Xu and Taylor 2014). That is, numerous CpG sites consistently show variation in methylation between young and old ages. Many sites also show heritable patterns of methylation, where the measured level of methylation is more similar between closely related than unrelated individuals (Bell et al. 2012; McRae et al. 2014; Day et al. 2016). This suggests that a genetic component underlies the variation in methylation at these CpG sites.
An environmental or stochastic component to the variation in methylation is also suggested, as the nearly identical DNA methylation patterns of monozygotic twins at birth have been observed to diverge with age (Fraga et al. 2005; Martin 2005; Zampieri et al. 2015). This age-related divergence in the methylation patterns of relatives has been termed epigenetic drift (Teschendorff et al. 2013; Issa 2014; Sun and Yi 2015). The exact mechanisms driving these changes are not yet understood, but both external environmental and internal cellular events, such as imperfect methylation maintenance, have been hypothesized to contribute (Fraga and Esteller 2007; Hannum et al. 2013). The age-associated DNA methylation changes of epigenetic drift are suggested to be acquired stochastically (Jones et al. 2015), and align with DS (Kirkwood 2005).
DNA methylation has been suggested as a biomarker of human aging, or an easily repeatable measure that is descriptive of biological age (Baker and Sprott 1998). The genome-wide patterns of methylation have been observed to be dynamic throughout life, with the methylation at numerous CpG sites shown to have strong associations with age. These age-associated changes in methylation have been reported at thousands of sites across the genome in human blood samples (Alisch et al. 2012; Hannum et al. 2013; Horvath 2013; Xu and Taylor 2014). Furthermore, chronological age can be accurately predicted from the methylation measurements at just a few 100 of these CpG sites (Hannum et al. 2013; Horvath 2013). DNA methylation at these sites shows consistent changes with age across individuals, as well as cell and tissue types, and forms an “epigenetic clock” (Jones et al. 2015). Estimates of biological age derived from the methylation measurements at the clock-like CpG sites have been found to predict mortality better than chronological age (Marioni et al. 2015). Together, these observations indicate that DNA methylation changes, both en masse and site-specific, reflect aspects of the aging process, and can be regarded as a biomarker of aging against which predictions of evolutionary theories can be tested.
Heritability of DNA methylation
In this study, we test the contrasting heritability predictions of MA and DS against familial DNA methylation data. We do not test heritability predictions of AP, as AP predicts a wide range of patterns of genetic variation, including patterns also expected under MA (Moorad and Promislow 2009). Unlike AP, specific predictions about genetic variation and heritability can be made for the theories of MA and DS, and these predictions are contrasting (Charlesworth and Hughes 1996; Kirkwood 2005; Moorad and Promislow 2009).
MA suggests the heritability of life span and other aging-related traits to increase with age. MA assumes aging to be caused by deleterious late-acting mutations that have accumulated in a population’s germline over many successive generations due to a decline in strength of selection with age (Medawar 1952). Because of this weakening selection, the equilibrium population frequency of deleterious mutations is expected to increase with the age of onset of mutation action (Charlesworth 1980; Partridge and Barton 1993). Increases in the number of mutations in the population equate to increases in genetic variation. As a result of this increasing genetic variation, the heritability of life span and other features of aging is also predicted to increase with age (Charlesworth 1994; Gavrilova et al. 1998; Gavrilov and Gavrilova 2002). In contrast to MA, DS assumes aging to be caused by random somatic damage that accumulates over the lifetime of an individual (Kirkwood and Rose 1991). This damage results from random failure events of somatic maintenance and repair mechanisms and predicts an inherently stochastic process of aging (Kirkwood 2005). This stochasticity under DS is expected to cause the phenotypic variation of aging-related traits to increase with age, which in turn causes the heritability of these traits to decrease with age.
If age-associated methylation changes are consistent with MA, we suggest that they mediate the age-specific effects of deleterious genetic mutations, and that the heritability of DNA methylation will increase with age. In contrast, if age-associated methylation changes are consistent with DS, we suggest that they result from stochastic failures in maintenance and repair mechanisms, and that the heritability of DNA methylation will decrease with age. In this study, we test for increasing and decreasing heritability of DNA methylation at age-differentially-methylated CpG sites. Sites where the heritability of methylation increases with age will be considered consistent with MA, while sites where the heritability of methylation decreases with age will be considered consistent with DS.
The existence of widespread changes in DNA methylation with age is potentially consistent with both MA and DS. Our novel use of DNA methylation data in testing these theories allows us to categorize individual CpG sites across the genome as consistent with either MA or DS, or inconsistent with both theories, and to assess the ability of each theory to explain the DNA methylation changes observed in aging (Robins and Conneely 2014).
Additionally, we test for the existence of a heritable rate of aging, which is consistent with both MA and DS. Under DS, genes regulating the accuracy of somatic maintenance and repair are suggested to dictate the rate of somatic damage accumulation and imply a heritable rate of aging that is constant throughout life. Under MA, deleterious genes with age-specific effects are suggested to cause aging, and imply a heritable rate of aging that is potentially variable throughout life (e.g., slow at young ages and fast at older ages). The prediction of a heritable rate of aging can be tested using a methylation-derived measure of the aging rate. An individual’s age can be estimated from DNA methylation levels at 353 CpG sites via a predictive linear model developed by Horvath (2013). The difference between an individual’s methylation-estimated age and chronological age provides a measure called age acceleration that describes that individual’s rate of aging (i.e., fast or slow). Here, we estimate the heritability of age acceleration using familial DNA methylation data. A significant nonzero heritability of age acceleration will indicate that changes in DNA methylation across a few hundred CpG sites are consistent with evolutionary models of aging. This will allow us to see if the patterns observed at the level of single CpG sites extend to a larger scale across the genome.
Materials and Methods
Overview of hypotheses to be tested
We tested for increasing or decreasing heritability of methylation with age using a variance components model. At each CpG site we tested three specific hypotheses:
Age is a predictor of methylation level.
Methylation level has a heritable component.
The heritability of methylation has an age-dependent component.
Testing the first and second hypotheses allowed us to define a set of age-differentially-methylated CpG sites and a set of CpG sites with heritable methylation levels for further investigation. To test the third hypothesis, we restricted the analyses to sites that are age-differentially-methylated and have heritable methylation levels. Testing this hypothesis allows us to directly test for increasing or decreasing heritability of methylation with age, and to determine which CpG sites have age-associated methylation changes that are consistent with MA or DS.
Familial DNA methylation data
DNA methylation was measured in a sample of 610 individuals from 176 different families recruited for the Brisbane Systems Genetics Study (BSGS) (Powell et al. 2012). These families are all of European descent, and are comprised of adolescent monozygotic and dizygotic twin pairs, their siblings, and their parents. The age distribution for these individuals ranges from 10 to 75 years, and has a mean age of 21 years (Supplemental Material, Figure S1 in File S1).
Measuring DNA methylation
DNA methylation was measured from blood samples using the Illumina Infinium HumanMethylation450 Beadchip (Bibikova et al. 2011). This array interrogates a total of 482,421 CpG sites and 3156 non-CpG sites across the genome using a bisulfite DNA treatment and two sets of site-specific probes binding associated methylated and unmethylated sequences (Triche et al. 2013). The proportion of DNA strands methylated at any particular site was estimated as the measured intensity of fluorescent signal from methylated probes relative to the intensity of fluorescent signal from both methylated and unmethylated probes. This ratio, with the addition of a stabilizing constant of 100 to the denominator, is referred to as a -value. Each individual sample was measured on a randomly-assigned chip and at a randomly-assigned position within that chip to avoid potential confounding due to family membership (McRae et al. 2014).
DNA methylation data quality control
The measured methylation state of a CpG site can be directly affected by the underlying DNA sequence. If there is a genetic variant at the cytosine or guanine of a CpG site, for instance, the site cannot be methylated. Furthermore, a genetic variant in the sequence probed by the array can impact array-binding affinity and bias the measured level of methylation. To minimize the impact of these direct genetic effects on our estimates of DNA methylation heritability, we removed CpG sites with SNPs present on the 50-base CpG site probe before performing heritability analyses. Sites with underlying SNPs were identified based on data from the 1000 Genomes Project phase I release, as annotated by (Barfield et al. 2014). We further cleaned the data by removing probes annotated as binding to multiple chromosomes, probes without CpG sites, and probes with > 11 individuals with missing data or > 5 individuals with detection P-values > 0.001 (McRae et al. 2014). After cleaning, a total of 373,006 probes remained for testing. A chart illustrating the data cleaning process is provided in the supplemental information (Figure S4 in File S1).
Each probe was residualized using a generalized linear model with a logistic link function similar to that used by McRae et al. (2014). The covariates in our model included chip, position on chip, and estimated proportions of the following cell types: CD8-positive T cells; CD4-positive T cells; natural killer cells; B-cells; monocytes; and granulocytes. The cell type proportions were estimated from the methylation array data using a method proposed by Houseman et al. (2012) and reference data on the methylation signatures of purified cell types (Reinius et al. 2012). We included these estimated cell type proportions as covariates in the model to avoid potential confounding due to the heterogeneous and changing cellular composition of whole blood. After residualizing each probe, we removed outlying measurements > 5 interquartile ranges for the nearest quartile. McRae et al. (2014) found these outlying data points, likely caused by rare genetic variants or measurement errors, to have a large influence on heritability estimates. After removing outliers, the residuals from the above model were used as the phenotype for all heritability analyses.
Modeling changes in the heritability of DNA methylation with age
To identify CpG sites consistent with each evolutionary theory, we fitted the familial BSGS data to a model of age and methylation that takes into account family structure and other relevant covariates.
For each CpG site, we implemented this model in SOLAR (Almasy and Blangero 1998), a statistical genetics software package, and tested our specific hypotheses by adding restrictions to the general model described above. Each hypothesis was tested using a likelihood ratio test (LRT), comparing the fit of a full model to the fit of a restricted model. At each CpG site we tested three specific hypotheses: (1) age is a predictor of methylation level; (2) methylation has a heritable component; and (3) the heritability of methylation has an age-dependent component.
To test the first two hypotheses, we did not partition the genetic and environmental variance into baseline and age-dependent terms as shown in the general model above. The full and restricted models used to test these hypotheses included and with no age-dependence (i.e., and set to 0), while the full and restricted models used to test the third hypothesis included and with age-dependence as shown in Equation (2). To test hypothesis 1, we tested the restriction that = 0. Sites where was found to be significantly nonzero after multiple test correction [false discovery rate (FDR < 0.05)] were defined as age-differentially-methylated. To test hypothesis 2, we tested the restriction that = 0. Sites where was found to be significantly nonzero after multiple test correction (FDR < 0.05) were designated as heritable.
We limited tests of hypothesis 3 to CpG sites that are both age-differentially-methylated and moderately heritable, with h2 > 0.2. To test hypothesis 3, we fitted two separate restricted models, with and independently restricted to zero. These two restrictions allowed us to test for age-dependent components in both genetic and environmental variance. This is necessary as heritability depends on both genetic and environmental variance, and age-dependent changes in either genetic or environmental variance will cause age-dependent changes in heritability.
We performed simulations to estimate the type I error rate and our power to detect age-dependent changes in genetic and environmental variance (for more information see File S1). Our results indicate that our modeling approach has appropriate levels of type I error and that our power to detect age-dependent changes is approximately equivalent for the genetic and environmental variances (that is, power is similar for tests of the restrictions = 0 and = 0).
Estimating the heritability of rate of aging using DNA methylation
We used a linear model developed and tested by Horvath (2013) to predict age using DNA methylation data from 353 CpG sites. The sites included in the model were selected using elastic net regression, and have been shown to accurately predict age across many cell and tissue types. We estimated methylation-derived ages for all 610 BSGS individuals with Horvath’s model and unresidualized -values. After estimating methylation age, we calculated age acceleration, defined as the difference between methylation age and chronological age. Positive values of age acceleration suggest an increased rate of aging (i.e., fast aging), while negative values suggest a decreased rate of aging (i.e., slow aging).
To estimate the heritability of rate of aging, we modified the model shown in Equations (1) and (2) to include age acceleration as the outcome. As described earlier, we tested for a heritable component by restricting to zero and comparing the full and restricted models with an LRT. The model was implemented in SOLAR (Almasy and Blangero 1998).
Annotation for genomic features
All CpG sites were annotated with respect to the following genomic features: CpG islands, CpG shores, CpG shelves, strong promoters, weak promoters, poised promoters, strong enhancers, weak enhancers, insulators, transcription factor-binding sites (TFBS), and CTCF-binding sites. For this annotation, we used three data sets downloaded from the University of California, Santa Cruz (UCSC) table browser for GRCh37/hg19 (ENCODE Project Consortium 2012) (Karolchik et al. 2004): (1) CpG Islands (Gardiner-Garden and Frommer 1987); (2) Broad ChromHMM for GM12878 (Ernst and Kellis 2010); and (3) Transcription factor ChIP V3 (TFBS) (ENCODE Project Consortium 2012). Each CpG site was annotated based on overlaps between the CpG location and the intervals of the genomic features provided by the UCSC data sets. We defined CpG island shores to be 1.5 kb out from CpG islands, and CpG island shelves to be 1.5 kb out from shores. For all other genomic features we adopted corresponding ChromHMM category definitions presented in Ernst et al. (2011).
Replication data
Familial DNA methylation data from the Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study, previously described by Corella et al. (2007), Irvin et al. (2010), and Day et al. (2016), were used to replicate the results of our analyses. Families with at least two siblings were recruited from participants in the National Heart, Lung, and Blood Institute Family Heart Study in Minneapolis, MN and Salt Lake City, UT (Hidalgo et al. 2014). DNA methylation data were available for 1050 individuals from 182 families. The age distribution for these individuals ranges from 18 to 88 years, and has a mean of 49 years (Figure S5 in File S1).
DNA methylation was measured from isolated CD4+ T cells using the Illumina Infinium HumanMethylation450 Beadchip (Bibikova et al. 2011). This chip was also used to measure DNA methylation from the BSGS blood samples, and is described in more detail in the “Measuring DNA methylation” section above. -values with a detection P-value > 0.01, samples with > 1.5% of probes missing, probes for which > 10% of the samples had inadequate intensity, and probes mapped to more than one location or a location not matching the annotation file were removed prior to analysis (Hidalgo et al. 2014). After these quality control steps, 991 individuals and 461,281 CpG sites remained. As these data were originally prepared for another study, the quality control criteria and specific sites considered suitable for analysis slightly differ between the GOLDN data and our original BSGS data. Only GOLDN data that passed the described quality control criteria were available for our replication analyses.
Expression data quality control and annotation
Gene expression levels were measured from the BSGS blood samples using Illumina HT12-v4.0 bead arrays (Powell et al. 2012, 2013). These arrays contain 47K probes designed to cover all well-characterized genes, gene candidates, and splice variants (Illumina 2011). Expression was measured for all 610 individuals for whom DNA methylation levels were also measured. Each individual sample was measured on a randomly-assigned chip and at a randomly-assigned position to avoid any potential confounding.
Before beginning our analyses, we removed probes where < 10% of the samples had a detection P-value < 0.05, as well as probes with overlapping SNPs and probes of low quality (i.e., probes unlikely to match the target transcript due to sequence mismatches or sequence matches at multiple locations), as annotated by Barbosa-Morais et al. (2010). After cleaning, the expression values of the remaining 13,222 probes were log transformed for analysis.
Gene information was annotated to each expression probe using Refseq transcript exon intervals downloaded from the UCSC table browser for hg19. For exons of the same gene with overlapping intervals, the union of the intervals was taken for consistency. Refseq gene information was annotated to an expression probe using the R Bioconductor package GenomicRanges (Lawrence et al. 2013), if there was more than a 25 bp overlap between the probe and exon interval.
Identifying changes in expression associated with age-related changes in DNA methylation
Data availability
Data from BSGS are archived at the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), accession numbers GSE56105 (DNA methylation) and GSE53195 (gene expression). DNA methylation data from GOLDN are archived on the database of genotypes and phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap), accession number phs000741.v1.p1.
Results and Discussion
Age-differentially-methylated CpG sites
In total, 91,261 CpG sites (24% of CpG sites tested) had significant association with age after multiple test correction (FDR < 0.05). Of these sites, 47% (43,029 CpG sites) had significant increases in methylation with age, while 53% (48,232 CpG sites) had significant decreases in methylation with age (Figure 1). The large number of CpG sites showing age-differential-methylation is consistent with results observed in other epigenome-wide association studies for age. For example, we tested 600 CpG sites previously shown to have significant association with age across four independent data sets (Xu and Taylor 2014). Of these 600 CpG sites, 582 (97%) were age-differentially-methylated in a concordant direction in our data after multiple test correction. Additionally, we tested 290 of the 353 CpG sites that were included in the age prediction model of (Horvath 2013). Of these 290, 144 (∼50%) of these sites were significantly age-differentially-methylated after multiple test correction.
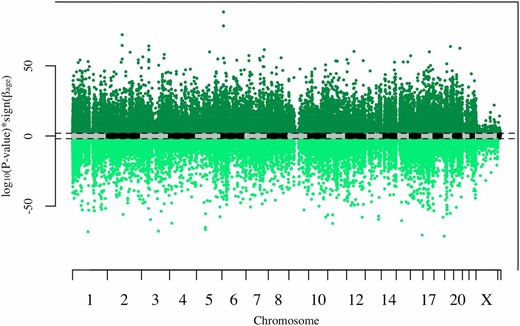
Manhattan plot of P-values with sign of estimate. Dashed lines represent genome-wide significance (false discovery rate < 0.05). Each point represents one CpG site. CpG sites with significant increases in methylation with age are colored dark green, while CpG sites with significant decreases in methylation with age are colored light green.
Heritability of DNA methylation
We estimated the average heritability of DNA methylation across all CpG sites to be 0.177 (Figure 2). A heritability of 0 was estimated at 20% of sites (72,927 sites), and significant nonzero heritability was estimated at 38% of sites after multiple test correction (142,169 sites, FDR < 0.05). After restricting to age-differentially-methylated CpG sites, the average estimated heritability of methylation increased to 0.272 (Figure 2). These heritability estimates are consistent with the results previously reported by McRae et al. (2014).
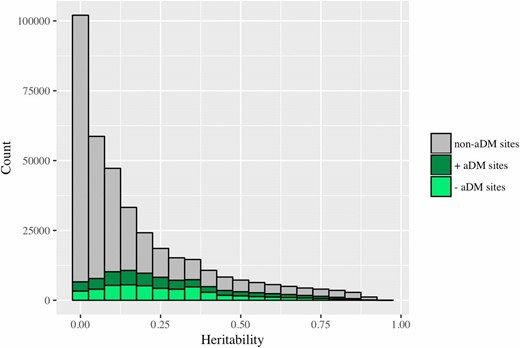
Distribution of heritability estimates for DNA methylation levels at age-differentially-methylated (aDM) and non-age-differentially-methylated sites (non-aDM). The average estimated heritability of methylation across all sites is 0.177. The average estimated heritability of methylation at aDM CpG sites is 0.272.
We performed Fisher’s exact tests to test if age-differentially-methylated sites are more likely than other sites to have heritable methylation levels. Specifically, we examined the overlap between sets of (1) age-differentially-methylated CpG sites and (2) CpG sites with heritable methylation, and tested whether this overlap is greater than expected by chance. These sets were defined after multiple test correction at varying significance levels, to ensure that results do not depend on a specific -level (Figure 3). We observed significant overlap between the sets of age-differentially-methylated sites and sites with heritable methylation at all significance levels, indicating that age-differentially-methylated CpG sites are more likely to have heritable methylation than other sites. This observed enrichment indicates a potential genetic basis for age-related DNA methylation changes.
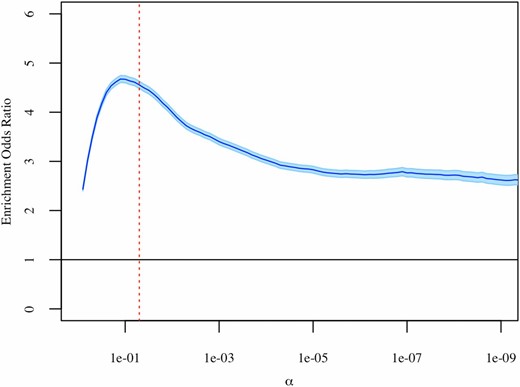
Results of Fisher’s exact tests at varying significance levels. Sets of age-differentially-methylated CpG sites and heritable CpG sites were defined after multiple test correction. The estimated enrichment odds ratio is shown in dark blue, with the 95% C.I. shown in light blue. The dashed red line represents α = 0.05.
Our models adjusted for both age and family structure simultaneously, so the overlap between age-differentially-methylated and heritable sites is unlikely to be due to confounding between age and family structure. Furthermore, to ensure that this overlap did not reflect differences in statistical power among CpG sites with high vs. low variability, we compared the variance of methylation across sites. We observed similar distributions for the variance of methylation in the following sets of CpG sites: age-differentially-methylated sites with heritable methylation vs. age-differentially-methylated sites with nonheritable methylation; and non-age-differentially-methylated sites with heritable methylation vs. non-age-differentially-methylated sites with nonheritable methylation (Figure S6 in File S1). Importantly, sites that were both age-differentially-methylated and heritable did not appear to have increased variability in methylation. This shows that the overlap between age-differentially-methylated sites and sites with heritable methylation is not simply driven by differences in phenotypic variation.
Changes in the heritability of DNA methylation with age
Our confidence in the estimated direction of heritability change with age for each site, and the resulting theory classification, increases with the significance of the and terms. We have increased confidence in the classification of a site as consistent with MA when that site has a significantly nonzero and positive (FDR < 0.05). At these sites, there are significant increases in the genetic variation underlying the variation in DNA methylation with age. Similarly, we have increased confidence in the classification of a site as consistent with DS when that site has a significantly nonzero and positive (FDR < 0.05). At these sites, there are significant increases in the environmental variation underlying the variation in DNA methylation with age. We are most confident in the classification of sites where both and are significant (FDR < 0.05), as in these cases we are confident in the direction of change for both the genetic and environmental variation underlying the variation in DNA methylation at those sites.
Because of this, we have divided the sites we found to be consistent with MA and DS into three different groups on the basis of the significance of the and terms (Table 1). Group 1 contains all sites consistent with MA or DS, irrespective of significance. In total, 13,467 sites were found to be consistent with MA, while 30,749 sites were found to be consistent with DS. Group 2 contains sites that are consistent with MA and have a significant term (FDR < 0.05), and sites that are consistent with DS and have a significant term (FDR < 0.05). A total of 102 sites found to be consistent with MA have significant terms, while a total of 2266 sites found to be consistent with DS have significant terms. Group 3 contains sites that are consistent with MA or DS and have significant and terms (FDR < 0.05). A total of 70 sites found to be consistent with MA have significant and terms, while a total of 203 sites found to be consistent with DS have significant and terms.
Categorization of heritable CpG sites
| CONSISTENT WITH MA | CONSISTENT WITH DS | |
|---|---|---|
| Increasing with age | Decreasing with age | |
| Group | > | < |
| 1: All consistent sites | 13,467 | 30,749 |
| 2: Significant (if MA) or (if DS) | 102 | 2,266 |
| 3: Significant and | 70 | 203 |
| CONSISTENT WITH MA | CONSISTENT WITH DS | |
|---|---|---|
| Increasing with age | Decreasing with age | |
| Group | > | < |
| 1: All consistent sites | 13,467 | 30,749 |
| 2: Significant (if MA) or (if DS) | 102 | 2,266 |
| 3: Significant and | 70 | 203 |
Counts of significant sites were determined after multiple test correction (false discovery rate < 0.05). MA, mutation accumulation; DS, disposable soma.
| CONSISTENT WITH MA | CONSISTENT WITH DS | |
|---|---|---|
| Increasing with age | Decreasing with age | |
| Group | > | < |
| 1: All consistent sites | 13,467 | 30,749 |
| 2: Significant (if MA) or (if DS) | 102 | 2,266 |
| 3: Significant and | 70 | 203 |
| CONSISTENT WITH MA | CONSISTENT WITH DS | |
|---|---|---|
| Increasing with age | Decreasing with age | |
| Group | > | < |
| 1: All consistent sites | 13,467 | 30,749 |
| 2: Significant (if MA) or (if DS) | 102 | 2,266 |
| 3: Significant and | 70 | 203 |
Counts of significant sites were determined after multiple test correction (false discovery rate < 0.05). MA, mutation accumulation; DS, disposable soma.
Figure 4 compares the significance and sign of and at each site, and shows the separation of sites into different groups. In this figure, the dotted lines represent the genome-wide significance levels for and These significance levels, along with the axes, divide the graph into 16 sections that can be used to visualize the categorization of sites. For instance, sites with significant positive values and significant negative values fall into the upper left section. All these sites are consistent with DS and included in the counts for groups 1, 2, and 3. Counts for each section have been superimposed on the graph, and each section has been color-coded to show theory classification and group inclusion. Red denotes sections with sites that are consistent with MA, while blue denotes sections with sites that are consistent with DS. Group membership based on significance is designated by color shade and numbered brackets. The dashed line represents the = identity line, and serves to separate counts in sections where there are sites that are consistent with both MA and DS.
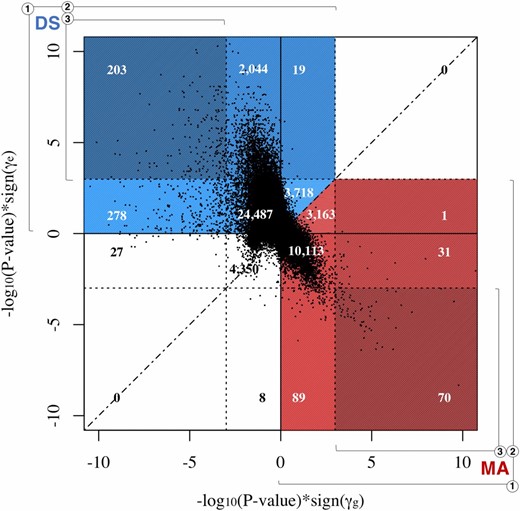
Scatterplot visualization of categorization of mutation accumulation (MA)- and disposable soma (DS)-consistent sites. The significance and sign of the estimated values of (x-axis) are plotted against the significance and sign of the estimated values of (y-axis). Each point represents one CpG site. The dotted lines represent the genome-wide significance levels for and and divide the graph into 16 sections used to visualize the categorization of sites. Counts are superimposed onto color-coded sections to show theory classification and group inclusion. Red indicates a section with MA-consistent sites. Blue indicates a section with DS-consistent sites. Group membership based on significance is indicated by color shade and numbered brackets. The dashed line represents the = identity line, and serves to separate counts in sections where there are sites that are consistent with both MA and DS.
Comparison of sites consistent with MA and DS
To gain a better understanding of what distinguishes MA and DS sites, we analyzed the locations, genomic features, and gene ontology associated with the CpG sites we found to be consistent with each theory.
Location:
The locations of CpG sites found to be consistent with MA do not notably differ from the locations of CpG sites found to be consistent with DS. This was observed for all three significance groups, as shown in Figure 5.
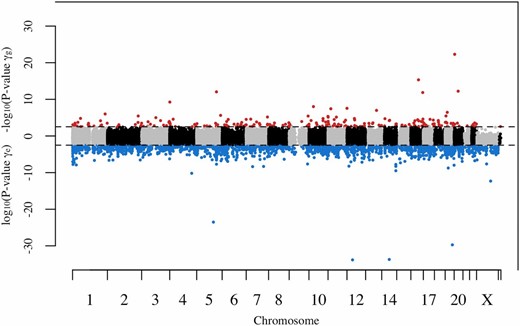
Manhattan plot of and P-values. Each point represents one CpG site consistent with either MA or DS. The dashed lines represent genome-wide significance (FDR < 0.05). All CpG sites consistent with MA and belonging to significance groups 2 and 3 are in red. All CpG sites consistent with DS and belonging to significance groups 2 and 3 are in blue. DS, disposable soma; FDR, false discovery rate; MA, mutation accumulation.
Enrichment for age-methylation or age-demethylation:
Fisher’s exact tests were performed on the sets of sites found to be consistent with MA and DS to test if these sites are more likely than others to be age-methylated or age-demethylated. To increase the power of these tests, we compared all sites found to be consistent with MA or DS, irrespective of significance (group 1), to the set of all sites tested for age-differential methylation. We defined age-methylated sites as those with increases in DNA methylation with age ( > 0), and age-demethylated sites as those with decreases in DNA methylation with age ( < 0).
We found MA sites to have significant depletion for age-methylation (OR = 0.86, P = 1.03 × 10−16), which is equivalent to significant enrichment for age-demethylation. In contrast, we found DS sites to have significant enrichment for age-methylation (OR = 1.16, P = 7.64 × 10−34) (Figure 6A). This means that DS-consistent sites are more likely than other sites to be age-methylated, while MA-consistent sites are more likely to be age-demethylated.
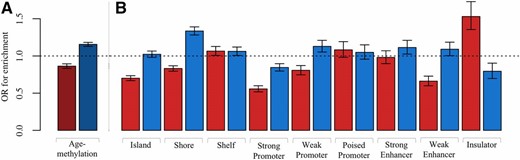
Histogram of enrichment in MA and DS sites. (A) Enrichment for age-methylation. MA sites are shown in red and DS sites are shown in blue. (B) Enrichment for genomic features. For each feature, MA sites are shown in red and DS sites are shown in blue. DS, disposable soma; MA, mutation accumulation; OR, odds ratio.
Enrichment for genomic features:
To better understand the genomic context of MA and DS sites, Fisher’s exact tests were performed to test for enrichment of the following genomic features: CpG islands, CpG island shelves, CpG island shores, strong promoters, weak promoters, poised promoters, strong enhancers, weak enhancers, and insulators. We compared all sites found to be consistent with MA or DS irrespective of significance (group 1), to the set of all sites tested for changes in heritability of DNA methylation with age. Previous work has shown age-methylated sites to be more likely to be located within CpG islands, and age-demethylated sites to be more likely to be located outside CpG islands (Christensen et al. 2009). We found MA sites to be significantly depleted in CpG islands (OR = 0.70, P = 1.43 × 10−51), which is consistent with previous work given our finding that MA sites are enriched for age-demethylation.
The enrichment pattern of MA and DS sites that was observed for CpG islands extended to CpG shores, defined to be 1.5 kb out from CpG islands. We found DS sites to be significantly enriched in CpG shores (OR = 1.33, P = 8.81 × 10−45), and MA sites to be significantly depleted (OR = 0.83, P = 6.98 × 10−17; Figure 6B). However, this pattern did not extend to CpG shelves, defined to be 1.5 kb out from CpG shores. We found no notable difference between MA and DS sites in enrichment for shelves; both sets of sites were enriched in shelves.
Weak promoters and weak enhancers were found to have similar enrichment patterns for MA and DS sites. We found MA sites to be significantly depleted in weak promoters and weak enhancers (OR = 0.81, P = 2.47 × 10−8 and OR = 0.66, P = 3.75 × 10−18, respectively), and DS sites to be slightly enriched (OR = 1.13, P = 5.74 × 10−14 and OR = 1.09, P = 0.04, respectively).
The one genomic feature we found to be significantly enriched in MA sites (OR = 1.53, P = 1.21 × 10−12) and significantly depleted in DS sites (OR = 0.79, P = 3.54 × 10−4) was insulators (Figure 6B). Previous work has suggested that the DNA methylation status of an insulator affects the binding of the transcriptional repressor CTCF, which may preferentially bind to unmethylated sequences (Kang et al. 2015). We found MA sites to be more likely to be located in insulators and to lose methylation with age. These results, combined with the findings of previously published work, suggest that MA sites may directly influence changes in transcription with age.
Gene ontology:
Gene ontology analyses were performed to gain a better understanding of the specific genes or type of genes that are associated with DNA methylation changes at the CpG sites consistent with MA and DS. We used the GOstats package in R (Falcon and Gentleman 2007) to assess whether any terms describing biological processes, molecular functions, or cellular components associate with the sets of genes closest to the CpG sites consistent with MA or DS. The gene closest to each CpG site was defined based on distance to transcription start site, as in Barwick et al. (2016). We analyzed all sites found to be consistent with MA or DS, irrespective of significance (group 1). Table S2 in File S1 shows the top five significant terms after multiple test correction for the MA and DS sets for each ontology category (FDR < 0.05), as well as the total number of significant terms associated with each set. Overall, the genes closest to DS sites are enriched for fewer molecular functions (3 vs. 2), cellular components (2 vs. 0), and biological processes (19 vs. 5) than the genes closest to MA sites. This is evidenced by fewer significant terms and higher P-values associated with the genes closest to DS sites (Table S2 in File S1). A lack of enrichment for functionality is consistent with stochastic age-associated DNA methylation changes and the stochastic process of aging that is suggested by DS.
Replication of results
To assess the generality of our results, we tested for changes in the heritability of DNA methylation with age in an independent data set from the GOLDN study. Our analysis was restricted to sites found to be consistent with MA or DS in the BSGS data set at the group 2 level of significance (significant for MA sites and significant for DS sites). After quality control, data from the GOLDN study were available for 101 of the 102 MA sites, and 2164 of the 2266 DS sites.
In the GOLDN data, 958 CpG sites were found to be consistent with MA and 1266 CpG sites were found to be consistent with DS, irrespective of significance (group 1). Of these sites, 56 sites were found to be consistent with MA and 1221 sites were found to be consistent with DS in both the GOLDN and BSGS data sets. This relates to a 55% replication for MA sites and 56% replication for DS sites.
When we restricted to sites with significant or values (group 2), 15 CpG sites were found to be consistent with MA and 247 sites were found to be consistent with DS. Of these sites, four sites were found to be consistent with MA and 229 sites were found to be consistent with DS in both the GOLDN and BSGS data sets.
Across significance groups (i.e., groups 1 and 2), more sites were found to be consistent with DS than with MA in both the GOLDN and BSGS data sets. Although this general trend replicated well, there was little replication at the CpG site level. The low replication rates we observed are likely due to differences in data composition. BSGS measured methylation from whole blood, while the GOLDN study measured methylation from isolated CD4+ T cells. To avoid potential confounding due to the heterogeneous cellular composition of whole blood, the BSGS methylation β-values were residualized on cell type proportions before running analyses. Since the methylation of a single cell type was measured in the GOLDN study, raw β-values were used in analyses. Additionally, the BSGS families are comprised of adolescent twins, their siblings, and their parents, while the GOLDN study families are comprised of adult siblings. This results in a bimodal age distribution with a mean age of 21 for the BSGS subjects (Figure S1 in File S1), and a unimodal age distribution with a mean age of 49 for the GOLDN subject (Figure S5 in File S1).
Replicated MA sites with significant and expression analysis
To help deepen our understanding of what characterizes CpG sites with MA-consistent age-associated DNA methylation changes, we investigated the attributes of sites found to be consistent with MA in both the BSGS and GOLDN data sets. Only MA-consistent sites were tested, as we expect the DNA methylation changes at DS-consistent sites to be stochastic and to associate with a random set of genes. We limited our investigation to the four MA-consistent sites with significant values in both data sets, as we are the most confident in the theory categorization at these sites. Table 2 lists the observed direction of change in methylation with age, and the annotated location, nearest gene, and genomic features and states of these four sites. Notably, half of the replicated MA sites are located within a TFBS. This indicates that MA sites may directly influence changes in transcription with age.
Attributes and features of the replicated MA sites with significant
| CpG Site | Location | Methylation Change with Age | Nearest Gene | Genomic Features and States |
|---|---|---|---|---|
| cg02914422 | Chr 7: 32110145 | Age-methylated | PDE1C | CpG island |
| TFBS | ||||
| Polycomb repressed | ||||
| CTCF-binding site | ||||
| cg05691152 | Chr 22: 38092978 | Age-methylated | TRIOBP | TFBS |
| Weak/poised enhancer | ||||
| CTCF-binding site | ||||
| cg13672736 | Chr 9: 135114066 | Age-demethylated | NTNG2 | CpG shelf |
| Weakly transcribed | ||||
| cg25038330 | Chr 10: 463561 | Age-demethylated | DIP2C | CpG shelf |
| Hetrochromatin; low signal |
| CpG Site | Location | Methylation Change with Age | Nearest Gene | Genomic Features and States |
|---|---|---|---|---|
| cg02914422 | Chr 7: 32110145 | Age-methylated | PDE1C | CpG island |
| TFBS | ||||
| Polycomb repressed | ||||
| CTCF-binding site | ||||
| cg05691152 | Chr 22: 38092978 | Age-methylated | TRIOBP | TFBS |
| Weak/poised enhancer | ||||
| CTCF-binding site | ||||
| cg13672736 | Chr 9: 135114066 | Age-demethylated | NTNG2 | CpG shelf |
| Weakly transcribed | ||||
| cg25038330 | Chr 10: 463561 | Age-demethylated | DIP2C | CpG shelf |
| Hetrochromatin; low signal |
Chr, chromosome; TFBS, transcription factor-binding site.
| CpG Site | Location | Methylation Change with Age | Nearest Gene | Genomic Features and States |
|---|---|---|---|---|
| cg02914422 | Chr 7: 32110145 | Age-methylated | PDE1C | CpG island |
| TFBS | ||||
| Polycomb repressed | ||||
| CTCF-binding site | ||||
| cg05691152 | Chr 22: 38092978 | Age-methylated | TRIOBP | TFBS |
| Weak/poised enhancer | ||||
| CTCF-binding site | ||||
| cg13672736 | Chr 9: 135114066 | Age-demethylated | NTNG2 | CpG shelf |
| Weakly transcribed | ||||
| cg25038330 | Chr 10: 463561 | Age-demethylated | DIP2C | CpG shelf |
| Hetrochromatin; low signal |
| CpG Site | Location | Methylation Change with Age | Nearest Gene | Genomic Features and States |
|---|---|---|---|---|
| cg02914422 | Chr 7: 32110145 | Age-methylated | PDE1C | CpG island |
| TFBS | ||||
| Polycomb repressed | ||||
| CTCF-binding site | ||||
| cg05691152 | Chr 22: 38092978 | Age-methylated | TRIOBP | TFBS |
| Weak/poised enhancer | ||||
| CTCF-binding site | ||||
| cg13672736 | Chr 9: 135114066 | Age-demethylated | NTNG2 | CpG shelf |
| Weakly transcribed | ||||
| cg25038330 | Chr 10: 463561 | Age-demethylated | DIP2C | CpG shelf |
| Hetrochromatin; low signal |
Chr, chromosome; TFBS, transcription factor-binding site.
MA proposes aging to be caused by deleterious genes with age-specific effects confined to only late in life. We suggest that the effects of such genes may be influenced by age-associated changes in DNA methylation, and that these changes may be accompanied by changes in gene expression. We tested for associations between gene expression and the methylation levels at MA-consistent sites. All 13,222 expression probes passing quality control were tested against the four replicated MA sites with significant values. After multiple test correction, 11 genes showed a significant change in expression that was associated with a change in methylation level at one of three CpG sites (FDR < 0.05; Table S3 in File S1). For 10 of these associations, expression was found to decrease with increasing methylation, indicating that methylation at these CpG sites may be interfering with transcription. Interestingly, all 11 of the genes with significant methylation-associated expression changes are located on a different chromosome from the CpG site. This suggests that the methylation of some MA-consistent CpG sites may interfere with the transcription of distal genes. Separate chromosomes can physically interact in the 3D space of the nucleus, and the transcription of some genes has been shown to be regulated by elements (e.g., enhancers) located on separate chromosomes though such interactions (Miele and Dekker 2008). The exact role of methylation in long-range expression control has not yet been well characterized, but it is possible that the associations we observe result from a physical interruption of interchromosomal interactions by methylation. Alternatively, the associations we observe could be indirect, such that methylation interferes with the transcription of a local gene and only influences the expression of distal genes indirectly, through a common pathway.
Heritability of rate of aging
We observed age acceleration, which describes an individual’s rate of aging, to have a significant nonzero estimated heritability of 0.63. This result is consistent with both MA and DS evolutionary models of aging. Horvath found the heritability of age acceleration in twins to be 100% for newborns and 39% for adults, suggesting that the importance of environmental factors increases with age (Horvath 2013). Given that the median age of the subjects in the BSGS data set is 14, our result is in line with these findings. Marioni et al. (2015) estimated the heritability of age acceleration to be 0.43 in the BSGS data after standardizing the methylation-age estimates to correct for differences in the age-prediction ability between adolescents and adults. The disparity between these results highlights the changing predictive ability of the Horvath model based on development phase, i.e., childhood vs. adulthood.
A study by Gentilini et al. (2013) similarly investigated the relationship between methylation and an individual’s rate of aging by comparing the methylation patterns of the offspring of centenarians and the offspring of nonlong-lived individuals. They found global methylation levels to decrease with age across all individuals, but found centenarians and their offspring to have significantly less global methylation loss than the offspring of nonlong-lived individuals. This result suggests that a genetic component underlies the preservation of methylation patterns and that the rate of biological aging is heritable. This study also identified 217 CpG sites that are differentially methylated in the offspring of centenarians compared to the offspring of nonlong-lived individuals. We compared these 217 CpG sites to the sites we found to be consistent with MA or DS irrespective of significance (group 1); we found 11 of these sites to be MA-consistent and 38 of these sites to be DS-consistent. This suggests that heritability in the rate of aging could be the result of both deleterious genes with age-specific effects, as suggested by MA, and genes regulating the accuracy of somatic maintenance and repair, as suggested by DS.
Using the familial BSGS data, we were able to test for the existence of a heritable rate of aging but unable to test for age-related changes in this rate. Methylation changes resulting from drift should occur at a constant rate throughout an individual’s life, while targeted methylation changes should occur at specific times and show age-dependent rates of change. This can be examined in the future using longitudinal DNA methylation data and can potentially be used to investigate if the epigenetic clock sites that have underlying changes are targeted and MA-consistent, or stochastic and DS-consistent.
Conclusions
We observed age-dependent changes in the heritability of methylation at age-differentially-methylated CpG sites consistent with both MA and DS. Both theories play a role in explaining human aging and the aging-related changes we observe. The number of sites found to have decreasing heritability of methylation that is consistent with DS was roughly three times the number of sites found to have increasing heritability of methylation that is consistent with MA. Decreases in the heritability of methylation with age, where the DNA methylation levels of relatives diverge with age, have previously been reported and described as epigenetic drift. DS and epigenetic drift are consistent with each other, and suggest that age-associated DNA methylation changes are stochastic and may be caused by both internal and external factors. Increases in the heritability of methylation with age, where the DNA methylation levels of relatives converge with age, have not previously been reported. The existence of such sites indicates that not all age-associated DNA methylation changes are stochastic and caused by epigenetic drift. Age-related methylation changes at these sites may instead be targeted changes that mediate the effects of deleterious age-specific mutations, as suggested by MA. Enrichment and expression analyses suggest that methylation changes at MA sites may do this by influencing transcription. Further work is needed to connect methylation changes at the CpG sites found to be consistent with MA to specific genes or gene networks and to elucidate the role that epigenetic drift plays in the aging process.
Additionally, we found an individual’s rate of aging to be heritable using a methylation-derived measure of biological age, which considers the DNA methylation at hundreds of CpG sites across the genome. A heritable rate of aging is consistent with both MA and DS. This result indicates a general agreement between the patterns observed at the small single-site scale and the larger many-site scale.
To validate our results, we repeated our analyses in an independent data set. The number of sites found to have decreasing heritability of methylation that is consistent with DS was greater than the number of sites found to have increasing heritability of methylation that is consistent with MA. Although this general trend of DS-consistent sites outnumbering MA-consistent sites was replicated between studies, little replication was seen between the results at the CpG site level. This was likely due to differences in data composition between our original and replication data sets. The original BSGS data we analyzed was from families with adolescent twins (age range:10–75; mean age: 21), while the replication GOLDN study data were from families with adult siblings (age range: 18–88 years; mean age: 49).
The age distributions of our original and replication data sets are the main limitations of our study. However, loci with significant age-associated DNA methylation changes have been shown to have significant overlap in pediatric and adult populations (Alisch et al. 2012), indicating that our age-differential methylation results should not be specific to the predominant age class of the data set analyzed. Nevertheless, having an age distribution with an abundance of individuals of young ages may have biased our theory classification results against the MA model. Changes associated with the deleterious age-specific genes suggested by MA occur only late in life, and may be missed in a data set that has predominantly adolescent individuals. Additionally, since the DS model works throughout an individual’s entire life, associated changes should be detectable even at young ages. This may inflate our estimate of the relative contribution of DS in explaining the aging-related methylation changes we observe. In future work, we hope to identify additional sources of family-based data to test our hypotheses against more uniform distributions and wider age ranges. Further work using data sets with more ideal compositions will help us to better understand and differentiate between biological aging- and development-related DNA methylation changes.
In this paper, we have developed and implemented a novel approach to testing the MA and DS evolutionary models of aging using DNA methylation data. The availability of genome-wide DNA methylation data has allowed us to investigate age-related changes at sites across the genome, and to better understand their connection to the MA and DS evolutionary models of aging. However, our approach restricts our focus and results to only CpG sites with heritable DNA methylation levels. It is possible that age-related methylation changes at non-heritable CpG sites are also consistent with the MA and DS evolutionary models, and that the distributions of MA- and DS-consistent changes at these sites differ from those at sites with heritable methylation levels. It should also be noted that DNA methylation data differs in its suitability for testing MA and DS. DS specifically predicts epigenetic changes, such as DNA methylation changes, to be one of many types of somatic damages that occur throughout an individual’s life. However, MA predicts only the existence of germline mutations, and does not predict any specific epigenetic effects. Therefore, DNA methylation data can only test the hypothesized epigenetic impact of genes that are directly predicted by MA.
Taken together, the results of this study suggest a role for both DS and MA in explaining patterns of epigenetic change with age. We suggest that many of the methylation changes that we observe with age are acquired stochastically and equivalent to epigenetic drift, but not all. Some aging-related methylation changes may be targeted. That is, aging-related methylation changes are likely to be caused by more than one process, and not equivalent throughout the genome. Furthermore, we believe our work demonstrates the utility of DNA methylation data in evolutionary investigations of human aging. We believe that the results of this study suggest that DNA methylation data will be useful in future investigations of evolutionary theories of aging.
Acknowledgments
The authors thank Benjamin Barwick for helpful discussions and for sharing his annotation and code for the gene ontology analysis. This research was supported by grants from the National Institute on Aging (NRSA 0000030423 to C.R.), the National Heart, Lung, and Blood Institute (RO1 HL104135 to D.K.A. and D.M.A.), and the National Health and Medical Research Council (grants 613608, 1046880, and 1078037 to P.M.V.; fellowship 1010374 and grant 1083656 to A.F.M.; and fellowship 110759 to J.E.P.). The authors have no conflicts of interest to declare.
Footnotes
Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.117.300217/-/DC1.
Communicating editor: P. Scheet

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}