




Abstract
The ARHGEF5/TIM oncogene belongs to the Dbl family of guanine nucleotide exchange factors (GEFs) for Rho GTPases. It is well established that Rho-GEFs play an important role in tumorigenesis and metastasis through the activation of their substrates, the Rho GTPases. Little is known about ARHGEF5/TIM oncogene expression and cellular functions. Because of its localization close to the common fragile site FRA7I, which has been shown to be responsible for an inverted duplication of the 7q34–q35 region in breast carcinoma cells, we examined the expression of the ARHGEF5/TIM oncogene in normal and tumoral breast tissue. We report here the identification of five novel ARHGEF5/TIM alternative transcripts specifically expressed in breast tumors. These variant transcripts were characterized by the absence of one or several exons, all coding for the catalytic Dbl-homology domain and generating modified or truncated predicted variant proteins. The variant transcripts were predominantly expressed in breast carcinoma cell lines and in the most aggressive primary breast carcinomas, suggesting they may play a role in breast tumor progression. Moreover, we demonstrate that the expression of recombinant ARHGEF5/TIM protein in transfected COS-7 and NIH-3T3 cells generated a loss of actin stress fibers and the formation of membrane ruffles and filopodia. This pattern suggests that ARHGEF5/TIM activates Rac1, Cdc42 or RhoG rather than RhoA, as previously demonstrated in in vitro guanine nucleotide exchange assays. We anticipate that the activation of the ARHGEF5/TIM oncogene, possibly by the variant isoforms detected here, may play an important role in proliferative breast disease.
INTRODUCTION
Breast cancer is one of the most common malignancies in Western countries and has a high mortality rate (1,2). Aside from a small subset of patients (∼5%) with hereditary genetic alterations, sporadic breast cancer accounts for the majority of all breast cancers and little is known about breast carcinogenesis. It is widely accepted that breast cancer, like most other cancers, develops through the accumulation of genetic aberrations (3). Some of these changes involve specific genetic loci, determining the activation of oncogenes or the inactivation of tumor-suppressor genes, while others confer genetic instability, which increases the possibility of acquiring additional genetic lesions relevant to tumorigenesis.
In this regard, our previous work has focused on the study of the PIP (prolactin-inducible protein) gene which is localized on the long arm of chromosome 7 at 7q34 (UCSC Genome Browser, http://genome.ucsc.edu/). This gene encodes a secreted factor known as prolactin-inducible protein (PIP) (4), which binds to CD4 (5–7), exerts a potent inhibition on T lymphocyte apoptosis mediated by CD4/TCR activation (8) and carries a fibronectin-specific aspartyl protease activity (9). We have found that the PIP gene exhibits various rearrangements in prostate and breast cancers (10,11). It is also overexpressed in about 60% of primary and metastatic breast cancers (4,12), as well as in some breast carcinoma cell lines. In one of them, T47D, that constitutively overexpresses PIP, we have recently demonstrated that the 7q34–q35 region containing the PIP gene exhibited an inverted duplication through the breakage–fusion–bridge (BFB) cycle mechanism initiated within the common fragile site FRA7I (13).
Based on these findings and on human genome sequencing data (UCSC Genome Browser, http://genome.ucsc.edu/), we looked for genes within the PIP-FRA7I region possibly involved in tumor development. We identified the ARHGEF5 (Rho guanine nucleotide exchange factor 5)/TIM (Transforming Immortalized Mammary) oncogene (14), localized at 7q35 (15) and (UCSC Genome Browser, http://genome.ucsc.edu/). This oncogene has been isolated by an expression cloning strategy as a clone with transforming activity in NIH/3T3 fibroblasts and has been shown to be tumorigenic when injected into nude mice (14). The ARHGEF5/TIM predicted protein exhibits sequence similarities to the Dbl family of guanine nucleotide exchange factors (GEFs) for Rho GTPases. Indeed, both a Dbl homology (DH) domain and a pleckstrin homology (PH) domain in tandem were identified, followed by an additional src homology 3 (SH3) domain (16).
The Dbl family of GEFs, also called Rho-GEFs, comprises about 60 members in humans, one-half of them being fully or partially characterized (16). Through their common structural module consisting of the tandem DH-PH domains, Rho-GEFs catalyze the exchange of GDP for GTP on Rho GTPases which become activated (17). The DH domain is responsible for Rho GTPase binding and for catalytic activity, the PH domain for subcellular localization of the GEFs (18).
Numerous studies have clearly established that cellular activities induced by Rho-GEFs are associated with the activation of signaling pathways known to be mediated by active Rho GTPases or their effector targets (16,17,19). So far, 18 Rho GTPase members have been identified which can be subdivided into three prominent subgroups: RhoA, Rac1 and Cdc42 (20,21). Early studies have shown that Rho GTPases regulate cell morphology and the actin cytoskeleton (22,23) and it is now clear that they also affect gene expression, cell proliferation and survival (20,21). These functions suggest that they may be involved in tumorigenesis. However, no Rho GTPase-defective mutant—analogous to those in oncogenic Ras—has been found so far in tumors. Only aberrant activation or abnormal expression levels of Rho GTPases have been described in primary tumors and metastasis (21,24–26), emphasizing the potential role of upstream activators such as Rho-GEFs.
The ARHGEF5/TIM oncogene belongs to the Rho-GEF family. Few informations are available about it. A recent study indicated that ARHGEF5/TIM exerts its GDP-GTP exchange activity on RhoA (17). Because this oncogene is localized within the PIP-FRA7I region at less than 0.5 Mb centromeric to FRA7I, we looked for possible rearrangements affecting ARHGEF5/TIM in breast carcinoma cell lines and primary breast cancers.
We report here the identification of novel splice variants of ARHGEF5/TIM which are specific for malignant breast tissues and cells. These variants correspond to the absence of one or several exons, all coding for the DH domain of the protein. The resulting modified or truncated proteins may have drastic effects on Rho GTPase signalling pathways and play a role in breast tumorigenesis.
RESULTS
Expression of ARHGEF5 in breast carcinoma cell lines
The expression of ARHGEF5/TIM in breast carcinoma cells and normal mammary gland was examined by northern blot. Hybridization with a 600 bp cDNA probe amplified with primers 2F/2R (Fig. 1 and Table 1) detected a 6.5 kb transcript (Fig. 2A), as previously described (14). T47D, MCF7, VHB1 and ZR75-1 breast carcinoma cells expressed the ARHGEF5/TIM transcript at a comparable moderate level, while MDA-MB231 exhibited a low expression level (Fig. 2A). Nevertheless, as compared with a control hybridization with an actin probe (Fig. 2A, lower panel), the expression level of the ARHGEF5/TIM transcript in breast carcinoma cells was consistently lower than in normal mammary gland. Overall, the expression level of the 6.5 kb ARHGEF5/TIM transcript in both normal and tumoral mammary gland was much weaker than in placenta and pancreas (Fig. 2B) and at a lesser extent than in most other tissues except brain and skeletal muscle (Fig. 2B). Interestingly, some of these healthy tissues expressed two additional 5 and 2.7 kb minor transcripts: both forms were detected in placenta, liver and lung. The 2.7 kb transcript alone was observed in pancreas and kidney (Fig. 2B). These minor transcripts were undetectable in the normal mammary gland and breast carcinoma cells.
Identification of novel ARHGEF5/TIM splice variants in breast carcinoma cell lines
To determine whether the ARHGEF5/TIM sequence in normal mammary gland was identical to the full-length cDNA prototype clone BC014555 sequence (2459 bp), RT–PCR was performed from mammary gland RNA (Stratagene) with primers 1F/1R located within 5′- and 3′-UTR regions, respectively (Fig. 1 and Table 1). The 1845 bp amplified product had the same sequence as the BC014555 clone (data not shown). To analyze the sequences of ARHGEF5/TIM transcript in the five breast carcinoma cell lines, RT–PCRs were performed with the primer pair 2F/2R designed to generate a 600 bp product containing part of ARHGEF5/TIM ORF (exon 5–exon 10; Fig. 1 and Table 1). In addition to the product of expected size detected in all samples, PCR amplification generated two fragments of reduced size (524 and 439 bp) in four of five breast carcinoma cell lines (T47D, MCF7, VHB1 and MDA-MB231; Fig. 3A). Sequencing of the cloned fragments revealed that the 524 and 439 bp amplified products corresponded to ARHGEF5/TIM sequences lacking exons 9 and 6, respectively (Fig. 4A). In these splicing variants, named V1 and V2 respectively (Fig. 3A), the deletion of the exons exactly occurred at splice junctions. V1 and V2 were detected neither in ZR75-1 breast carcinoma cells nor in normal mammary gland samples from five distinct women (Fig. 3A). Similar results were obtained with various primer pairs (data not shown).
To analyze the tissue distribution of the variant transcripts, total or polyA+-RNAs from normal tissues and tumoral cell lines from various origins were screened by RT–PCR with primers 2F/2R (Table 1). All tissues expressed the wild-type (WT) ARHGEF5/TIM transcript and no variants were detected (Table 2A). A coexpression of WT and V1 and V2 ARHGEF5/TIM transcripts was detected in two out of 11 tumoral cell lines from various origins, the PC3 prostate carcinoma and JAR choriocarcinoma cells (Fig. 3B and Table 2B). The relative amounts of the variants as compared with WT transcripts amplified in each cell line were quantified by densitometry analysis of agarose gel photographs using the NIH Image software. The V1 variant intensity was 53, 46 and 41% of the one of the WT transcript in T47D, VHB1 and MCF7 breast carcinoma cells, respectively (Table 2B); the V2 variant intensity was slightly lower in these cell lines (34, 33, 26%, respectively). Among breast carcinoma cells, MDA-MB231 exhibited the weakest levels of V1 and V2 variants (27 and 12%, respectively), which were similar to V1 and V2 levels expressed in the choriocarcinoma cells JAR (Table 2B). In the prostate carcinoma cells PC3, both variants represented about 10% of the WT transcript intensity (Table 2B). These results indicate that the V1 and V2 splicing variants of the ARHGEF5/TIM oncogene are almost exclusively expressed in breast carcinoma cell lines and are specific for the tumoral phenotype of the mammary gland.
V1, V2 and additional ARHGEF5/TIM variant transcripts are expressed in primary breast tumors
To determine whether ARHGEF5/TIM alternative transcripts were also expressed in vivo, a panel of primary breast carcinomas was screened. Because of the limited amount of tumor RNA samples, RT–PCR with primers 2F/2R was followed by a nested PCR with primers 3F/3R (Table 1 and Fig. 1). We first controlled that nested PCR did not modify the results obtained in breast carcinoma cells and normal mammary gland: as expected, 551, 475 and 390 bp fragments corresponding to WT, V1 and V2 transcripts were amplified at the same relative levels as observed in the above RT–PCR experiments (data not shown). Out of 33 primary breast carcinomas, 17 (52%) expressed the WT transcript, 15 (45%) expressed ARHGEF5/TIM variant transcripts, and one patient was negative (Table 3). V1 and V2 variants or the V1 variant alone were found in 12/15 patients together with the WT transcript (Table 3). The expression level of the variants was comparable to that observed in breast carcinoma cell lines. Interestingly, three additional fragments were amplified: V3 (432 bp) and V4 (271 bp) were found in two patients, V5 (163 bp) in one patient (Fig. 3C). V3 and V4 were coexpressed with the WT transcript, V5 was expressed alone at a high level (Fig. 3C). Amplification of the β-actin transcript confirmed the integrity of RNAs (Fig. 3C).
All fragments were cloned into pCR2.1 and sequenced. V1 and V2 were identical to their homologues in breast carcinoma cells and lacked exons 9 (V1) and 6 (V2). V3 lacked exon 8 and V4 exons 6 and 8 (Fig. 4A). As for V1 and V2, the deletion of the exons in the V3 and V4 splicing variants occurred at splice junctions. By contrast, in the V5 variant the large deletion, involving part of exon 6, exons 7 and 8 and part of exon 9, did not correspond to exon splice junctions (Fig. 4A).
Predicted variant ARHGEF5/TIM protein sequences exhibit alterations of the DH domain
Strikingly, all skipped exons encode parts of the ARHGEF5/TIM DH domain (Fig. 4B). Predicted protein sequences indicate that two variants, V1 and V5, may produce shortened proteins, while V2, V3 and V4 generate truncated proteins (Fig. 4C). The V1 variant lacking exon 9 may result in a 494 amino acid protein with an in-frame loss of 25 amino acids affecting the end of the DH domain; V5 lacking part of exon 6 to part of exon 9 (Fig. 4A) might generate a 386 amino acid protein with an in-frame deletion of 133 amino acids suppressing the major part of the DH domain and a Glu262-to-Gln mutation (Fig. 4C). The absence of exon 6, alone (V2) or together with exon 8 (V4), resulted in a frameshift leading to a putative truncated protein of 108 amino acids with eight C-terminal residues differing from those of the WT protein. This predicted protein lacks the whole catalytic DH domain, only retaining the N-terminal part of the ARHGEF5/TIM protein. The absence of exon 8 in variant V3 also resulted in a frameshift possibly generating a 206 amino acid protein with three C-terminal mutations (Fig. 4C).
In order to visualize more precisely the effects of V1 and V5 variant-induced modifications, a model of the DH domain of ARHGEF5/TIM (ARH5, Swiss-Prot Entry name) was generated by homology with the DH domain of Vav (Fig. 5A and B). The homology model was based on the sequence alignment shown in Figure 5C using the Vav NMR structure as a template (Protein Data Bank id code 1F5X) (27). Its three-dimensional structure is composed of a flattened, elongated bundle of 11 α-helices (Fig. 5A). As it has been shown that ARHGEF5/TIM catalyzes in vitro the guanine nucleotide exchange for RhoA (17), interactions of RhoA with the DH domain of ARH5 were modeled based on the interactions observed in the Dbs–RhoA complex (PDB id code 1LB1) (17). It clearly appears that V1 variant-induced changes affect the two last helices J and K (Fig. 5A), which do not participate in the formation of the GTPase interaction pocket. By contrast, the interaction pocket with RhoA is completely disrupted in the V5 variant protein (Fig. 5B).
Analysis of the ARHGEF5/TIM gene in variant-positive breast carcinoma cells and tumors
In order to determine whether ARHGEF5/TIM variant transcripts resulted from genomic changes, PCR analysis and sequencing of ARHGEF5/TIM gene regions surrounding the missing exons were performed using DNA derived from healthy donor leukocytes, the four breast carcinoma cells and from 16 patients with available DNA samples. In all cases, the fragments amplified with primer pairs 4F/4R (435 bp), 5F/5R (372 bp) or 4F/5R (1855 bp) (Fig. 1 and Table 1) exhibited the expected size (data not shown) according to the known gene sequence (UCSC Genome Browser, http://genome.ucsc.edu/), indicating that no deletion accounted for the variant transcripts. Sequencing of the amplified fragments detected the presence of four point mutations that corresponded to single-nucleotide polymorphism (SNP) already described in NCBI SNP database (www.ncbi.nlm.nih.gov/SNP/index.html) as SNP clusters rs2699508, rs1208161, rs1208162 and rs179368. Both homozygous and heterozygous genotypes were observed for these SNPs as revealed by sequence chromatogram analysis of the uncloned PCR fragments. No correlation was found between any of these SNPs and the presence of the variant transcripts. Interestingly, SNP cluster rs1208161 corresponded to a G to A conversion at the acceptor splicing site of intron 5 resulting in the replacement of AG by AA, possibly responsible for exon 6 skipping as observed in the V2 variant. Two cell lines, T47D and VHB1, exhibited an heterozygous AG/AA genotype, that might support the presence of both WT and V2 transcripts. However, this cannot be held true for MCF7, VHB1 and all patients with a V2 transcript (n=5) who were homozygous for this AG splice site. This indicates that the V2 variant did not result from the impairment of the acceptor splicing site of intron 5 and suggests that the alternative ARHGEF5/TIM transcripts result from a transcriptional event which remains to be elucidated.
Distribution of ARHGEF5/TIM splice variants according to tumor classification
Interestingly, the variants were found in five out of eight (63%) and 10 out of 25 (40%) patients with lobular and ductal or ductotubular breast carcinomas, respectively (Table 3). Moreover, in the group of ductal carcinomas the variants were found in seven out of 14 grade III–IV tumors (50%) and in three out of 11 (27%) grade I–II tumors. These results suggest that the presence of the variants correlates with the most aggressive tumor type and grade. In contrast, no correlation was found between the presence of the variants and tumor staging or node involvement: five out of 12 (42%) patients with node-negative carcinoma expressed either variant or did not express ARHGEF5/TIM (one patient), while 10 out of 21 (48%) patients with node-positive breast cancer expressed the variants. Surprisingly, four out of six patients (67%) with benign fibroadenoma expressed V1 and V2 together with the WT transcript or the V5 variant alone; one patient did not express ARHGEF5/TIM (data not shown). Three out of the four variant positive patients exhibited a marked ductal hyperplasia. These results contrast with the lack of variant expression in normal mammary gland from five distinct individuals.
Expression of recombinant wild-type ARHGEF5/TIM protein in COS-7 and NIH-3T3 cells
The unique function for ARHGEF5/TIM known to date is a transforming activity in NIH-3T3 cells (14). However, no information is available related to its in vivo Rho-GEF activity. In order to gain better insights into such cellular functions, we examined the role of ARHGEF5/TIM expression. COS-7 and NIH-3T3 cells were transfected with the recombinant pcDNA–DEST47::ARHGEF5 vector engineered to produce a chimeric protein fused to the green fluorescent protein (GFP) at its C-terminus. Forty-eight hours after transfection, ARHGEF5-GFP expressing cells exhibited both a cytoplasmic and nuclear fluorescence (Fig. 6A and D). As GFP is known to distribute to both the cytoplasmic and nuclear compartments (Fig. 6C and F), we used the pcDNA–DEST40::ARHGEF5 construct, engineered to produce a chimeric protein fused to V5 epitope–6xHis at its C-terminus. Staining of transfected COS-7 and NIH-3T3 cells with an anti-V5 monoclonal antibody (Invitrogen) and FITC-conjugated goat anti-mouse Ig antibody showed that ARHGEF5/TIM expressing cells exhibited an identical fluorescence pattern (Fig. 6B and E). This indicates that the nuclear localization of the protein was not due to the GFP tag and confirms that ARHGEF5/TIM is specifically expressed in both the nucleus and cytoplasm.
Effect of ARHGEF5/TIM on actin organization in COS-7 and NIH-3T3 cells
To investigate the potential role of ARHGEF5/TIM on actin cytoskeleton, transfected cells were stained with tetramethyl-rhodamine (TRITC)-conjugated phalloidin and observed under confocal laser microscope. About 92% of ARHGEF5–GFP expressing COS-7 cells (Fig. 7A, a–d) exhibited profound changes in actin fiber organization (a). Particular actin condensation (arrows, a) and structures resembling membrane ruffles were observed at the edges and dorsal surface of the cells (a and d, arrows). Filopodia and pseudopodia were also visible (arrowheads, a and d). The mean number of such structures in a cell was 20±18. ARHGEF5–GFP partially colocalized with condensed actin present in the cell (b and c, arrows) and in ruffles (d, arrows). No change in actin fiber organization was seen in control EGFP-expressing COS-7 cells (Fig. 7A, e–g).
Actin cytoskeleton modifications were even more remarkable in ARHGEF5–GFP expressing NIH-3T3 cells (Fig. 7B, a–g). Most cells (82%) exhibited a loss of actin stress fibers either complete (a and c) or partial (d), as shown by TRITC–phalloidin staining. Cytoskeleton remodeling was associated with appearance of various structures: most cells (77%) exhibited actin-containing filopodia (a, c, d and f) and membrane ruffles (f) together or not with a cortical condensation of filamentous actin (d). The mean number of such structures in a cell was 15±18. In all cases, colocalization of ARHGEF5–GFP and condensed actin was visible (Fig. 7B, b, e and g, arrows and arrowheads). Expression of control EGFP did not generate changes of actin organization (Fig. 7B, h and i).
Given the previous observation that membrane ruffling and filopodia formation are through a Rac1/Cdc42-mediated pathway, respectively (28), our results suggest that ARHGEF5/TIM activates either endogenous Rac1/cdc42 or RhoG (29) in transfected cells, resulting in this typical actin reorganization.
DISCUSSION
Based on our previous observation that a break within the common fragile site, FRA7I generated a BFB-mediated inverted duplication of the 7q34–q35 region in the breast carcinoma cells T47D (13), we examined the expression of the ARHGEF5/TIM oncogene which maps within this region. We demonstrated in this work that five novel ARHGEF5/TIM alternative transcripts were specifically expressed in the tumoral mammary gland. Strikingly, all variant transcripts exhibited a loss of exons encoding the DH domain of the protein and were more frequently expressed in the most aggressive lobular than in ductal breast carcinomas. These results suggest that ARHGEF5/TIM alternative transcripts may play a role in breast tumor progression. Moreover, we found that the expression of recombinant ARHGEF5/TIM protein generated a loss of actin stress fibers and the formation of membrane ruffles and filopodia.
ARHGEF5/TIM is a member of the Rho-GEF family (14,17). It is now well established that these proteins play an important role in tumorigenesis and metastasis through the activation of their substrates, the Rho GTPases (16,21). Although the ARHGEF5/TIM oncogene was identified almost 10 years ago (14), little is known about its expression and function in normal cells and tumors. By northern blot, we confirmed that a major 6.5 kb ARHGEF5/TIM transcript was ubiquitously expressed except in brain (14). In addition to this major transcript, we observed two minor 5 and 2.7 kb ARHGEF5/TIM transcripts expressed in some tissues. According to the human genome sequence data (UCSC Genome Browser, http://genome.ucsc.edu/) and to the recently published sequence of chromosome 7 (30), the size of the 6.5 kb ARHGEF5/TIM transcript does not fit with any of the ARHGEF5/TIM sequences present in these databases. By contrast, the 2.7 kb minor ARHGEF5/TIM transcript might correspond to the full-length cDNA prototype clone BC014555 (GenBank accession number) and to the unique ARHGEF5/TIM gene. According to the Acembly Gene Prediction program (www.acedb.org/Cornell/acembly/), a 4036 base transcript has been predicted, in which the 5′-UTR region seems to be incomplete or missing. This transcript might correspond to the major 6.5 kb ARHGEF5/TIM transcript detected in northern blot by us and others (14). Alternatively, either the 6.5 kb transcript or the minor transcripts may derive from the duplicated ARHGEF5/TIM region, which has been recently identified (30) and maps about 80 kb centromeric to the classical transcription unit. In all cases, these results suggest that the sequence of the ARHGEF5/TIM gene reported in the databases is incomplete and requires further studies.
Although the ARHGEF5/TIM oncogene has been isolated from a cDNA expression library derived from a human mammary epithelial cell line (14), its expression in breast tissue has been poorly documented. We show here by northern blot analysis that the major 6.5 kb ARHGEF5/TIM transcript is expressed at low level in the normal mammary gland. All breast carcinoma cell lines also expressed the ARHGEF5/TIM transcript but at a 3- to 4-fold weaker level than normal breast tissue. In particular, ARHGEF5/TIM expression level in the T47D breast carcinoma cell line was not higher than in other cell lines, although a duplicated FISH signal was detected with a BAC clone containing the ARHGEF5/TIM oncogene (MAD, DPT unpublished data). This contrasts with the important increase of PIP expression level observed in this cell line which displays an inverted duplication of the 7q34–35 region (13). Neither in the mammary gland nor in the breast carcinoma cells the minor ARHGEF5/TIM transcripts were detectable. This may be due to the weak ARHGEF5/TIM expression level in this tissue or to regulation events to be determined.
Most interestingly, we identified by RT–PCR in four of five breast tumor cells five novel alternative ARHGEF5/TIM transcripts, V1–V5, all of them being characterized by the loss of one or several exons encoding the DH domain. These variants were also expressed in 15/33 primary breast carcinomas, most of them being coexpressed with the wild-type transcript. Their absence in the normal mammary gland as well as in other tissues and tumoral cells, except in PC3 and JAR, two cell lines in which very low levels were detected, confirmed their specificity for tumoral breast tissue. No deletion was found in genomic DNA surrounding the exons skipped in the variants, indicating that these alternative transcripts resulted from aberrant pre-mRNA processing, a phenomenon frequently observed in human cancer cells related to mutations or altered gene expression affecting the splicing machinery (31).
The ARHGEF5/TIM variant transcripts were more frequently found in the most aggressive breast tumors, i.e. lobular rather than ductal breast carcinomas. Surprisingly, they were also detected in four of six breast fibroadenomas with a high level of ductal hyperplasia. These results suggest that these alternative transcripts are associated with a high rate of proliferation in breast tissue and with breast tumorigenesis.
Breast tumors have been frequently shown to be associated with an overexpression of genes encoding members of the Rho GTPases family (21) or with overexpressed splice variants, such as Rac1b (25). By contrast, both up- and down-regulation of the expression of Rho-GEF encoding genes, such as ßPix (32) and Tiam1 (33), respectively, have been reported in various tumors. Here, we found that ARHGEF5/TIM expression was down-regulated in breast carcinoma cells as compared with the normal mammary gland. However, it is as yet unknown whether ARHGEF5/TIM transcript levels parallel the expression level of the protein. Moreover, rather than the expression level of the transcripts, a role for the breast tumor specific splice variants and their products has to be considered in breast carcinogenesis.
The five alternative ARHGEF5/TIM transcripts expressed in breast tumors were predicted to generate either truncated proteins (V2, V3 and V4) only retaining the N-terminal part of ARHGEF5/TIM or modified proteins (V1 and V5) lacking parts of the DH domain. According to the model of the ARH5 DH domain-RhoA complex depicted in Figure 5, V5 exhibited a complete disruption of the interaction pocket with RhoA. By contrast, the binding pocket was conserved in V1, suggesting that it may retain its GEF activity. However, in both cases, modifications of the orientation and/or the structure of the C-terminal adjacent PH domain may also occur. These changes in the functional DH–PH module may result in the inactivation of the GEF activity by preventing the binding of the Rho GTPase substrate. Alternatively, a constitutive activation of ARHGEF5/TIM may be induced. Indeed, the DH–PH module has been reported to regulate Rho-GEF catalytic activity (reviewed in 16). In this regard, V1-induced modifications might be responsible for the relief of DH domain autoinhibition, generating a constitutively activated ARHGEF5/TIM variant protein. Similarly, the truncated variants only retaining ARHGEF5/TIM N-terminal region may control ARHGEF5/TIM activity through interactions with the WT protein. Rho-GEF N-terminal regions have indeed been shown to establish intramolecular contacts with the PH domain or to recruit inhibitory cellular factors (reviewed in 19). Truncated ARHGEF5/TIM variants might thus compete with such interactions and/or inhibitory proteins and stimulate ARHGEF5/TIM activity (reviewed in 16,19). In both cases a constitutive activation of the cognate Rho GTPase will promote deregulated proliferation and modifications of the actin cytoskeleton, that contribute to tumorigenesis (21).
Little is known about ARHGEF5/TIM cellular functions. Here we demonstrate that overexpression of recombinant wild-type ARHGEF5-GFP in COS-7 and NIH-3T3 cells induced cytoskeletal remodeling with appearance of membrane ruffles and filopodia. Concomitantly, a loss or strong decrease of actin stress fibers was observed in both cell lines. It has been established that RhoA activation induces the formation of stress fibers (28) whereas activation of Rac1 and Cdc42 generates the formation of membrane ruffles and filopodia, respectively (28). More recently, it was reported that the RhoG GTPase also generates such structures through Rac1 and Cdc42 activation (29). Hence, the pattern observed in transfected cells suggests that ARHGEF5/TIM activates Rac1 and Cdc42, either directly or indirectly through RhoG. By contrast, our results do not support ARHGEF5/TIM-mediated RhoA activation, as previously shown in in vitro guanine nucleotide exchange assays (17). This discrepancy may rely on intrinsic differences between in vitro and in vivo assays. Alternatively, we may hypothesize that ARHGEF5/TIM may contain two separate GEF domains as shown for Trio (34). Although the presence of two domains does not match the structure of the protein expressed in this work, we cannot exclude that a long wild-type isoform, derived from the 6.5 kb ARHGEF5/TIM transcript, may contain two GEF domains. In this regard, the short recombinant ARHGEF5/TIM protein, expressed herein, might interfere with the endogenous long isoform, blocking the activation of RhoA. Additional experiments using pull-down assays and specific antibodies will be required for identifying the substrate(s) and functions of the endogenous ARHGEF5/TIM protein(s) in normal and tumor cells, respectively.
In conclusion, our results demonstrated for the first time the existence of five alternative ARHGEF5/TIM transcripts that affect the catalytic DH domain and are specifically expressed in proliferative breast disease. In addition, we demonstrated that the ARHGEF5/TIM oncogene exhibits a Rac1/Cdc42 or RhoG activity in vivo when expressed in transfected cells. Our results together with the initial demonstration that ARHGEF5/TIM induced altered growth properties in vitro and was tumorigenic when injected into nude mice (14) emphasize the role of this Rho-GEF at different stages of breast tumorigenesis.
MATERIALS AND METHODS
Cell lines and breast carcinoma tissue samples
All cell lines were from American Type Culture Collection, except VHB1 (35) and Jurkat which were provided by J. Soudon (Hopital Saint-Louis, Paris, France) and A. Alcover (Institut Pasteur, Paris, France), respectively. The cells were grown in RPMI or DMEM (GIBCO/BRL Life Technologies, Paisley, UK) supplemented with 10% fetal calf serum (Perbio Sciences, Helsingborg, Sweden), penicillin (100 IU/ml) and streptomycin (100 µg/ml) in a 5% CO2 incubator.
Tumor samples were obtained from 33 unselected primary breast tumors surgically removed from patients at the Department of Surgery of the National Cancer Institute Fondazione Pascale (Naples, Italy). None of the patients had undergone previous treatment. Tumors were histopathologically classified according to the UICC system. Immediately after surgery, tumors samples were stored at −80°C until RNA or DNA extraction.
RNA and DNA preparation
RNAs derived from tumors and cell lines were prepared using TRI reagent solution (Sigma, St Louis, MO, USA) and RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions, respectively. The quality of RNAs was controled with the Agilent Bioanalyseur 2100 (Agilent Technologies, Böblingen, Germany). Total RNA (Stratagene Europe, Amsterdam, Netherlands) and mRNA (BD Biosciences Clontech, Palo Alto, CA, USA) from various healthy tissues were obtained from commercial sources.
DNA from tumors and breast carcinoma cell lines was extracted using a DNA extraction kit (Roche Molecular Biochemicals, Roche Diagnostics Corp., Meylan, France) according to the manufacturer's instructions.
RT–PCR and PCR analysis
Reverse transcription of 2.5 µg RNA was performed with oligo(dT)12–18 and SuperscriptTM II Rnase H− reverse transcriptase (GIBCO/BRL Life Technologies) according to the manufacturer's instructions. For PCR reactions, 2 µl of the resulting cDNA were incubated with 1.5 U Expand High Fidelity Taq polymerase (Roche Molecular Biochemicals), 200 µM dNTP, 1.5 mM MgCl2, and 300 nM of each ARHGEF5/TIM primer (Fig. 1, Table 1). The amplification was carried out in a Gene Amp PCR system 9600 cycler (Perkin Elmer Cetus, Boston, MA, USA) for 35 cycles at 94°C for 30 s, 55–62°C for 30 s according to primer Tm, 72°C for 1 min, followed by a final elongation at 72°C for 7 min. For nested PCR reactions, 1 µl of the first PCR diluted at 1/50 was used and amplified for 30 cycles as above. RT–PCR with primers specific for β-actin was performed as control. Genomic DNA (100 ng) extracted from breast carcinoma cell lines and tumors was amplified as above with specific primers (Table 1).
The PCR products were resolved on agarose gel and stained with ethidium bromide. They were purified using the High Pure PCR Purification Kit (Roche Molecular Biochemicals), subcloned into pCR2.1 vector using the TA Cloning Kit (Invitrogen, Cergy-Pontoise, France). Plasmid DNA was extracted (DNA purification System Wizard® Plus SV Minipreps, Promega, Charbonnieres, France) and sequenced (Sequentia, www.sequentia.fr/).
Northern blot analysis
Total RNAs (15–50 µg) from breast carcinoma cell lines were electrophoresed in a 1.6% formaldehyde agarose gel and transferred onto Hybond-N nylon membranes (Amersham Biosciences, Buckinghamshire, UK) according to standard techniques (36). A multitissue northern blot was purchased from BD Biosciences Clontech. cDNA probes were labeled with [α-32P] dCTP (3000 Ci/mmol; Amersham Biosciences) using Random Primed DNA Labeling kit (Roche Molecular Biochemicals). Northern blots were hybridized at 68°C for 16 h with 32P-labeled probes (1.5×106 cpm/ml) in ExpressHybTM Hybridization solution (BD Biosciences Clontech), washed twice in 2×SSC/0.05% SDS at room temperature for 30 min and twice in 0.1×SSC/0.1% SDS at 50°C for 45 min. Membranes were autoradiographed at −80°C on Kodak X-Omat AR X-ray films (Kodak, Rochester, NY, USA).
Modeling of DH domain
Multiple alignment of DH domains of Vav, β-Pix, Tiam, Dbs (Protein Data Bank id code 1F5X, 1BY1, 1FOE, 1LB1, 1DBH, respectively) was performed using PipeAlign (37) and was manually modified based on the structure. The models were generated using the Meta-Server at CBS (http://bioserv. infobiosud.univ-montp1.fr/) (38). Ribbon diagrams of ARH5 DH domain and interactions with RhoA were modeled using Vav NMR structure (Protein Data Bank id code 1F5X) and Dbs-RhoA complex structure [PDB id code 1LB1 (22)], respectively.
Expression plasmids and reagents
ARHGEF5/TIM coding sequence was amplified with attB1 and attB2 primers (Invitrogen; Table 1) and cloned into the pDONRTM221 vector by recombinational cloning according to Gateway Cloning Technology (Invitrogen). Control sequencing of the recombinant pDONR221 clone was carried out (Sequentia) before recombinational transfer into pcDNA-DEST47 or pcDNA-DEST40 mammalian expression vectors (Invitrogen) for expression of GFP- or V5-epitope-tagged ARHGEF5 C-terminal fusion. TRITC-conjugated phalloidin was from Sigma. Monoclonal antibody to V5-epitope and FITC-conjugated goat anti-mouse IgG antibody were purchased from Invitrogen and Caltag (Burlingame, CA), respectively.
Cell transfection and microscopy
COS-7 (4×106 cells) were transfected by electroporation with 5 µg of recombinant ARHGEF5 plasmid and 15 µg of pUC18 carrier DNA. NIH-3T3 plated on glass coverslips were transfected with 1 µg recombinant DNA in the presence of 6 µl of lipofectamine reagent (GIBCO/BRL Life Technologies). Cells were analyzed 48 h after transfection for expression of recombinant ARHGEF5-GFP protein under fluorescence microscope (Axioplan, Zeiss, Esslingen, Germany). For intracellular staining transfected cells were fixed with 3.7% paraformaldehyde (Merck, Haar, Germany) in PBS for 10 min, permeabilized with 0.1% Triton X100 in PBS for 10 min and incubated for 30 min at room temperature with the specific antibodies or with TRITC-phalloidin. Stacks of confocal images were collected with a LSM 510 laser scanning confocal microscope (Zeiss) using a 63×1.4 NA apochromat plan objective. The excitation wavelengths for GFP and TRITC were 488 and 543 nm, respectively. Image analysis was performed using LSM Image Examiner software (Zeiss).
ACKNOWLEDGEMENTS
We are grateful to J. Soudon and A. Alcover for providing human cell lines. This work was supported by the Centre National de la Recherche Scientifique (CNRS) and by grants from Association pour la Recherche sur le Cancer (ARC) to D.P.T.; the Associazione Italiana per la Ricerca sul Cancro (AIRC) to J.G. M.A.D. is a fellow of the French Government (MENRT) and the Ligue Nationale Française contre le Cancer; M.C. is a fellow of ARC and AIRC.
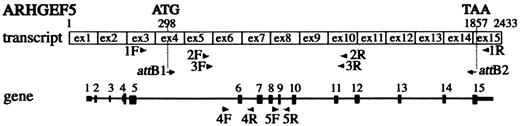
Figure 1. Schematic representation of the localization of ARHGEF5/TIM primers derived from the transcript and the gene. Primers are represented by arrow heads. Exons 1–15 are numbered and represented by black boxes in the gene. Initiation and stop codons are indicated.
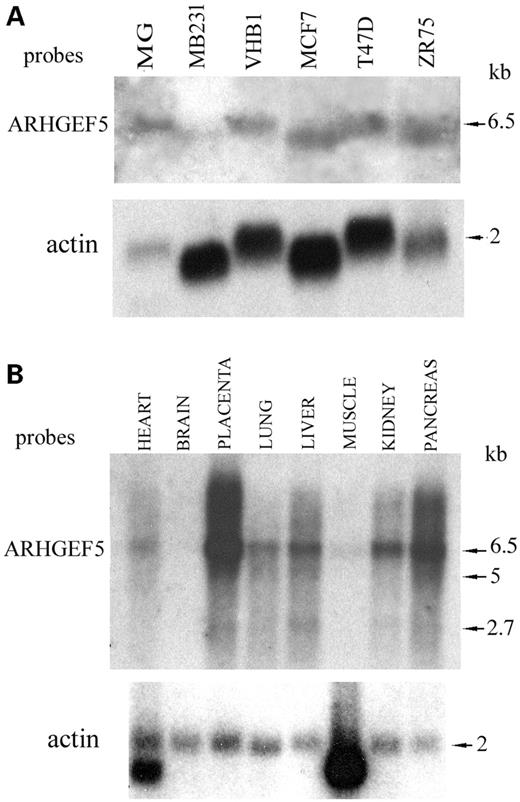
Figure 2. Northern blot analysis of ARHGEF5/TIM transcripts in breast carcinoma cells and healthy tissues. (A) Total RNAs extracted from healthy mammary gland (MG, 15 µg) and breast carcinoma cell lines (50 µg) were electrophoresed and transferred to a nylon membrane. (B) Multitissue northern (Clontech) with mRNAs from various healthy tissues. The filters were probed with a 600 bp ARHGEF5/TIM cDNA probe amplified with primers 2F/2R (A, B upper panels) and with a β-actin probe (A, B lower panels). The size of the transcripts is indicated.
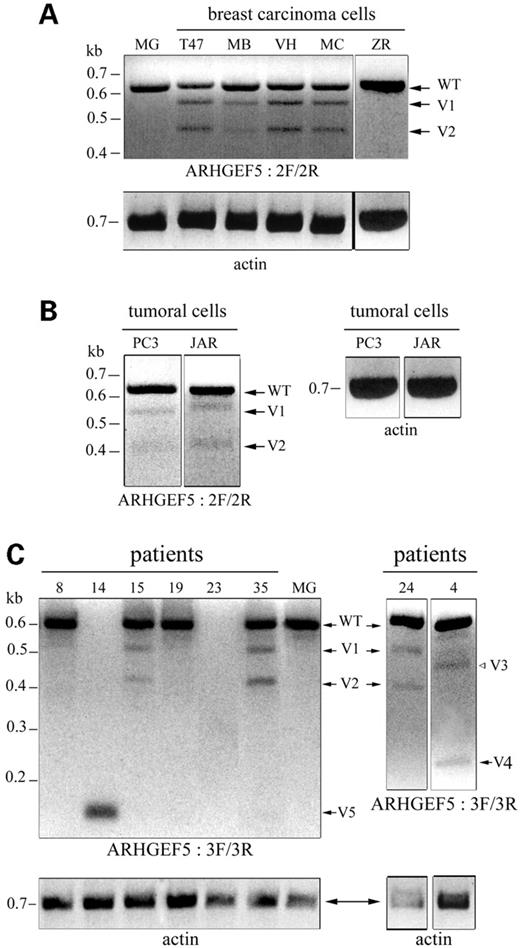
Figure 3. Identification of novel ARHGEF5/TIM alternative transcripts amplified from tumoral breast cells and tissues. RT–PCRs were performed with the primer pair 2F/2R (A, B) and followed by a nested PCR with primers 3F/3R (C). The amplified products were resolved on a 2% agarose gel stained with ethidium bromide and negative images of the photographs are shown. (A) normal mammary gland (MG) and breast carcinoma cell lines T47D (T47), MDA-MB231 (MB), VHB-1 (VH), MCF7 (MC), ZR-75 (ZR): three fragments corresponding to wild-type (WT), V1 and V2 variant ARHGEF5 transcripts were detected in 4/5 breast tumor cells. (B) tumoral cells from various origins: 2/11 cell lines, PC3 (prostate carcinoma) and JAR (choriocarcinoma), expressed the V1 and V2 variants at a low level. (C) Breast carcinoma samples: in 15/33 patients, V1, V2 and three additional variants (V3–V5) were observed associated or not with the WT transcript. Patient 23 did not express ARHGEF5/TIM. RT-PCRs with primers derived from β-actin, performed as controls, are shown.
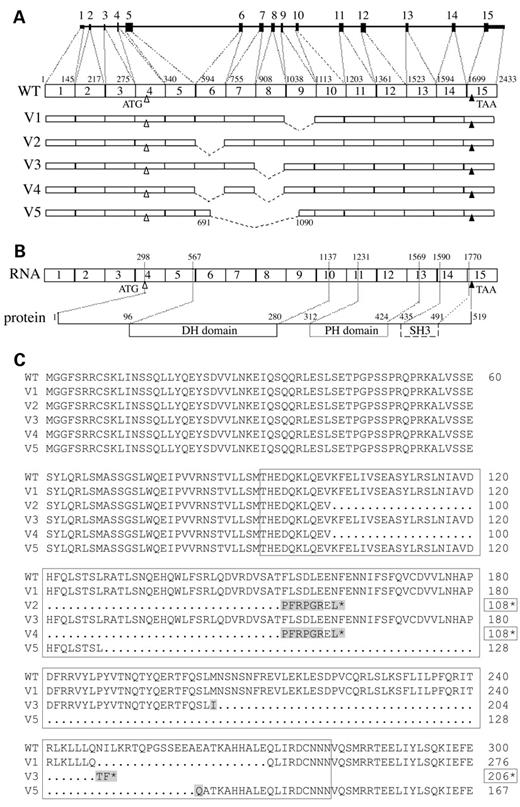
Figure 4. Structure of the ARHGEF5/TIM variant transcripts and sequences of the predicted variant proteins. Amplified products depicted in Figure 3 were cloned and sequenced. (A) Schematic representation of the variant transcripts V1 to V5. (B) Schematic representation of the protein encoded by the WT transcript. (C) Sequence alignment of the N-terminal region and DH domain of WT and predicted variant proteins. The length of V2, V3 and V4 truncated proteins is indicated by boxed numbers followed by a star. Boxed area denotes the ARHGEF5/TIM DH domain. Mutations are shaded.
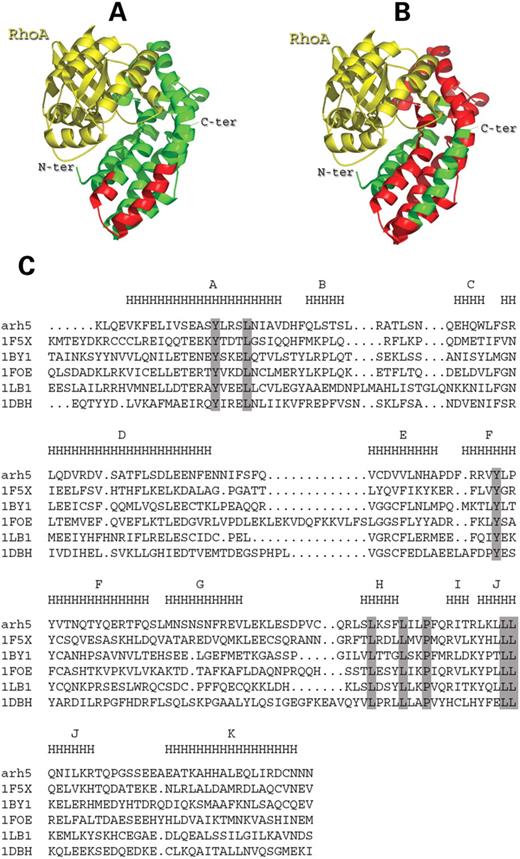
Figure 5. Illustration of the structural modifications in the DH domain of ARHGEF5 (ARH5) variant proteins V1 and V5. (A, B) Ribbon diagram of the ARH5 DH domain (green) complexed with RhoA (yellow). The deleted regions in the DH domain of V1 (A) and V5 (B) are shown in red. (C) Multiple alignment of the known DH domains. Sequences are designated by their Protein Data Bank id code (1F5X, Vav; 1BY1, β-Pix; 1FOE, Tiam; 1LB1, Dbs; 1DBH, Sos1). Residues strictly conserved in the six sequences are boxed in grey. Consensus secondary structure is shown above the sequence (H indicates α-helices) with α-helix nomenclature from Vav and β-Pix structures.
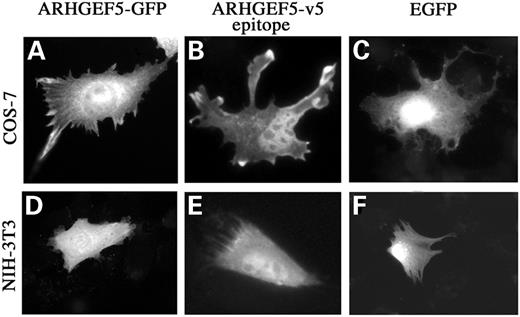
Figure 6. Cellular expression of recombinant ARHGEF5/TIM. Exponentially growing COS-7 and NIH-3T3 cells were transfected with plasmids encoding ARHGEF5-GFP (A and D), V5-epitope-tagged ARHGEF5 (B and E) or EGFP (C and F). Cells were fixed 48 h after transfection and monitored for GFP fluorescence. For each panel, cells shown are representative of more than 100 observed cells.
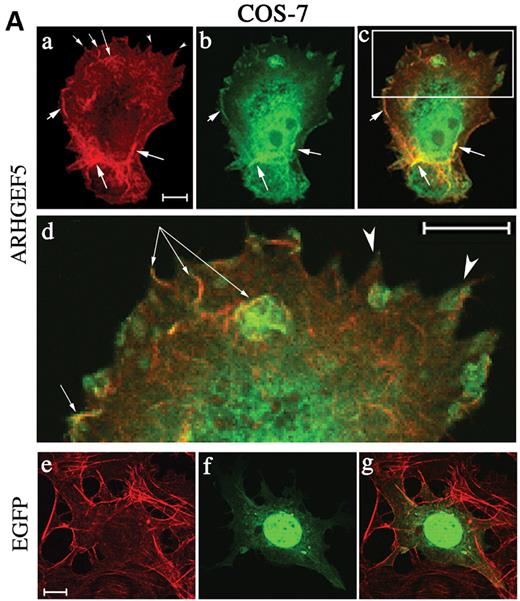
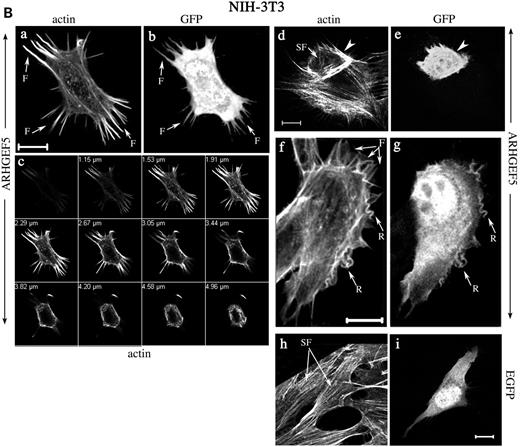
Figure 7. Actin reorganization induced by ARHGEF5-GFP protein expression. COS-7 (A) and NIH-3T3 cells (B) were transfected with plasmids encoding ARHGEF5-GFP (A, a–d; B, a–g) or EGFP (A, e–g; B, h and i) proteins. Cells were fixed, permeabilized, stained with TRITC-conjugated phalloidin and observed under confocal microscopy. Selected slices are shown in separate staining and as merged images (A, c, d and g). (A) Panel (d) represents a magnification of a delimited area (rectangle) of the cell shown in (c). Arrows indicate dorsal ruffles with colocalized ARHGEF5-GFP (d). (B) Panel (c) represents selected TRITC-phalloidin stained sections of the cell in (a). Filopodia, membrane ruffles and stress fibers are indicated by arrows designated by F (a and f), R (f), SF (a, d and h), respectively; arrowheads show a cortical actin condensation (d); colocalized ARHGEF5/TIM in these structures is shown by identical symbols (b, e and g). For each panel, cells shown are representative of more than 100 observed cells. Bar, 10 µm.
Sequences of ARHGEF5/TIM primers
| Name | 5′–3′ sequence | Name | 5′–3′ sequence |
|---|---|---|---|
| 1F | CAGATTCAAGAGGTCCAGCC | 1R | CAAAGTCCCTCGAAATCCCC |
| 2F | CTCCATGACCCATGAAGACC | 2R | TCTGTCCGTCGCATACTCTG |
| 3F | CCAAAAGCTGCAAGAG | 3R | GACATTGTTATTGCAGTCC |
| 4F | TCTGGTGTGACTGTGAAC | 4R | GGAATCATGGAAGAAACTGA |
| 5F | CCCTCAGGAAGACCCACAAC | 5R | GCAGGGAGGTGGCTGAGCTC |
| attB1 | ggggacaagtttgtacaaaaaagcaggcttc | attB2 | ggggaccactttgtacaagaaagctgggtcGGC |
| GAGGTGATGGGAGGCTTTTC | TTGCTGTTCCACCAGCTGTAG | ||
| AAGACGCTGCTC |
| Name | 5′–3′ sequence | Name | 5′–3′ sequence |
|---|---|---|---|
| 1F | CAGATTCAAGAGGTCCAGCC | 1R | CAAAGTCCCTCGAAATCCCC |
| 2F | CTCCATGACCCATGAAGACC | 2R | TCTGTCCGTCGCATACTCTG |
| 3F | CCAAAAGCTGCAAGAG | 3R | GACATTGTTATTGCAGTCC |
| 4F | TCTGGTGTGACTGTGAAC | 4R | GGAATCATGGAAGAAACTGA |
| 5F | CCCTCAGGAAGACCCACAAC | 5R | GCAGGGAGGTGGCTGAGCTC |
| attB1 | ggggacaagtttgtacaaaaaagcaggcttc | attB2 | ggggaccactttgtacaagaaagctgggtcGGC |
| GAGGTGATGGGAGGCTTTTC | TTGCTGTTCCACCAGCTGTAG | ||
| AAGACGCTGCTC |
F, forward primer; R, reverse primer; attB1 and attB2, forward and reverse primers for recombinational cloning into pDONR221 (Gateway System, Invitrogen). Recombination sequences are shown in italic small letters.
Sequences of ARHGEF5/TIM primers
| Name | 5′–3′ sequence | Name | 5′–3′ sequence |
|---|---|---|---|
| 1F | CAGATTCAAGAGGTCCAGCC | 1R | CAAAGTCCCTCGAAATCCCC |
| 2F | CTCCATGACCCATGAAGACC | 2R | TCTGTCCGTCGCATACTCTG |
| 3F | CCAAAAGCTGCAAGAG | 3R | GACATTGTTATTGCAGTCC |
| 4F | TCTGGTGTGACTGTGAAC | 4R | GGAATCATGGAAGAAACTGA |
| 5F | CCCTCAGGAAGACCCACAAC | 5R | GCAGGGAGGTGGCTGAGCTC |
| attB1 | ggggacaagtttgtacaaaaaagcaggcttc | attB2 | ggggaccactttgtacaagaaagctgggtcGGC |
| GAGGTGATGGGAGGCTTTTC | TTGCTGTTCCACCAGCTGTAG | ||
| AAGACGCTGCTC |
| Name | 5′–3′ sequence | Name | 5′–3′ sequence |
|---|---|---|---|
| 1F | CAGATTCAAGAGGTCCAGCC | 1R | CAAAGTCCCTCGAAATCCCC |
| 2F | CTCCATGACCCATGAAGACC | 2R | TCTGTCCGTCGCATACTCTG |
| 3F | CCAAAAGCTGCAAGAG | 3R | GACATTGTTATTGCAGTCC |
| 4F | TCTGGTGTGACTGTGAAC | 4R | GGAATCATGGAAGAAACTGA |
| 5F | CCCTCAGGAAGACCCACAAC | 5R | GCAGGGAGGTGGCTGAGCTC |
| attB1 | ggggacaagtttgtacaaaaaagcaggcttc | attB2 | ggggaccactttgtacaagaaagctgggtcGGC |
| GAGGTGATGGGAGGCTTTTC | TTGCTGTTCCACCAGCTGTAG | ||
| AAGACGCTGCTC |
F, forward primer; R, reverse primer; attB1 and attB2, forward and reverse primers for recombinational cloning into pDONR221 (Gateway System, Invitrogen). Recombination sequences are shown in italic small letters.
Expression of ARHGEF5/TIM splice variants in normal tissues and tumoral cell lines
| Transcripts | |||
|---|---|---|---|
| WT | V1+V2 | ||
| (A) Normal tissues | |||
| Mammary gland | + | − | |
| Prostate | + | − | |
| Liver (fetal/adult) | + | − | |
| Lung (fetal/adult) | + | − | |
| Heart (fetal/adult) | + | − | |
| Placenta | + | − | |
| Uterus | + | − | |
| Testis | + | − | |
| Spleen | + | − | |
| Salivary gland | + | − | |
| (B) Cell lines (tumor origin) | |||
| HepG2; HA22T; Malhavu; PLC-PRF (liver hepatoma) | + | − | |
| H3E5 (colon carcinoma) | + | − | |
| HT1080 (fibrosarcoma) | + | − | |
| HeLa (cervical carcinoma) | + | − | |
| Jurkat (T-cell leukemia) | + | − | |
| Raji (burkitt lymphoma) | + | − | |
| ZR75-1 (breast carcinoma) | + | − | |
| T47D (breast carcinoma) | + | + (53, 34) | |
| MCF7 (breast carcinoma) | + | + (41, 26) | |
| VHB1 (breast carcinoma) | + | + (46, 33) | |
| MDA-MB231 (breast carcinoma) | + | + (27, 12) | |
| PC3 (prostate carcinoma) | + | + (13, 11) | |
| JAR (choriocarcinoma) | + | + (21, 25) | |
| Transcripts | |||
|---|---|---|---|
| WT | V1+V2 | ||
| (A) Normal tissues | |||
| Mammary gland | + | − | |
| Prostate | + | − | |
| Liver (fetal/adult) | + | − | |
| Lung (fetal/adult) | + | − | |
| Heart (fetal/adult) | + | − | |
| Placenta | + | − | |
| Uterus | + | − | |
| Testis | + | − | |
| Spleen | + | − | |
| Salivary gland | + | − | |
| (B) Cell lines (tumor origin) | |||
| HepG2; HA22T; Malhavu; PLC-PRF (liver hepatoma) | + | − | |
| H3E5 (colon carcinoma) | + | − | |
| HT1080 (fibrosarcoma) | + | − | |
| HeLa (cervical carcinoma) | + | − | |
| Jurkat (T-cell leukemia) | + | − | |
| Raji (burkitt lymphoma) | + | − | |
| ZR75-1 (breast carcinoma) | + | − | |
| T47D (breast carcinoma) | + | + (53, 34) | |
| MCF7 (breast carcinoma) | + | + (41, 26) | |
| VHB1 (breast carcinoma) | + | + (46, 33) | |
| MDA-MB231 (breast carcinoma) | + | + (27, 12) | |
| PC3 (prostate carcinoma) | + | + (13, 11) | |
| JAR (choriocarcinoma) | + | + (21, 25) | |
RT–PCR was performed with primers 2F/2R. Numbers in parentheses correspond to the mean percentage of V1 and V2 variant levels related to WT transcript level, as determined by densitometry using the NIH Image software.
Expression of ARHGEF5/TIM splice variants in normal tissues and tumoral cell lines
| Transcripts | |||
|---|---|---|---|
| WT | V1+V2 | ||
| (A) Normal tissues | |||
| Mammary gland | + | − | |
| Prostate | + | − | |
| Liver (fetal/adult) | + | − | |
| Lung (fetal/adult) | + | − | |
| Heart (fetal/adult) | + | − | |
| Placenta | + | − | |
| Uterus | + | − | |
| Testis | + | − | |
| Spleen | + | − | |
| Salivary gland | + | − | |
| (B) Cell lines (tumor origin) | |||
| HepG2; HA22T; Malhavu; PLC-PRF (liver hepatoma) | + | − | |
| H3E5 (colon carcinoma) | + | − | |
| HT1080 (fibrosarcoma) | + | − | |
| HeLa (cervical carcinoma) | + | − | |
| Jurkat (T-cell leukemia) | + | − | |
| Raji (burkitt lymphoma) | + | − | |
| ZR75-1 (breast carcinoma) | + | − | |
| T47D (breast carcinoma) | + | + (53, 34) | |
| MCF7 (breast carcinoma) | + | + (41, 26) | |
| VHB1 (breast carcinoma) | + | + (46, 33) | |
| MDA-MB231 (breast carcinoma) | + | + (27, 12) | |
| PC3 (prostate carcinoma) | + | + (13, 11) | |
| JAR (choriocarcinoma) | + | + (21, 25) | |
| Transcripts | |||
|---|---|---|---|
| WT | V1+V2 | ||
| (A) Normal tissues | |||
| Mammary gland | + | − | |
| Prostate | + | − | |
| Liver (fetal/adult) | + | − | |
| Lung (fetal/adult) | + | − | |
| Heart (fetal/adult) | + | − | |
| Placenta | + | − | |
| Uterus | + | − | |
| Testis | + | − | |
| Spleen | + | − | |
| Salivary gland | + | − | |
| (B) Cell lines (tumor origin) | |||
| HepG2; HA22T; Malhavu; PLC-PRF (liver hepatoma) | + | − | |
| H3E5 (colon carcinoma) | + | − | |
| HT1080 (fibrosarcoma) | + | − | |
| HeLa (cervical carcinoma) | + | − | |
| Jurkat (T-cell leukemia) | + | − | |
| Raji (burkitt lymphoma) | + | − | |
| ZR75-1 (breast carcinoma) | + | − | |
| T47D (breast carcinoma) | + | + (53, 34) | |
| MCF7 (breast carcinoma) | + | + (41, 26) | |
| VHB1 (breast carcinoma) | + | + (46, 33) | |
| MDA-MB231 (breast carcinoma) | + | + (27, 12) | |
| PC3 (prostate carcinoma) | + | + (13, 11) | |
| JAR (choriocarcinoma) | + | + (21, 25) | |
RT–PCR was performed with primers 2F/2R. Numbers in parentheses correspond to the mean percentage of V1 and V2 variant levels related to WT transcript level, as determined by densitometry using the NIH Image software.
Amplification of ARHGEF5/TIM transcripts in primary breast carcinoma samples
| ARHGEF5 fragments | Total | Breast carcinoma (n=33) | ||
|---|---|---|---|---|
| Ductal/ductotubular (n=25) | Lobular (n=8) | |||
| High grade (n=14) | Low grade (n=11) | High grade (n=8) | ||
| WT | 17 | 7 (50%) | 8 (73%) | 2 (25%) |
| WT+V1+V2 | 8 | 4 | 2 | 2 |
| WT+V1 | 4 | 3 | — | 1 |
| WT+V3 | 1 | — | 1 | — |
| WT+V3+V4 | 1 | — | — | 1 |
| V5 | 1 | — | — | 1 |
| None | 1 | — | — | 1 |
| Total variants | 15 (45%) | 7 (50%) | 3 (27%) | 5 (63%) |
| ARHGEF5 fragments | Total | Breast carcinoma (n=33) | ||
|---|---|---|---|---|
| Ductal/ductotubular (n=25) | Lobular (n=8) | |||
| High grade (n=14) | Low grade (n=11) | High grade (n=8) | ||
| WT | 17 | 7 (50%) | 8 (73%) | 2 (25%) |
| WT+V1+V2 | 8 | 4 | 2 | 2 |
| WT+V1 | 4 | 3 | — | 1 |
| WT+V3 | 1 | — | 1 | — |
| WT+V3+V4 | 1 | — | — | 1 |
| V5 | 1 | — | — | 1 |
| None | 1 | — | — | 1 |
| Total variants | 15 (45%) | 7 (50%) | 3 (27%) | 5 (63%) |
Results from RT–PCR and nested PCR performed with primers 2F/2R and 3F/3R. Distribution of the ARHGEF5/TIM splicing variants related to type and malignancy grade of 33 primary breast carcinomas.
Amplification of ARHGEF5/TIM transcripts in primary breast carcinoma samples
| ARHGEF5 fragments | Total | Breast carcinoma (n=33) | ||
|---|---|---|---|---|
| Ductal/ductotubular (n=25) | Lobular (n=8) | |||
| High grade (n=14) | Low grade (n=11) | High grade (n=8) | ||
| WT | 17 | 7 (50%) | 8 (73%) | 2 (25%) |
| WT+V1+V2 | 8 | 4 | 2 | 2 |
| WT+V1 | 4 | 3 | — | 1 |
| WT+V3 | 1 | — | 1 | — |
| WT+V3+V4 | 1 | — | — | 1 |
| V5 | 1 | — | — | 1 |
| None | 1 | — | — | 1 |
| Total variants | 15 (45%) | 7 (50%) | 3 (27%) | 5 (63%) |
| ARHGEF5 fragments | Total | Breast carcinoma (n=33) | ||
|---|---|---|---|---|
| Ductal/ductotubular (n=25) | Lobular (n=8) | |||
| High grade (n=14) | Low grade (n=11) | High grade (n=8) | ||
| WT | 17 | 7 (50%) | 8 (73%) | 2 (25%) |
| WT+V1+V2 | 8 | 4 | 2 | 2 |
| WT+V1 | 4 | 3 | — | 1 |
| WT+V3 | 1 | — | 1 | — |
| WT+V3+V4 | 1 | — | — | 1 |
| V5 | 1 | — | — | 1 |
| None | 1 | — | — | 1 |
| Total variants | 15 (45%) | 7 (50%) | 3 (27%) | 5 (63%) |
Results from RT–PCR and nested PCR performed with primers 2F/2R and 3F/3R. Distribution of the ARHGEF5/TIM splicing variants related to type and malignancy grade of 33 primary breast carcinomas.
References
McPherson, K., Steel, C.M. and Dixon, J.M. (
Harbeck, N., Thomssen, C., Berger, U., Ulm, K., Kates, R.E., Hofler, H., Janicke, F., Graeff, H. and Schmitt, M. (
Brenner, A.J. and Aldaz, C.M. (
Murphy, L.C., Lee-Wing, M., Goldenberg, G.J. and Shiu, R.P.C. (
Autiero, M., Abrescia, P. and Guardiola, J. (
Autiero, M., Cammarota, G., Friedlein, A., Zulauf, M., Chiappetta, G., Dragone, V. and Guardiola, J. (
Basmaciogullari, S., Autiero, M., Culerrier, R., Mani, J.C., Gaubin, M., Mishal, Z., Guardiola, J., Granier, C. and Piatier-Tonneau, D. (
Gaubin, M., Autiero, M., Basmaciogullari, S., Métivier, D., Mishal, Z., Culerrier, R., Oudin, A., Guardiola, J. and Piatier-Tonneau, D. (
Caputo, E., Manco, G., Mandrich, L. and Guardiola, J. (
Autiero, M., Culerrier, R., Bouchier, C., Basmaciogullari, S., Gaubin, M., El Marhomy, S., Blanchet, P., Paradis, V., Jardin, A., Guardiola, J. et al. (
Autiero, M., Camarca, A., Ciullo, M., Debily, M.A., El Marhomy, S., Pasquinelli, R., Capasso, I., D'Aiuto, G., Anzisi, A.M., Piatier-Tonneau, D. et al. (
Clark, J.W., Snell, L., Shiu, R.P., Orr, F.W., Maitre, N., Vary, C.P., Cole, D.J. and Watson, P.H. (
Ciullo, M., Debily, M.A., Rozier, L., Autiero, M., Billault, A., Mayau, V., El Marhomy, S., Guardiola, J., Berheim, A., Coullin, P. et al. (
Chan, A.M., McGovern, E.S., Catalano, G., Fleming, T.P. and Miki, T. (
Takai, S., Chan, A.M., Yamada, K. and Miki, T. (
Schmidt, A. and Hall, A. (
Snyder, J.T., Worthylake, D.K., Rossman, K.L., Betts, L., Pruitt, W.M., Siderovski, D.P., Der, C.J. and Sondek, J. (
Blomberg, N., Baraldi, E., Nilges, M. and Saraste, M. (
Zheng, Y. (
Van Aelst, L. and D'Souza-Schorey, C. (
Fritz, G., Just, I. and Kaina, B. (
Schnelzer, A., Prechtel, D., Knaus, U., Dehne, K., Gerhard, M., Graeff, H., Harbeck, N., Schmitt, M. and Lengyel, E. (
Pruitt, K. and Der, C.J. (
Aghazadeh, B., Lowry, W.E., Huang, X.Y. and Rosen, M.K. (
Nobes, C.D. and Hall, A. (
Gauthier-Rouvière, C., Vignal, E., Mériane, M., Roux, P., Montcourier, P. and Fort, P. (
Hillier, L.W., Fulton, R.S., Fulton, L.A., Graves, T.A., Pepin, K.H., Wagner-Mcpherson, C., Layman, D., Maas, J., Jaeger, S., Walker, R. et al. (
Wang, Z., Lo, H.S., Yang, H., Gere, S., Hu, Y., Buetow, K.H. and Lee, H.M. (
Ahn, S.J., Chung, K.W., Lee, R.A., Park, I.A., Lee, S.H., Park, D.E. and Noh, D.Y. (
Engers, R., Zwaka, T.P., Gohr, L., Weber, A., Gerharz, C.D. and Gabbert, H.E. (
Debant, A., Serra-Pages, C., Seipel, K., O'Brien, S., Tang, M., Park, S.H. and M., S. (
Vandewalle, B., Collyn d'Hooghe, M., Savary, J.B., Vilain, M.O., Peyrat, J.P., Deminatti, M., Delobelle-Deroide, A. and Lefebvre, J. (
Maniatis, T., Fritsch, E.F. and Sambrook, J. (
Plewniak, F., Bianchetti, L., Brelivet, Y., Carles, A., Chalmel, F., Lecompte, O., Mochel, T., Moulinier, L., Muller, A., Muller, J. et al. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}