




Abstract
Hyperphosphorylation of tau at multiple sites has been implicated in the formation of neurofibrillary tangles in Alzheimer’s disease; however, the relationship between toxicity and phosphorylation of tau has not been clearly elucidated. Putative tau kinases that play a role in such phosphorylation events include the proline-directed kinases glycogen synthase kinase-3β (GSK-3β) and cyclin-dependent kinase 5 (Cdk5), as well as nonproline-directed kinases such as microtubule affinity-regulating kinase (MARK)/PAR-1; however, whether the cascade of events linking tau phosphorylation and neurodegeneration involves sequential action of kinases as opposed to parallel pathways is still a matter of controversy. Here, we employed a well-characterized Drosophila model of tauopathy to investigate the interdependence of tau kinases in regulating the phosphorylation and toxicity of tau in vivo. We found that tau mutants resistant to phosphorylation by MARK/PAR-1 were indeed less toxic than wild-type tau; however, this was not due to their resistance to phosphorylation by GSK-3β/Shaggy. On the contrary, a tau mutant resistant to phosphorylation by GSK-3β/Shaggy retained substantial toxicity and was found to have increased affinity for microtubules compared with wild-type tau. The fly homologs of Cdk5/p35 did not have major effects on tau toxicity or phosphorylation in this model. These data suggest that, in addition to tau phosphorylation, microtubule binding plays a crucial role in the regulation of tau toxicity when misexpressed. These data have important implications for the understanding and interpretation of animal models of tauopathy.
INTRODUCTION
The microtubule-binding protein tau is a component of neurofibrillary tangles (NFT) in Alzheimer’s disease (AD) and related disorders that are collectively referred to as tauopathies (1,2). Tau in NFT is hyperphosphorylated and it is widely believed that the pathological action of tau kinases such as glycogen synthase kinase-3β (GSK)-3β, microtubule affinity-regulating kinase (MARK) and cyclin-dependent kinase 5 (Cdk5) plays a role in the formation of insoluble tau aggregates and NFT (3,4).
The host of kinases that phosphorylate tau in vivo can be divided into two main groups: the proline-directed protein kinases (PDPKs) and nonproline-directed protein kinases (NPDPKs), depending on the residues modified. PDPKs include GSK-3β, mitogen-activated protein kinase (ERK1/2), Jun N-terminal kinase (JNK1) and the cyclin-dependent protein kinases Cdk5 and Cdc2 (5–16). NPDPKs include cyclic AMP-dependent protein kinase A, MARK and casein kinase II (12,17–20).
We and others have utilized the model organism Drosophila melanogaster to model tauopathies (21–23). Direct misexpression of wild-type human tau in the fly retina resulted in early onset cell death in the larval eye disc, as evidenced by the formation of lamin-containing aggregates, a characteristic of caspase-dependent cell death (21). In the adult, neurodegeneration was manifest as a ‘rough’ eye phenotype with disordered ommatidia and bristle abnormalities; internal retinal architecture showed polarity defects and loss of photoreceptor neurons. We demonstrated that phosphorylation of wild-type human tau by Shaggy, the single fly homolog of GSK-3β, results in its phosphorylation and formation of NFT (21). Others have highlighted the role of PAR-1, the fly homolog of MARK (24), in the regulation of tau toxicity (25). A mutant tau construct resistant to PAR-1 phosphorylation has been shown to be less toxic than wild-type tau, suggesting a relatively more important role for PAR-1 in determining tau toxicity compared with GSK-3β (25). More recently, others have suggested that tau that cannot be phosphorylated is rendered nontoxic; nonetheless, the identification of the specific phosphorylation sites that regulate toxicity has proved elusive (26,27).
Here, we set out to examine the relative importance of tau phosphorylation by GSK-3β/Shaggy, MARK/PAR-1 and Cdk5 in the regulation of tau phosphorylation, toxicity and solubility. Although tau resistant to phosphorylation by PAR-1 is less toxic than wild-type tau, this is not related to its resistance to be subsequently phosphorylated by Shaggy. On the other hand, tau that is resistant to phosphorylation by Shaggy retains toxicity. Cdk5 does not play a major role in tau phosphorylation or toxicity in our model system. The toxicity of tau constructs appears to be closely related to their affinity for microtubules, suggesting that the gain of function phenotypes obtained by tau overexpression are related most strongly to microtubule-based transport. These studies have important implications for the development and interpretation of animal models of tauopathy.
RESULTS
Misexpression of PAR-1 but not Shaggy produces neurodegeneration
Drosophila PAR-1 and Shaggy were expressed using the binary GAL4/UAS system with the pan-retinal GMR-GAL4 driver. Compared with the driver-alone control (Fig. 1A), the eyes misexpressing PAR-1 (Fig. 1B) displayed a reduced, rough-eye phenotype with disordered ommatidia and missing bristles. Transgenics misexpressing Shaggy, on the other hand, displayed an eye phenotype more similar to the control (Fig. 1C). Compared with transgenics misexpressing PAR-1, the eyes of transgenics misexpressing Shaggy had a larger and more uniform appearance. In order to compare the internal retinal morphology, confocal imaging of adult retinas stained with TRITC-phalloidin was performed. Tangential optical sections revealed a near-normal trapezoidal array of rhabdomeres in the driver-alone controls (Fig. 1D). In contrast, PAR-1 produced disorganized ommatidia with marked disorganization of photoreceptor neurons (Fig. 1E). The retinas of transgenics misexpressing Shaggy (Fig. 1F) showed a relatively normal array of rhabdomeres other than some abnormal polarity.

Misexpression of PAR-1 (68) using GMR-GAL4 produces a rough eye with the disruption of retinal architecture, whereas Sgg (63) misexpression produces a relatively normal eye. (A–C) SEM images. The normal-eye phenotype observed using the GMR-GAL4 driver (A) is disrupted in eyes misexpressing PAR-1 (B) but not in eyes misexpressing Shaggy (C). (D–F) Retinal whole mounts stained with phalloidin-TRITC to identify rhabdomeres of photoreceptor neurons (single tangential confocal sections). At this apical section, normally, clusters of seven photoreceptors within each ommatidium form a characteristic chevron-shaped structure (D). Full genotypes: (i) w1118, GMR-GAL4/+ (A and D); (ii) w1118, GMR-GAL4/+, UAS-PAR-1/+ (B and E); (iii) w1118, GMR-GAL4/+, UAS-Sgg/+ (C and F). Scale bars: 100 µm (A–C); 10 µM (D–F).
We and others have shown that the expression of wild-type human tau in fly photoreceptor neurons causes marked neurodegeneration (21,28) and suggested that this neurodegeneration is linked to tau phosphorylation. We further evaluated two important serine/threonine kinases, PAR-1 and Shaggy, which are known to phosphorylate tau at multiple sites. In order to study the interaction of tau with these two kinases in more detail, we engineered two different tau substrates, TauS2A and TauS11A, which included mutations in the major PAR-1 and Shaggy phosphorylation sites, respectively. The two main PAR-1 phosphorylation sites are serines 262 and 356, both of which are located in the microtubule-binding region of tau (Fig. 2). This tau substrate has no accessible PAR-1 phosphorylation sites but can, in theory, be phosphorylated by Shaggy. The second tau substrate was constructed in which 11 of 14 major serine/threonine Shaggy phosphorylation sites were mutated to alanine. This particular substrate, TauS11A, was designed so that it would be phosphorylated by PAR-1 but resistant to Shaggy-induced phosphorylation at the AT8, AT180, PHF1 and AT100 epitopes.
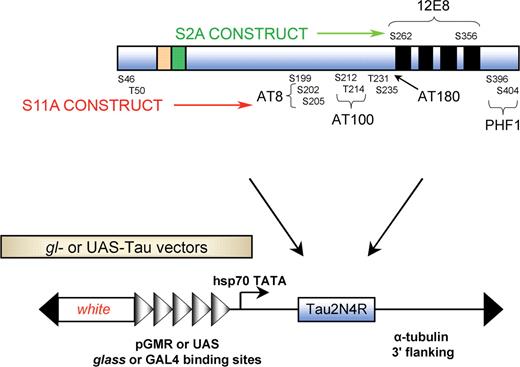
Schematic representation of the tau constructs used. TauS2A was constructed by mutating two serine residues (S262 and S356) to alanine; these are the phosphoepitopes recognized by the 12E8 antibody. TauS11A has 11 serine and threonine residues mutated to alanine. The epitopes recognized by phosphorylation-dependent antibodies, e.g. AT8, AT180, PHF1 and AT100, are shown. GMR-GAL4 driver was used to drive combinations of UAS-Tauwt, UAS-TauS2A and UAS-TauS11A along with PAR1 and Shaggy. gl drivers were also used for the same purpose.
Wild-type tau is more toxic than S2A tau in the fly eye
As a first step toward elucidating the relative importance of PAR-1 and Shaggy on tau toxicity and phosphorylation, we examined the effects produced by the misexpression of the wild-type and S2A tau transgenes alone. Compared with the driver-alone control, wild-type tau produced a reduced, rough-eye phenotype (Fig. 3A). SEM analysis revealed fused and disordered ommatidia with missing and irregular bristles. In contrast to wild-type tau, the eyes misexpressing S2A tau under GMR-GAL4 control appeared relatively normal (Fig. 3B). Although mild bristle abnormalities were apparent using SEM, the eyes were larger compared with wild-type tau, with relatively normal ommatidia. Thus, S2A Tau was less toxic compared with wild type. This relatively normal-eye phenotype was observed for several independently derived UAS-TauS2A lines. We also generated direct fusion gl-TauS2A lines, which also consistently had more normal phenotypes compared with those derived using wild-type tau (Supplementary Material, Fig. S1). We further examined retinal morphology using confocal imaging of adult retina stained with TRITC-phalloidin. Those retinas expressing wild-type tau showed marked ommatidial disorganization and cell loss (Fig. 3C), whereas S2A had a much more modest effect on retinal morphology (Fig. 3D) and resembled the driver control (Fig. 1D).
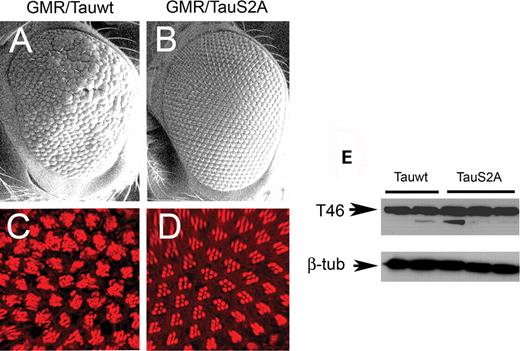
The retinal phenotype of wild-type tau (Tauwt) under the control of GMR-GAL4 is more severe than that of S2A tau. (A and B) SEM images. The mild rough-eye phenotype produced by Tauwt (A) is not observed with TauS2A (B). Scale bars: 100 µm. (C and D) Confocal images of adult retina stained with TRITC-phalloidin (red). Ommatidial disorganization and cell loss are apparent in the eyes overexpressing the Tauwt transgene (C) compared with eyes expressing TauS2A (D), which displays a largely normal trapezoidal array of rhabdomeres. Genotypes: (i) GMR-GAL4/+, UAS-Tauwt/+ (A and C); (ii) GMR-GAL4/+, UAS-TauS2A/+ (B and D). (E) Immunoblot using T46 demonstrates that several independently derived Tauwt and TauS2A lines express comparable amounts of total tau protein. β-Tubulin-loading control for total protein in head extracts is shown below. Scale bars: 100 µm (A and B); 10 µm (C and D).
Given the dramatically different phenotypes produced by wild-type and S2A tau, we compared the expression of the transgenes using two independently derived wild-type (Tauwt-1 and Tauwt-2) and three different S2A tau lines (TauS2A-1, TauS2A-2 and TauS2A-3) by immunoblotting using the T46 monoclonal antibody. Tau expression appeared to be similar between the two genotypes (Fig. 3E).
S2A but not wild-type tau is resistant to PAR-1-induced phosphorylation
PAR-1 has been suggested to play a central role in regulating tau phosphorylation and toxicity (25). However, Lu and colleagues did not extensively evaluate effects of PAR-1 coexpression on wild-type and S2A tau. We directly compared effects of PAR-1 misexpression on wild-type and S2A tau. The rough-eye phenotype produced by wild-type tau (Fig. 4A) was stronger when PAR-1 was coexpressed (Fig. 4B); the mild disorganization of the retina observed in wild-type tau eyes was dramatically increased, resulting in smaller eyes with more missing bristles and fused ommatidia. We note, however, that this may be an additive rather than a synergistic effect. When S2A was coexpressed with PAR-1 (Fig. 4D), there was only a modest worsening of the rough-eye phenotype compared with transgenics overexpressing S2A tau alone (Fig. 4C); the dual S2A/PAR-1 transgenics had slightly more irregular eyes with disordered and occasional missing bristles. Confocal analysis of adult retina revealed abnormalities of ommatidial polarity and rhabdomere loss with wild-type tau alone (Fig. 4E); these became more severe when PAR-1 was coexpressed (Fig. 4F). Compared with transgenics of the wild-type tau data set, the internal retinal morphology of flies coexpressing S2A tau and PAR-1 was only slightly worse than that of S2A tau alone, with slightly misshapen rhabdomeres and a moderate loss of photoreceptor neurons (Fig. 4H).
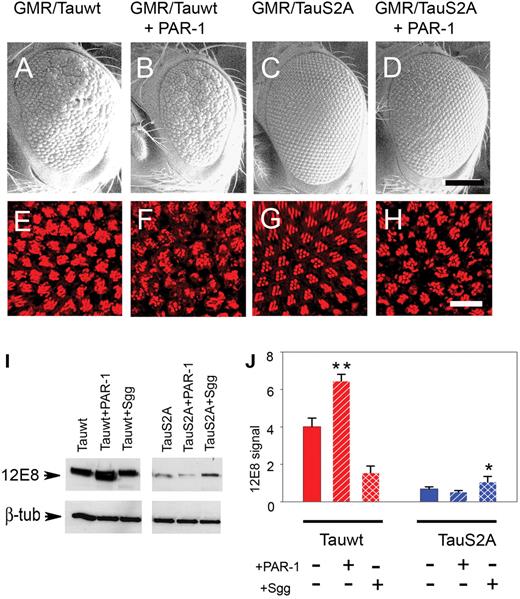
S2A is less toxic than wild-type Tau and relatively resistant to PAR-1-induced phosphorylation. (A–D) SEM images. The rough-eye phenotype of transgenics misexpressing Tauwt (A) is severely enhanced when coexpressed with PAR-1 (B), whereas the phenotype of TauS2A (C) is only mildly enhanced by PAR-1 overexpression (D). (E–H) Phalloidin-TRITC staining of whole-mount retina. TauS2A (G) is less toxic than Tauwt (E), and the enhancement in response to PAR-1 is less severe in TauS2A (H) compared with Tauwt (F). Genotypes: (i) GMR-GAL4/+, UAS-Tauwt/+ (A and E); (ii) GMR-GAL4/+, UAS-Tauwt/+, UAS-PAR-1/+ (B and F); (iii) GMR-GAL4/+, UAS-TauS2A/+ (C and G); (iv) GMR-GAL4/+, UAS-TauS2A/+, UAS-PAR-1 (D and H). Scale bars: 100 µm (A–D); 10 µm (E–H). (I) Immunoblot analysis with 12E8 detects stronger immunoreactivity with Tauwt compared with TauS2A. (J) Histograms representing relative phosphorylation level changes after the coexpression of PAR-1 and Shaggy. Results shown are derived from densitometric analysis of three separate blots. Each bar represents mean ± SEM (n = 3). *P < 0.05; **P < 0.01 (ANOVA with the Newman–Keuls post hoc comparison).
Given that tau toxicity has been proposed to depend largely, if not entirely, on its propensity for phosphorylation (26,27), immunoblot analysis was performed using the 12E8 antibody, which detects tau phosphorylated at serine 262 and 356 residues, the two sites phosphorylated by PAR-1 in the microtubule-binding domains. As predicted, 12E8 immunoreactivity was increased in flies expressing both wild-type tau and PAR-1 compared with wild-type tau alone (Fig. 4I) or coexpressed with Shaggy. On the other hand, S2A tau flies displayed minimal 12E8 signal, which did not increase when PAR-1 was coexpressed. Quantitative analysis from three separate experiments revealed that the 12E8 intensity increased 1.7-fold in animals coexpressing wild-type tau and PAR-1 compared with transgenics overexpressing wild-type tau only (Fig. 4J). S2A tau, on the contrary, displayed no significant increase in 12E8 signal with kinase coexpression, consistent with the assertion that S2A tau is resistant to phosphorylation by PAR-1.
S2A tau is resistant to Shaggy-induced phenotypic enhancement but not phosphorylation
Another major source of tau phosphorylation in the brain is the proline-directed serine/threonine kinase, GSK-3β, the mammalian homolog of Drosophila Shaggy. As a next step, we evaluated phosphorylation and toxicity of wild-type and S2A tau transgenics coexpressed with Shaggy. The coexpression of Shaggy with wild-type tau produced a markedly reduced eye with abnormally fused ommatidia and complete loss of bristles (Fig. 5B), similar to our findings using a slightly different expression system (21). In contrast, the flies coexpressing S2A Tau and Shaggy displayed more normally sized eyes (Fig. 5D) with better organized ommatidial arrays and bristles. The internal retinal architecture of dual wild-type Tau/Shaggy transgenics was severely disrupted (Fig. 5F) compared with wild-type tau alone (Fig. 5E), whereas the animals coexpressing S2A tau and Shaggy displayed near-normal retinal morphology (Fig. 5H) compared with transgenics overexpressing S2A tau alone (Fig. 5G).
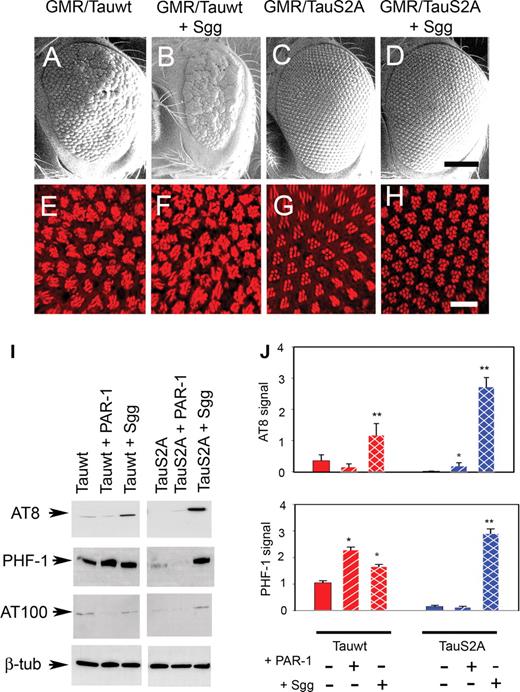
TauS2A is relatively resistant to Shaggy-induced toxicity but not phosphorylation. (A–D) SEM images. The rough-eye phenotype of Tauwt (A) is enhanced by the coexpression of Shaggy (B). The normal eye phenotype of TauS2A (C) remains unchanged when coexpressed with Shaggy (D). (E–H) Phalloidin staining. Shaggy expression enhances the phenotype of Tauwt (F) but not TauS2A (H). Genotypes: (i) GMR-GAL4/+, UAS-Tauwt/+ (A and E); (ii) GMR-GAL4/+, UAS-Tauwt/+, UAS-Sgg/+ (B and F); (iii) GMR-GAL4/+, UAS-TauS2A/+ (C and G); (iv) GMR-GAL4/+, UAS-TauS2A/+, UAS-Sgg (D and H). Scale bars: 100 µm (A–D); 10 µm (E–H). (I) Immunoblot analysis shows that priming of tau by PAR-1 is not required for subsequent phosphorylation by overexpressed Sgg. The magnitude of the increase in phosphorylation at the AT8 and PHF-1 epitopes when stimulated by Sgg is greater for TauS2A than for Tauwt. (J) Histograms show relative effects on phosphorylation derived from densitometric analysis of three separate blots. Each bar represents mean ± SEM (n = 3). *P < 0.05; **P < 0.01 (ANOVA with the Newman–Keuls post hoc comparison).
We then sought to correlate retinal phenotypes with abnormal modifications of tau as assessed by phosphorylation at AD-relevant epitopes known to be preferentially phosphorylated by GSK-3β (29). These are sites known to be phosphorylated in paired helical filamentous form of tau, and they are detected by well-characterized antibodies, including AT8, AT100, PHF-1 and AT180. As expected, compared with wild-type tau alone, we observed a moderate increase in immunoreactivity at these epitopes in transgenics coexpressing wild-type tau and Shaggy (Fig. 5I). Quantitative analysis of three separate experiments revealed increases of 3-fold in the case of AT8 and 1.5-fold in the case of PHF-1 phosphoepitopes when wild-type tau and Shaggy were coexpressed (Fig. 5J). In contrast to the wild-type tau data set, the basal phosphorylation of S2A Tau was scarcely detectable; however, when S2A tau was coexpressed with Shaggy, there was a dramatic increase in the level of phosphorylation at these particular phosphoepitopes. Quantitative analysis from three separate experiments displayed a relative enhancement of 9-fold and 6-fold at the AT8 and PHF-1 phosphoepitopes, respectively, compared with wild-type tau data set (Fig. 5J). A similar enhancement in phosphorylation level was observed for S2A Tau and Shaggy at the AT100 epitope (Fig. 5I; Supplementary Material, Fig. S2A and B). These data were somewhat perplexing given the more dramatic increases at these particular epitopes when Shaggy was coexpressed with S2A compared with when it was coexpressed with wild-type tau. We also probed the level of activated Shaggy (phosphotyrosine 214, corresponding to residue 216 of GSK-3β) for all genotypes and observed elevated immunoreactivity in dual S2A Tau/Shaggy transgenics compared with wild-type tau/Shaggy animals (Supplementary Material, Fig. S2C). Intriguingly, despite displaying dramatically elevated phosphorylation at GSK-3β-relevant epitopes, the external eye phenotypes and internal retinal morphology of dual S2A Tau/Shaggy transgenics were relatively normal. Hence, tau phosphorylation may be only one factor underlying tau toxicity in this experimental paradigm.
Dissociation of toxicity and phosphorylation of a Shaggy-resistant mutant tau construct
Phenotypes of the direct fusion wild-type tau data set (Fig. 6A–F) matched the data set obtained using GMR-GAL4/UAS-Tau coexpressed with PAR-1 and Shaggy (Figs 4 and 5). SEM analysis of the S11A animals showed reduced, rough eyes with fused ommatidia and missing bristles (Fig. 6G). This rough-eye phenotype was more severe when PAR-1 was also expressed, resulting in a reduction of the S11A eye size (Fig. 6H). However, when Shaggy was coexpressed with S11A tau, the eye phenotype was also enhanced (Fig. 6I). Internal retinal morphology of the S11A tau transgenics was moderately abnormal (Fig. 6J), whereas retinas from transgenics coexpressing S11A tau and PAR-1 revealed dramatic disruption (Fig. 6K). In the case of dual S11A tau/Shaggy transgenics, rhabdomeres showed disorganization with some photoreceptor loss (Fig. 6L).
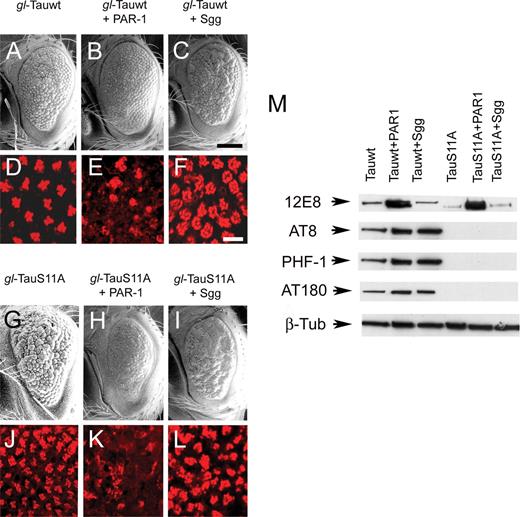
TauS11A retains toxicity despite being resistant to phosphorylation by Sgg. (A–C) and (G–I) SEM images. Direct fusion of gl-Tauwt (A–C) and gl-TauS11A (G–I) phenotypes is shown. TauS11A retains toxicity as evidenced by a severe rough-eye phenotype with disordered ommatidial morphology (G). However, the degree of enhancement by Sgg (I) is less than that observed with wild-type Tau (C). Scale bar: 100 µm. (D–F) and (J–L) TRITC-phalloidin staining indicates that TauS11A (J) retains some toxic effects on ommatidial morphology, although the response to Shaggy (L) is blunted compared with Tauwt. Scale bars: 100 µm (A–C and G–I); 10 µm (D–F and J–L). Genotypes: (i) GMR-GAL4/+, gl-Tauwt/+ (A and D); (ii) GMR-GAL4/+, gl-Tauwt/+, UAS-PAR-1/+ (B and E); (iii) GMR-GAL4/+, gl-Tauwt/+, UAS-Sgg/+ (C and F); (iv) GMR-GAL4/+, gl-TauS11A/+ (G and J); (v) GMR-GAL4/+, gl-TauS11A/+, UAS-PAR-1/+ (H and K); (vi) GMR-GAL4/+, gl-TauS11A/+, UAS-Sgg/+(I and L). (M) Immunoblot analysis showing decreased basal and induced phosphorylation at AT8, PHF1, AT180 and AT100 phosphoepitopes for TauS11A compared with Tauwt. Results obtained using the direct fusion gl-Tau construct in combination with GMR-GAL4 driving UAS-kinases in this figure differ somewhat from those in which GMR-GAL4 drives both tau and kinases owing to less amplification of Tau expression in the former case, i.e. greater tau expression is achieved when glass drives GAL4 expression, which drives Tau. This difference results in a lower ratio of Tau to kinase when gl-Tau is used.
We then sought to determine whether the relationships between the eye phenotypes produced by S11A tau correlated with their phosphorylation. Immunoblotting using 12E8 revealed a strong signal in dual S11A/PAR-1 eyes; thus, these eye phenotypes correlated with phosphorylation for S11A/PAR-1. However, S11A tau showed no immunoreactivity at AT8, PHF-1 or AT180 epitopes under basal or stimulated conditions (Fig. 6M). Thus, tau toxicity is not determined solely by phosphorylation.
Misexpression of Drosophila Cdk5/p35 does not modulate tau phenotypes
In order to elucidate the basis of S11A tau toxicity despite its resistance to phosphorylation by Shaggy, we tested the effects of another proline-directed serine/threonine kinase, Cdk5, which also is thought to play a role in tau hyperphosphorylation (30,31). Cdk5 is present in an active form in postmitotic neurons, where it remains associated with its regulator p35 (32). Cdk5/p35 is known to play an important role in neurite outgrowth during neuronal differentiation and also acts as a tau kinase. Under pathological conditions, p35 is thought to be proteolytically cleaved to p25, which then associates with Cdk5 to phosphorylate tau. We examined whether the coexpression of Cdk5 and p35 with wild-type, S2A and S11A tau results in more severe phenotypes. However, SEM analysis failed to identify substantial effects of Cdk5/p35 on the eye phenotypes produced by these different forms of tau (Supplementary Material, Fig. S3). Immunoblotting with AT8 and PHF-1 showed no effect of Cdk5/p35 on wild-type or S2A Tau transgenics. Basal or enhanced level of immunoreactivity was not detected in S11A transgenics at these phosphoepitopes. However, there was a modest Cdk5/p35-induced increase in AT270 immunoreactivity antibody for S11A tau compared with wild-type tau and S2A tau. Thus, Cdk5/p35 does not appear to play a substantial role in mediating tau toxicity in the fly eye compared with PAR-1 or Shaggy; moreover, the ability of S11A tau to produce abnormal retinal phenotypes despite being resistant to Shaggy-induced phosphorylation is unlikely to be due to responses to Cdk5.
Toxicity of S11A tau is not due to phosphorylation or oxidative stress
Further experiments were carried out to analyze additional factors that might contribute to the retinal neurodegeneration induced by S11A compared with wild-type and S2A tau. Thr181, which is phosphorylated by GSK-3β and found in PHF, has not been mutated in the S11A tau construct. This residue did not appear to contribute substantially to S11A toxicity, however; immunoreactivity for phosphorylated Thr181 was indistinguishable in the three transgenic lines (Supplementary Material, Fig. S4). The lower mobility shift observed in the S11A band compared with wild-type and S2A tau arises due to the lack of phosphorylation by endogenous Shaggy at the mutated serine and threonine residues.
We then evaluated the total endogenous level of some putative tau kinases along with their active forms. Although thus far we have found no evidence for Shaggy or Cdk5 phosphorylation of S11A tau, it remains possible that dysregulation of these kinases could in some way contribute toward S11A toxicity. The total levels of endogenous Cdk5 and Shaggy were indistinguishable between wild-type, S2A and S11A tau. Moreover, there was no change in the active form of Cdk5 at Tyr (15); however, interestingly, there was a marked increase in the level of activated Shaggy (i.e. Shaggy Y214) (Supplementary Material, Fig. S4). An increase in the activated form of GSK-3β could contribute toward promiscuous phosphorylation of other substrates, thereby indirectly mediating S11A tau-induced toxicity.
We further examined JNK signaling, which occurs as a consequence of oxidative stress in a related model of tauopathy (33). However, endogenous JNK expression was similar in wild-type, S2A and S11A tau transgenic lines (Supplementary Material, Fig. S4).
Toxicity of S11A tau is not due to formation of soluble- and insoluble-tau aggregates
We then examined sarcosyl extracts in conjunction with the PHF-1 antibody in an effort to determine whether the toxic phenotype of S11A tau was related to the formation of tau aggregates. PHF-1 detects tau phosphorylated at Ser 396 and 404 epitopes in disease forms. For equivalent amounts of input tau (Fig. 7A, upper panel), both wild-type and S2A tau formed high molecular weight aggregates in the soluble fraction, whereas in the insoluble fraction the tau protein detected was monomeric (Fig. 7A, lower panel). The level of insoluble tau was greater for wild-type tau compared with S2A. No immunoreactivity was detected for S11A tau in either the soluble or the insoluble fraction. In an independent immunoblot, sarcosyl-soluble and -insoluble fractions were tested using the T46 antibody, which detects total tau (Fig. 7A, middle panel). S11A displayed much less immunoreactivity in the soluble fraction and no immunoreactivity in the insoluble fraction compared with wild-type and S2A tau. Thus, the retained toxicity of S11A tau is not due to the formation of aggregates.
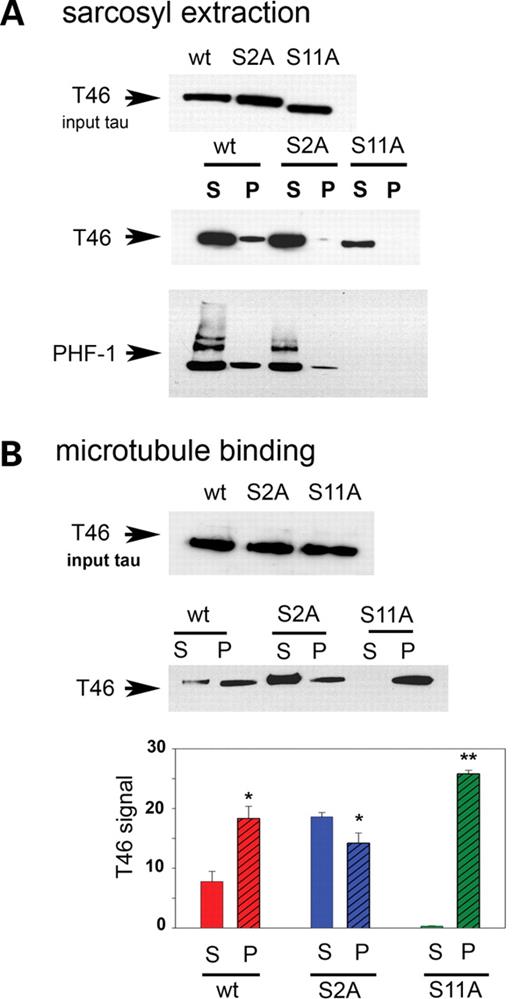
Toxicity of S11A and lack of effects of S2A tau correlate not with the formation of sarcosyl-soluble and -insoluble tau fractions, but with microtubule affinity. (A) Sarcosyl extracts of wild-type, S2A and S11A tau probed with T46 (total tau) and PHF-1 (a pathological phosphoepitope). Upper panel, input tau for the sarcosyl extraction; middle panel, sarcosyl-soluble and -insoluble fractions probed with T46 (total tau); lower panel, pellets and supernatants probed with PHF-1 (a pathological phosphoepitope). TauS11A does not form any soluble or insoluble aggregates compared with wild-type tau, whereas for TauS2A, the amount of insoluble material is much less compared with wild-type Tau. (B) Microtubule-binding assays. Upper panel, input tau for the microtubule-binding studies; middle panel, free or microtubule-bound tau. Compared with wild-type tau, TauS2A associates with microtubules somewhat less avidly, whereas TauS11A shows a markedly increased affinity for microtubules. T46 blotting indicates that the levels of input tau were the same for all three genotypes in both sarcosyl extraction and microtubule-binding experiments. All experiments used UAS constructs in trans to GMR-GAL4. Histograms represent the relative levels of free and bound tau in the supernatant (S) and pellet (P) fractions, respectively, for the three different transgenics. Each bar represents mean ± SEM (n = 3). *P < 0.05 for Tauwt and TauS2A, and **P < 0.001 for TauS11A. (One-way ANOVA with the Newman–Keul’s post hoc comparison).
Microtubule binding of S2A tau is reduced, whereas that of S11A is increased compared with wild-type tau
One of the important physiological functions of the tau protein is to promote the assembly and stabilize the structure of microtubules. Therefore, microtubule-binding affinity was tested for the three different tau constructs as an additional factor that could potentially affect tau-induced toxicity. We carried out microtubule-binding assays for wild-type, S2A and S11A tau by incubating equivalent amounts of input wild-type or mutant tau (Fig. 7B, upper panel) with paclitaxel-stabilized microtubules. Amounts of tau present in supernatant and pellet fractions were quantitated using the T46 antibody, recognizing total tau. Although more of the tau protein in S2A was found in the supernatant compared with wild-type tau, almost all of the S11A Tau remained bound to microtubules and was detected in the pellet fraction (Fig. 7B, middle panel). Quantitative analysis from three different experiments represents the relative level of tau in the supernatant and pellet fractions in the case of wild type, S2A and S11A. In the case of S11A, this difference in the level of ‘free’ and ‘bound’ tau was found to be highly significant (P < 0.001) compared with wild type and S2A (Fig.7B, lower panel). One theory underlying tau-associated neurodegeneration is that once hyperphosphorylated, tau detaches from microtubules, leading to microtubule destabilization and neurodegeneration (34,35); however, this idea has yet to be proven in intact animal or human studies. On the other hand, it is equally possible that abnormally high binding of tau to microtubules causes an imbalance in the structure and function of microtubules, thereby affecting various cellular transport processes eventually leading to neurodegeneration.
Shaggy is more effective than PAR-1 in releasing tau from the microtubules
Since both PAR-1 and Shaggy seem to play an important role in affecting the physiological properties of tau, our next step was to test the effects of the individual kinases on the microtubule-binding properties of wild-type and S2A tau. This experiment was done by coexpressing both PAR-1 and Shaggy with wild-type tau and S2A. We observed that when PAR-1 was coexpressed with wild-type tau and S2A, it was found to exert a modest effect on wild-type tau, with partial distribution of tau in both the supernatant and microtubule-bound pellet fractions, and no effect on S2A. On the contrary, the coexpression of Shaggy with wild-type tau and S2A caused most of the tau to be present in the supernatant fraction, unassociated with the microtubules, with very little in the pellet fractions (Supplementary Material, Fig. S5A and B, upper panel; one-way ANOVA with the supplementary Newman–Keuls test). Quantitative analysis of three different experiments reveals that when Shaggy is coexpressed with wild-type tau and S2A, there is a significant difference between the relative levels of tau in the supernatant and pellet fractions (**P < 0.001). However, the coexpression of PAR-1 produced a significant difference in the case of the wild type (*P < 0.05) but not S2A (Supplementary Material, Fig. S5A and B, lower panel). Hence we can conclude that although both PAR-1 and Shaggy were effective in releasing tau from microtubules, Shaggy seemed to exert stronger effects.
A similar experiment was carried out for S11A coexpressing both PAR-1 and Shaggy. Histograms obtained from three different blots revealed that despite the upregulation of PAR-1 and Shaggy, most of the tau in S11A was found in the pellet fractions associated with the microtubules (Supplementary Material, Fig. S6). The difference in the level of tau in the pellet and supernatant was significant for all three genotypes (**P < 0.001). Since Shaggy is more effective than PAR-1 in dislodging tau from the microtubules, it is not surprising that in the absence of Shaggy phosphorylation, most of the S11A tau remains tightly bound to the microtubules and is detected exclusively in the pellet fraction (Fig. 7B, middle panel). Our observations here are consistent with a mechanism underlying S11A tau toxicity that is associated not with phosphorylation but rather with markedly increased microtubule affinity.
DISCUSSION
We have demonstrated previously that the misexpression of wild-type tau alone induces neurodegeneration in Drosophila, and that this phenotype is exacerbated when tau is coexpressed with Shaggy, the single fly homolog of GSK-3β (21). On the other hand, Lu and coworkers reported that PAR-1 plays a primary role in regulating tau-induced toxicity in the fly (25), and that phosphorylation by PAR-1 is a prerequisite for tau to be subsequently phosphorylated by GSK-3β/Shaggy or Cdk5. Other investigators have suggested on the basis of findings in fly models that tau-induced neurodegeneration is caused by defects in microtubule-dependent axonal transport, leading to a loss of synaptic transmission and vesicle aggregation in the early stages of AD (36,37); nonetheless, recent evidence directly measuring axonal transport in vitro has questioned this mechanism (38).
Although the misexpression of kinases and/or human tau in the Drosophila eye undoubtedly causes neurodegeneration that differs in many important respects from that occurring in AD and related neurodegenerative tauopathies, our group and numerous others have repeatedly demonstrated that this model system can lead to powerful insights into pathophysiological mechanisms underlying these disorders (21,25–28,39–46). The ‘rough’ eye readout is relatively nonspecific in models of human neurodegenerative diseases and can result from the disruption of a host of developmental processes in addition to cell death per se. Nonetheless, compared with the eye phenotypes obtained in fly models of Huntington’s disease (47–51), as an example, the tau eye phenotype provides a highly sensitive and robust readout that makes it quite useful for assessing phenotypic effects of genetic manipulations.
Reduced toxicity in S2A compared with wild-type tau is not due to its inability to be phosphorylated by Shaggy
Lu and colleagues have suggested that TauS2A, which is resistant to PAR-1 phosphorylation, is also resistant to phosphorylation by Shaggy and Cdk5, implying that these proline-directed kinases act downstream of PAR-1. We observed that although TauS2A alone displayed lower immunoreactivity at a variety of AD-related phosphoepitopes compared with Tauwt, there was a dramatic increase in phosphorylation when Shaggy was coexpressed with TauS2A (Fig. 5I). We also examined total and activated Shaggy. Despite equivalent levels of endogenous Shaggy in wild-type and S2A tau (data not shown), activated Shaggy was much higher when Shaggy was coexpressed with TauS2A compared with Shaggy coexpressed with Tauwt (Supplementary Material, Fig. S2C). In presenilin-1 familial AD mutations, the PI3K/Akt pathway is inhibited, thus promoting GSK-3β activity and tau phosphorylation, and in turn promoting tau pathology (52). In the case of S2A tau, however, we could not correlate this Shaggy-induced hyperphosphorylation with neurodegeneration, suggesting dissociation in some instances between tau phosphorylation and neurodegeneration.
S11A is resistant to GSK-3β/Shaggy phosphorylation but retains toxicity
We provide further evidence for dissociation between tau phosphorylation and neurodegeneration using the construct TauS11A. Using multiple different insertions and expression systems, in several instances we observed that the external eye phenotype of TauS11A data set (Fig. 6G–I) is as severe as displayed by Tauwt data set (Fig. 6A–C). However, immunoblotting using Shaggy-dependent phosphoepitope antibodies fails to detect any signal for S11A, though both SEM analysis of external eye phenotypes and confocal imaging of internal retinal architecture confirm substantial toxicity of this construct despite its resistance to phosphorylation (Fig. 6M).
Formation of soluble and insoluble aggregates is more pronounced in wild type and S2A compared with S11A tau
Tau is abnormally phosphorylated in AD and is the main component of PHF and thus NFT. Biochemically, NFT can be isolated on the basis of insolubility in the detergent sarcosyl (53). Immunoblot analysis using T46 antibody that detects both phosphorylated and unphosphorylated tau reveals that in transgenic Drosophila, wild-type tau is primarily sarcosyl insoluble, whereas sarcosyl-soluble tau is present in all three transgenics. Further analysis of sarcosyl fractions with PHF-1 detected no signal for TauS11A, whereas hyperphosphorylated tau was localized to both soluble and insoluble fractions for wild-type and S2A tau transgenics. It is also interesting to note that the tau in the soluble fractions from wild-type and S2A tau transgenics forms high molecular weight aggregates. It is now speculated that certain soluble pathological forms of tau may be more detrimental than insoluble forms, but the correlation of tau aggregation and toxicity remains to be elucidated (54).
Microtubule-binding properties of S11A are different from wild-type and S2A tau
In the case of wild-type and S2A tau transgenics, we observed distribution in both supernatant (unbound tau) and pellet (bound tau) fractions, whereas for TauS11A almost all of the tau was detected in the pellet bound to the microtubules. This observation may help to explain TauS11A-induced toxicity and neurodegeneration in the absence of phosphorylation. One current hypothesis holds that the physiological function of tau is adversely affected due to excess tau phosphorylation. This hyperphosphorylated tau then gets displaced from the microtubules, aggregates and leads to microtubule disassembly such that axonal transport is disrupted. However, certain observations contradict this model. Tau-deficient transgenic mice show no major phenotype (55), perhaps since tau can be substituted for by other factors (e.g. MAP1b). Moreover, mice misexpressing tau show transport defects even though microtubules are intact and tau aggregates are absent (56,57); flies misexpressing tau also show defects in neuronal traffic without evidence of tau aggregation (37). These observations argue that even ‘normal’ tau may be detrimental when its expression becomes elevated. Moreover, there are further reports that prove that the coexpression of GSK-3β with the longest tau isoform actually serves to reduce axonal dilations and degeneration (58). From all these observations, it appears that an optimal level of tau phosphorylation is required to achieve the balance in the level of ‘free’ and ‘microtubule bound’ tau that is essential in maintaining microtubule dynamics and subsequent axonal transport.
PAR-1 and Shaggy act in a concerted fashion to release tau from microtubules with a more significant contribution from Shaggy
The respective contributions of the kinases PAR-1 and Shaggy toward microtubule-binding properties of tau were examined. Both PAR1 and Shaggy are instrumental in releasing tau from microtubules, but Shaggy is probably a more important contributor in this respect. In the case of TauS11A, since most Shaggy phosphorylation sites have been mutated, this form of tau may be irreversibly bound to microtubules, disrupting axonal transport and resulting in subsequent neurodegeneration.
Perspectives on the relationship between tau phosphorylation and toxicity in the context of animal models
Other recent work using a related Drosophila model of tauopathy has suggested that the phosphorylation of tau is the sine qua non for its toxicity (27). This observation was based on a tau construct which differed from the longest isoform of human tau not only in the presence of 14 serine/threonine residues which had been converted to alanine, but also in that this construct lacks the amino terminal inserts generated by alternative splicing of exons 2 and 3. The discrepancy between the lack of toxicity of this ‘S14A’ construct and the toxic S11A construct shown here might derive in part from this lack of amino terminal sequences that may positively regulate aggregation (59). More recently, Feany and colleagues reported that although single- or double-phosphorylation mutants did not appreciably alter toxicity compared with wild-type tau, one mutant consisting of five altered residues [Ser202/Thr205 (AT8), T212 (part of AT100) and T231/S235] did not exhibit substantially lower toxicity compared with wild-type tau (26). Whether this construct exhibited increased microtubule affinity as we observed for S11A tau transgenic was not reported. The most recent data of Feany and colleagues do, however, agree with our hypothesis that although tau phosphorylation, all other things being equal, does play a permissive role in neurodegeneration, phosphorylation is the only factor determining toxicity of tau. Recently Nixon and colleagues directly measured axonal transport in the optic nerve of animals misexpressing tau and failed to identify impairments using direct measures, as opposed to indirect markers of axonal transport blockage (38). Whether the toxicity of phosphorylation-resistant tau depends on impaired axonal transport due to irreversible microtubule binding, or possibly a nonspecific effect on microtubules without affecting axonal transport per se, as has been suggested using studies of squid, remains to be determined (60). Our data do, however, suggest that multiple factors potentially regulating tauopathy in addition to phosphorylation must be considered in the interpretation of animal models of tauopathy.
In a physiological milieu, tau remains bound to microtubules to maintain stability and undergoes a normal phosphorylation/dephosphorylation cycle so that it can transiently come off the microtubules. This process facilitates neurite outgrowth during development, as well as axonal transport throughout life, processes which would otherwise likely come to a complete halt in the presence of excess tau. Although both proline- and nonproline-directed kinases such as GSK-3β and MARK are effective in releasing tau from microtubules, our data suggest that the former exerts a more robust effect overall. Any disruption of the phosphorylation/dephosphorylation cycle can exert toxic effects on the Drosophila eye during development. Hyperphosphorylation of tau leads to the formation of soluble and insoluble aggregates which form a hallmark lesion of AD; this form of tau pathology is evident in the case of wild-type tau coexpressed with PAR-1 and Shaggy. On the other hand, the inability of tau to be phosphorylated by Shaggy can also be detrimental for the cells since this may cause tau to irreversibly bind to microtubules, thereby overstabilizing them and blocking cellular processes dependent upon microtubule-based transport. This form of tau pathology has been observed in the case of S11A tau and its coexpression with PAR-1 and Shaggy. Our observations therefore support a model in which a delicate balance exists between tau hyper- and hypophosphorylation, any deviation from which may lead to toxic effects of tau.
MATERIALS AND METHODS
Constructs, genetics and stocks
The UAS-Tauwt transgenic Drosophila lines were derived by subcloning a cDNA encoding wild-type htau4R into the Exelixis modification (61) of the pUAST vector (62). Seven transgenic lines were obtained using standard techniques. Lines were selected for use on the basis of convenience of chromosomal linkage; in no case were lines on the basis of strong or weak phenotypes. We chose the UAS-Tauwt line 11.1 on chromosome II, which shows a moderate phenotype, for use in the majority of our experiments. The UAS-TauS2A transgenics were derived by site-directed mutagenesis of the wild-type tau construct to transform serines 262 and 356 to alanine. Four such lines were obtained, of which lines 1.33 and 1.62, both located on chromosome II, were chosen for further study. These lines are similar to the UAS-TauS2A line reported by Lu and coworkers (25) except that they are based on the expression of the longest isoform of wild-type human tau, rather than the FTDP-17-associated R406W mutation. The UAS-TauS11A transgenics were derived using an oligonucleotide encoding wild-type tau with mutations at 11 residues: S46A, T50A, S199A, T202A, T205A, T212A, S214A, T231A, S235A, S396A and S404A. Four lines were obtained; we selected line 1.2 on chromosome II for most experiments. These lines are similar to the UAS-TauAP lines reported by Feany and colleagues (27) except that our S11A lines also include the two amino terminal inserts that are generated by alternative splicing of exons 2 and 3. Transgenic lines for UAS-Tauwt, UAS-TauS2A and UAS-TauS11A were matched for equivalent levels of total tau expression by immunoblot analysis using the T46 monoclonal antibody. UAS-Shaggy was obtained from Siegfried (63). Lu generously supplied the UAS-PAR1 line (25), and the UAS-Cdk5 and UAS-p35 lines were obtained from Giniger (64). The gl-TauS2A and gl-TauS11A transgenes were obtained by subcloning the appropriate inserts, as described previously, into the pExpress-gl modification (61) of the GMR expression vector (65). Overexpression of UAS transgenes was achieved using a relatively weak X-linked GMR-GAL4 driver (66). Flies were grown on standard cornmeal-based Drosophila medium at 23°C.
Histology and immunohistochemistry
For SEM, flies were dehydrated in ethanol, incubated overnight in hexamethyldisilazane, dried under vacuum, attached to stubs with black nail polish and analyzed using a Hitachi SEM (21). TRITC-phalloidin (Sigma, St Louis, MO, USA) whole-mount staining of adult retina was carried out as described previously (48) using a Zeiss Pascal laser scanning confocal microscope.
Immunoblotting
To analyze the phosphorylation status of tau, freshly eclosed adult flies were collected and heads were dissected and homogenized in a lysis buffer consisting of Tris–Cl (10 mm, pH 7.4), NaCl (150 mm), EDTA (5 mm), EGTA (5 mm), glycerol (10% v/v), NaF (50 mm), Na3VOF3 (1 mm), NaPPi (5 mm), DTT (5 mm), urea (4 M) and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN, USA). Extracts were mixed with SDS sample buffer, heated at 70°C, centrifuged at 4000 g for 5 min and separated by 10% SDS–PAGE. Proteins were transferred to nitrocellulose, blocked in 5% (w/v) nonfat dried milk in Tris-buffered saline containing 0.1% Tween 20 and immunoblotted using the following antibodies: T46 (Invitrogen, Carlsbad, CA, USA; 1:1000), AT8, AT180, AT270 and AT100 (Pierce Biotechnology, Rockford, IL, USA; all used at 1:2000 except for AT100 (1:500), anti-Thr 181 (Promega, Madison, WI, USA; 1:200), anti-active JNK (Promega, 1:500), PHF-1 (1:5000), 12E8 (1:2000), anti-Sgg/GSK-3β (clone 0.T.56; US Biological, Swampscott, MA, USA; 1:500), anti-GSK3βY216 (clone 0.T.57; US Biological; 1:500; corresponding to SggY214), anti-CDK5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:500), anti-Cdk5Y (15) (Santa Cruz; 1:500) and anti-active JNK (Promega 1:500). The membranes were incubated with peroxidase-labeled anti-mouse or anti-rabbit IgG and signals were detected using chemiluminescence. Films derived from immunoblots were scanned using a Powerlook 1000 transmissive flatbed scanner (UMAX, Dallas, TX, USA). Densities of bands were measured with NIH ImageJ (http://rsb.info.nih.gov/ij). Graphical and statistical analyses were performed using SigmaPlot 9.0 and SigmaStat 3.1 (Systat, San Jose, CA, USA).
Microtubule-binding assay
Determination of tau binding to exogenously added microtubules was performed according to the manufacturer’s protocol (Cytoskeleton, Denver, CO, USA) with some modifications (67). Whole-head extracts were homogenized in a lysis buffer consisting of HEPES (5 mm, pH 7.4) and NaCl (100 mm) and including protease inhibitors. Extracts were clarified with a low spin at 4000 g for 5 min followed by a high spin of 7500 g for 20 min at 4°C. The protein content of the high-spin supernatant was determined by the Bradford method, and extracts containing equivalent amount of total protein were incubated with paclitaxel-stabilized, in vitro polymerized microtubules following the manufacturer’s instruction. Following incubation, fractions were loaded onto a 50% glycerol gradient and centrifuged at 110 000 g (TLA 55) for 45 min. The supernatant and pellet fractions were collected and analyzed by immunoblotting with T46 to estimate the amount of tau bound to microtubules.
Sarcosyl extraction
Fifty fly heads were homogenized in 100 µl of homogenization buffer consisting of Tris–Cl (10 mm, pH 7.4), NaCl (0.8 M), NaCl (1 mm), EGTA (1 mm) and sucrose (10% w/v). The homogenates were centrifuged at 21 000 g for 30 min. The supernatant was brought to 1% N-lauroylsarcosinate and incubated 1 h at room temperature while shaking gently. After a 1 h spin at 90 000 g at 4°C, the supernatants were collected as the sarcosyl-soluble fraction; the sarcosyl-insoluble pellets were suspended in Tris–Cl (50 mm) and stored at 4°C. These samples were later used for immunoblot analysis.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
This work was supported by grants from the National Institutes of Health (NS046489; AG016570) and American Health Assistance Foundation. Funding to pay the open access charges was provided by the National Institutes of Health.
ACKNOWLEDGEMENTS
Thanks to Bingwei Lu, Esther Siegfried, Ed Giniger and the Bloomington Stock Center for fly strains. Thanks also to Peter Davies and Peter Seubert for antibodies.
Conflict of Interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}