




Abstract
Can Drosophila models be engineered that accurately reflect Huntington's disease (HD) and other neurological diseases and can they contribute to the search for treatments and cures? A number of publications seem to provide a resounding yes to that question. Here we seek to review some of the salient features of these models.
SEARCHING FOR CURES
The quest to find cures for human diseases involves two major objectives: understanding the molecular and cellular mechanisms of the disease process and finding drugs and treatments that will alleviate the disease and/or its symptoms. Although it can be argued that it is not absolutely necessary to understand the disease process before finding a treatment for it, it is also true that with understanding comes opportunity. Currently, screening for drugs and therapeutics takes place sequentially, with the first steps carried out in a cell-free or cultured cell setting that typically reflects only one part of a myriad of characteristics of a particular disease process. Identification of lead compounds then proceeds to animal testing, typically mice, where a large number drop out either because they do not ameliorate the disease process in vivo or because of unwanted side effects. This sequence of events is slow and expensive and has a major influence on the time and cost of drug development. However, it could proceed more rapidly and at lower cost by using nonvertebrate organisms that can be genetically engineered and have short generation times that allow rapid identification of the most promising strategies for testing in mice. The question is, can nonmammalian organisms be engineered to accurately reflect the human disease process?
THE FRUIT FLY IS WELL SUITED FOR MODELING
The fly is one of the best invertebrates for modeling higher organisms. Comparative genome analysis reveals that at least 50% of fly genes have similar genes in man (blast cutoff value of E<10−10) (1). Among those human genes known to be associated with disease, ∼75% have a Drosophila ortholog (2).
The fly is also an excellent choice for modeling neurodegenerative diseases because it contains a fully functional nervous system with an architecture that separates specialized functions such as vision, olfaction, learning and memory in a manner not unlike that of mammalian nervous systems (3–5). Further, the compound eye of a fruit fly is made up of hundreds of repeating constellations of photoreceptor neurons such that any perturbation in the pattern is quite evident. Most importantly, in Drosophila foreign genes can be engineered to be expressed in tissue-specific and temporally regulated patterns and an impressive array of genetic tools are available.
HD AND RELATED DISEASES ARE ASSOCIATED WITH ABNORMAL PROTEIN ACCUMULATIONS
Huntington's disease is a now classic example of a family of at least nine dominant, late-onset diseases that are caused by expanded CAG triplet repeat sequences that encode expanded polyglutamine repeats (poly Q) in the affected protein (Q is the single letter code for glutamine). The polyQ diseases are part of a much larger family of protein conformation diseases, many of which also cause dominant, late-onset neurodegeneration. A key feature of these disorders is that they are caused by mutations or cellular events that lead to accumulation of abnormal structural forms of a particular protein. The polyQ diseases produce nuclear inclusions; Alzheimer's disease (AD) is associated with β amyloid plaques and neurofibrillary tangles and Parkinson's disease (PD) is typified by the formation of Lewy bodies. These, along with diseases such as prion disease, Pick's disease, other tauopathies and amyotrophic lateral sclerosis (ALS) comprise the majority of the protein conformation diseases. What is clear is that these altered proteins can be toxic. What is not yet clear is why. However, from a therapeutic perspective, it is encouraging that many of these protein conformation diseases may share common toxic cellular processes.
KEY HALLMARKS OF HD AND polyQ DISEASES
The signature profile of HD and other polyQ diseases is that they are dominant, of late onset, cause progressive degeneration, are associated with abnormal protein aggregates and lead to motor function loss, early death and other symptoms. Huntington's disease is one of the few truly dominant inherited diseases with full penetrance. It is caused by a polyQ repeat expansion in the HD gene which encodes a large (∼350 kDa) protein (Huntingtin, Htt) of as yet unknown biochemical function that is expressed in essentially all cells, beginning during embryogenesis (9–12). A single copy of the abnormal gene invariably causes disease and rare individuals with two affected copies exhibit the same age of onset as those with one, although once symptoms begin, homozygotes may have a more rapid progression than heterozygotes (13,14). The difference between disease state and normal shows a sharp threshold, with expansions above ∼39 Qs invariably leading to disease while individuals with ≤35 Qs are disease free unless further expansion occurs (8,15). The onset of clinical symptoms typically occurs late in the fourth or fifth decade of life (for expansions of ∼40–60 Qs) although the onset of clinical symptoms occurs earlier in individuals with ≥∼60 polyQs (16).
OVERVIEW OF THE FLY SYSTEM
The dominant neurodegenerative diseases are particularly well suited for modeling in Drosophila because they are caused by gain of function mutations in single genes that can readily be engineered to be expressed in flies and cause phenotypes that closely mimic the human disease.
Drosophila embryogenesis spans approximately one day with neurogenesis beginning at about 5 hours and completing by about 15 hours. First instar larvae (instar refers to the larval stages) hatch from the egg and molt to the second instar and to the third instar after a day at each stage. During the third larval stage (from 3 to 4.5 days post fertilization), the eye imaginal discs complete their growth, and photoreceptor neurons are born (17,18). The larvae form pupae and begin a five day period of metamorphosis (19), during which time, parts of the central nervous system (CNS) are retained as a scaffold and other parts are replaced by new rounds of neurogenesis (20). In addition, the photoreceptor cells that were born in the imaginal disc now become organized into the adult eye. Approximately 10 days after the initial laying of an egg, the adult fly ecloses (emerges) from the pupal case.
Foreign genes are expressed using a bipartite gene expression system (21,22) in which genes inserted behind the yeast upstream activator sequence (UAS) are activated by the yeast Gal4 protein. Genes fused to UAS and injected into embryos with a helper element integrate into the chromosome producing transgenic lines carrying the UAS>transgene. Unlike DNA integration events in some other systems that lead to multiple tandem insertions, the Drosophila system favors the insertion of a single transgene at a random site. Transgenic animals are then crossed to ‘driver lines’ that express Gal4 in a variety of tissue-specific patterns. A large collection of ‘Gal4 drivers’ is available (23). For any given Gal4 driver/UAS>transgene combination, it is frequently observed that using the same driver to drive different isolates of the same uas>transgene gives somewhat different phenotypes, often referred to as strong, medium and weak (24). It is assumed that these differences are due to position effects, where the different transgene insertion sites are expressed at higher or lower levels due to surrounding chromatin sequences, although this assumption is rarely tested experimentally. In addition, the level of expression from the Gal4/UAS combination is highly sensitive to temperature (25).
Many measures of neuronal dysfunction are possible, with some of the most common ones being climbing ability (motor function) or integrity of photoreceptor cells of the eye. The ommatidia of the eye are precisely organized in a repeating pattern and are made up of nine neuronal cells (eight photoreceptors, one mechanosensory) and 11 support cells including the primary, secondary and tertiary pigment cells and the cone cells that make the lens. Each photoreceptor cell produces a highly reticulated membrane (the rhabdomere) that carries light-gathering rhodopsins. It is this trapezoid of seven visible rhabdomeres that one observes in sectioned material or with the pseudopupil technique of shining a light through the back of the head (Fig. 1A). Only seven rhabdomeres are visible because R7 and R8 sit on top of one another. Cells are born as photoreceptor neurons during a morphogenetic event in the eye disc (18,26,27).
One widely used driver for neurodegeneration studies is elav, which expresses Gal4 in every cell of the nervous system from embryogenesis onward (28) and another is gmr, which expresses in all cells of the eye including both neurons and surrounding supporting cells (29). Expression of both elav and gmr is activated at the front of a morphogenetic wave that occurs in the eye disc and creates a gradient of neurons that have been exposed to toxic polyQ proteins for defined periods of time. Expression of polyQ containing proteins by elav can lead to the degeneration of the neurons, but this is not accompanied by any overt external dysmorphology. On the other hand, expression of transgenes with the gmr driver leads to extensive degeneration in the eye and is often evident as external dysmorphology (27,30–33). A caveat in the interpretation of these phenotypes is that the development of the eye depends on the stepwise specification of particular cell fates, which requires the continued contact between cells of different fates (34). Consequently, care must be exercised in interpreting neuronal cell death in settings in which the support cells are also subject to degeneration.
Motor function is readily addressed by exploiting the negative geotropic behavior of flies and counting the number of flies that can climb to the top of a tube in a specified amount of time (35,36).
THE FLY MIMICS HUMAN DISEASE
How accurately do engineered Drosophila mimic the key features of human disease? Expression of pathogenic forms of Htt (37), ataxin-1 (SCA1), ataxin-3 (SCA3/MJD) and AR (Kennedy's disease) all cause neuropathology in Drosophila that exhibits most of the features of human disease (27,30–33,38–40). In all these models, it has been found that pathology exhibits a polyQ length dependency similar to humans. Further, no evidence of neurodegeneration has been described early in the larval stages, but clear evidence of degeneration occurs in mature larvae, in pupae and in aging adults (Fig. 2). The severity of neuropathology is progressive (Fig. 3A) with animals at 3 and 7 days of age showing more severe neuropathology than at one day post eclosion (27,39,40). Thus, by every measure, flies expressing mutant human genes or polyQ peptides alone present with pathology that mimics the human disease in every important way, for example,
polyQ causes cellular pathology;
pathology is a function of polyQ length;
pathology is late onset (late in larval/pupal life);
pathology is progressive;
pathology leads to loss of motor function; and
pathology causes early death.
WHAT HAVE WE LEARNED?
Having a genetically tractable model of human disease allows one to test hypotheses regarding mechanism and to perform genetic screens to identify pathways that may affect polyQ pathogenesis. Genetic screens identify genes, which when reduced cause the phenotype to get worse (enhancers) or better (suppressors). Such experiments have been carried out in Drosophila models of several polyQ diseases as well as in worm and yeast models of polyQ diseases (41–44). Several promising treatment targets have emerged from these studies.
The cloning and expression pattern of the HD gene did not provide immediate clues to pathogenesis. However, it was noted that although normal Htt is cytoplasmic (45), mutant Htt was progressively localized to the nucleus and large aggregates (inclusions) were found in neurons (45,46). Several subsequent studies suggested that transcriptional dysregulation might be contributing to pathogenesis (47). The availability of nonmammalian models of HD has proved a rapid means of testing some of the hypotheses raised by these studies in vivo. For example, nuclear inclusions of mutant Htt were found to contain transcriptional co-activators such as CBP, an acetyl transferase (AT) (48–50). CBP and other (histone) acetyl transferases typically act as co-activators of transcription by modifying histones and other proteins to increase transcription. The possibility that sequestration and direct inhibition of AT protein activities might be contributing to pathogenesis in vivo was tested in Drosophila and other models by inhibiting the counteracting activity of Histone DeACetylases (HDACs) both genetically and pharmacologically (39,42,51,52). Independently, genetic screens and other studies identified genes involved in transcriptional dysregulation as well as other overlapping sets of genes as relevant to polyQ pathology (32,33,53,54). Such results confirm the critical nature of balanced protein acetylation and deacetylation in polyQ pathogenesis and provided a potential pharmacologic therapy that has subsequently proved effective in mammals (55). The speed and methods that were available to test these hypotheses illustrate the value of invertebrate models of human disease in rapidly identifying therapeutic strategies that are promising enough to test in mice.
Nuclear inclusions in several polyQ diseases are ubiquitinated and sequester molecular chaperones, underscoring a role for protein processing and degradative pathways in pathogenesis. Overexpression of chaperones had reduced polyQ aggregates in cultured cells (56–59). However, the in vivo role of the proteosome and chaperone pathways in neurotoxicity cannot be readily assessed in cell assays. Using a Drosophila model of Machado Joseph disease, it was shown that increased chaperone activity could suppress pathology (60). Genetic screens for suppressors and enhancers of a SCA1 model in Drosophila, also identified chaperones as modifiers of polyQ pathology (32,33) as well as Parkinson's pathology (61). Subsequent studies showing that overexpression of Hsp70 reduced pathology in a mouse model of SCA 1 (62) confirmed the significance of chaperones that were identified in the Drosophila studies. Again, the value of invertebrate models in testing hypotheses and identifying relevant pathways is evident.
As stated earlier, polyQ diseases are one of several ‘protein conformation’ diseases. Since aggregates are such ubiquitous hallmarks of neurodegenerative diseases, polyQ aggregates are a tempting target for high-throughput screening of pharmacologic agents that might block or disrupt aggregate formation, and many cell-free and cell-based screens have been developed (42,63–66) (Diamond, personal communication). However, the potential efficacy of aggregate disrupting/preventing compounds in relieving pathology must be addressed in vivo. Again, Drosophila models have proven effective in rapidly allowing the efficacy of various pharmacologic and synthetic peptide agents on neuropathology to be tested (30,66,67). Some of these suppressors show visible effects upon aggregation in flies (67), while others show no visible change but may affect the composition of aggregates (68).
Exciting recent findings using a fly model of SCA1 have implicated phosphatidylinositol 3-kinase/AKT signaling and 14-3-3/ataxin1 protein interactions in vivo in neurotoxicity (69). Modulation of levels of these cellular proteins modifies neurodegenerative phenotypes and highlights a completely novel target for therapeutic intervention. Another screen has identified Drosophila VCP, an AAA+ ATPase superfamily member, as a dominant suppressor of polyQ pathology (70).
The fly model has also been useful in addressing the question whether neurodegeneration is due to altered activity of the mutant proteins or to an intrinsic pathology of expanded polyQ itself. Indeed, all of the symptoms above are evident in Drosophila expressing several mutant forms of human genes that have expanded polyQ peptides and are also evident when polyQ peptides alone are expressed that are free of any disease gene context (31,32). These observations argue that at least a large part of pathology is due to a dominant activity of the expanded polyQ itself. This is encouraging because it suggests that the pathogenic mechanism of many or all of the polyQ diseases may share some common biochemical features that allow therapy for one disease to be effective in the others. Such hope is bolstered by the recent demonstration that many neurological disorders, including those caused by polyQ-containing peptides, may share a common structural epitope that is toxic (71).
The concordance of compounds that are effective in both fly and mouse models of HD underscores the utility of using fly models of human disease to screen for target pathways. It also argues that wider use of invertebrate systems to screen directly for compounds that lead to functional neurologic improvement may be effective (55,61,63,66,72–74). Aside from cell survival assays, all cell and cell-free based screening strategies must be based on some assumptions about the disease mechanisms. To the extent that those mechanisms may not be fully understood (a common situation) live animal screens can identify compounds that are effective even if the mechanism is not fully understood. On the downside, live animal screens are inherently lower throughput than cell or cell-free based screens. However, they can filter out a large number of false leads in the early phases of screening. Efforts to automate and improve the throughput of live animal screens are under way in several sites.
FUTURE GOALS AND MAJOR UNRESOLVED QUESTIONS
The full potential of the Drosophila model systems will only be realized when models are made for the majority of the human degenerative diseases and such models are appearing more and more frequently (41,61,75,76). As each disease model is studied, it is hoped that the comparisons between the pathways identified in genetic screens and the efficacy of therapeutic strategies in different models will allow one to identify the commonalities between and the unique features of the different diseases. Another area of endeavor will be the effort to make Drosophila models more amenable to high-throughput and automated screening for therapeutics. In this regard, practical hurdles to be overcome are the automated manipulation and scoring of flies and the fact that the animal is not accessible to externally administered drugs and compounds during the five-day pupal period nor during embryogenesis. Are drugs that are discovered first in flies the best candidates for testing in mice? It is too soon to tell, but early indications of concordance are good.
SUMMARY
It has been well documented by now that one can engineer Drosophila to mimic several important neurodegenerative diseases including HD and the polyQ diseases in general as well as other late-onset neurodegenerative diseases such as Parkinson's and tauopathies (61,75,77,78). These models provide the tools to investigate the mechanisms of disease and to develop screens and cures. Genetic screens have been used to look for modifiers of the mutant phenotype. Such screens can point to cellular pathways that influence the severity of a particular disease, for example, the proteosome pathway, transcriptional regulating proteins, and so on. These models can also be used to test hypotheses of the pathology, for example, the role of transcription in disease, and potentially to find promising leads for pharmacologic cures or relief. The list currently includes HDAC inhibitors, several chemical or peptide inhibitors of aggregation, and drugs that target cellular stress responses (30,55,66,67,79) (unpublished observations).
ACKNOWLEDGEMENTS
This work was supported by NIH awards HD36081 and HD36049 to J.L.M., a Cure HD Initiative of the Hereditary Disease Foundation (to L.M.T. and J.L.M.), Coalition for the Cure of the Huntington's Disease Society of America (to L.M.T.), and Human Frontiers Science Program grant (to L.M.T.).
To whom correspondence should be addressed at: Department of Developmental and Cell Biology, 4244 McGaugh Hall, University of California, Irvine, CA 92697-2300, USA. Tel: +1 9498246677; Email: jlmarsh@uci.edu
These authors contributed equally to this work.
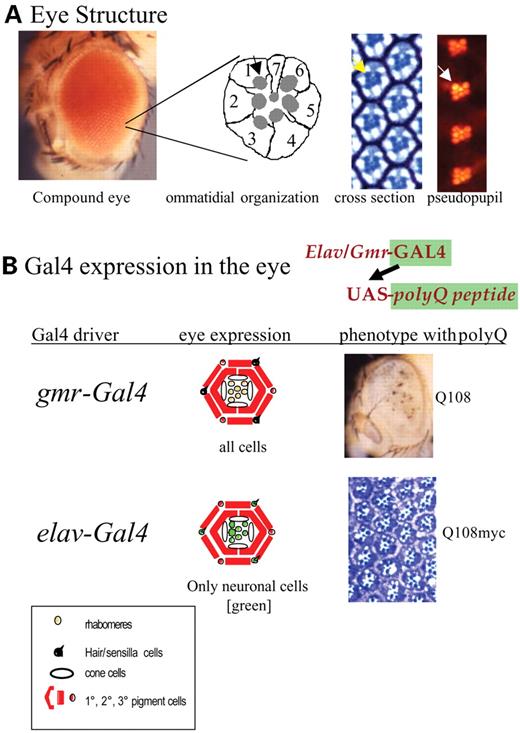
Figure 1. (A) Structure of the adult eye showing the external eye, a diagram of the structure of the photoreceptor cells in an ommatidium, and a section of an eye showing ommatidia in cross-section and by the pseudopupil technique. The rhabdomeres (arrows) can be seen in each panel. (B) Expression of polyQ peptides with the gmr-Gal4 and elav-Gal4 drivers. The phenotype obtained by expressing strongly cytotoxic polyQ peptides is shown. Note the external rough eye caused by expression with gmr-Gal4 and the degeneration of the non-neuronal pigment cells. Expression with elav-Gal4 gives no external phenotype but causes modest loss of photoreceptors when 48 Qs are expressed and significant loss of photoreceptor neurons when 108 Qs are expressed (see Fig. 2).
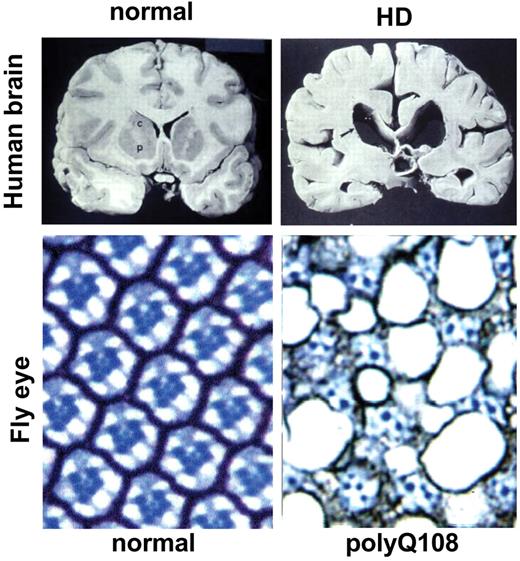
Figure 2. Neurodegeneration in flies mimics man. Cross-sections through a normal and postmortem HD patient brain demonstrate the dramatic degeneration and loss of neuronal tissue. Cross-sections through they eye of a fly expressing polyQ108 in the photoreceptor neurons show similar significant loss of neuronal tissue. Photos of human brain courtesy of Drs P. Harper and J. Neal, Cardiff.
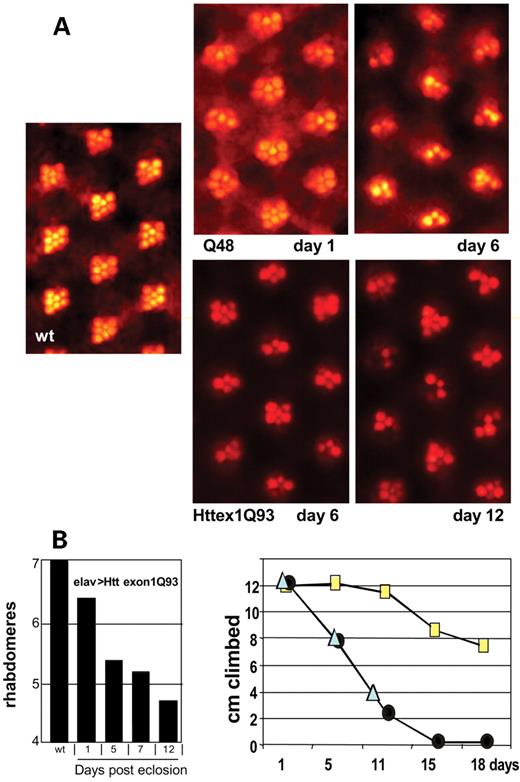
Figure 3. Degeneration is progressive. (A) The rhabdomeres at different ages are shown for flies expressing a pure polyQ peptide (Q48) and expressing a mutant exon1 fragment of a human Htt gene with 93Qs (Httex1Q93). Note that the rhabdomere constellations get progressively worse. Note also that the severity of the effect is greater with the pure polyQ than with the pathogenic human Htt protein fragment. (B) The progressive loss of rhabdomeres in flies expressing Htt ex1Q93 over 12 days is shown compared to wild-type eyes that exhibit seven throughout their life. Motor function is also impaired and is progressively lost as shown by the climbing assay. Flies exhibit negative geotropism. The distance climbed in 20 seconds was measured for flies expressing Q48 (circles) and Htt ex1Q93 (triangles) under the control of elav-Gal4 and compared to the nonexpressing sibs Q48/CyO (squares). Note that the climbing ability progressively declines for both genotypes.
References
Rubin, G.M., Yandell, M.D., Wortman, J.R., Gabor Miklos, G.L., Nelson, C.R., Hariharan, I.K., Fortini, M.E., Li, P.W., Apweiler, R., Fleischmann, W. et al. (
Reiter, L.T., Potocki, L., Chien, S., Gribskov, M. and Bier, E. (
Wong, A.M., Wang, J.W. and Axel, R. (
Marin, E.C., Jefferis, G.S., Komiyama, T., Zhu, H. and Luo, L. (
Rein, K., Zockler, M., Mader, M.T., Grubel, C. and Heisenberg, M. (
Zoghbi, H.Y. and Orr, H.T. (
Ross, C.A. (
Bates, G., Harper, P. and Jones, L. (
Group, T.H.s.D.C.R. (
Duyao, M.P., Auerbach, A.B., Ryan, A., Persichetti, F., Barnes, G.T., McNeil, S.M., Ge, P., Vonsattel, J.P., Gusella, J.F., Joyner, A.L. et al. (
Li, S.H., Schilling, G., Young, W.S., III, Li, X.J., Margolis, R.L., Stine, O.C., Wagster, M.V., Abbott, M.H., Franz, M.L., Ranen, N.G. et al. (
Strong, T.V., Tagle, D.A., Valdes, J.M., Elmer, L.W., Boehm, K., Swaroop, M., Kaatz, K.W., Collins, F.S. and Albin, R.L. (
Wexler, N.S., Young, A.B., Tanzi, R.E., Travers, H., Starosta-Rubinstein, S., Penney, J.B., Snodgrass, S.R., Shoulson, I., Gomez, F., Ramos Arroyo, M.A. et al. (
Squitieri, F., Gellera, C., Cannella, M., Mariotti, C., Cislaghi, G., Rubinsztein, D.C., Almqvist, E.W., Turner, D., Bachoud-Levi, A.C., Simpson, S.A. et al. (
Penney, J.B., Jr, Vonsattel, J.P., MacDonald, M.E., Gusella, J.F. and Myers, R.H. (
Tomlinson, A. and Rady, D.F. (
Cagan, R.L. and Ready, D.F. (
Truman, J.W., Taylor, B.J. and Awad, T.A. (
Brand, A.H. and Perrimon, N. (
Brand, A.H. and Dormand, E.L. (
Bonini, N.M. and Fortini, M.E. (
Duffy, J.B. (
Ma, C., Zhou, Y., Beachy, P.A. and Moses, K. (
Warrick, J.M., Paulson, H.L., Gray-Board, G.L., Bui, Q.T., Fischbeck, K.H., Pittman, R.N. and Bonini, N.M. (
Robinow, S. and White, K. (
Ellis, M.C., O'Neill, E.M. and Rubin, G.M. (
Nagai, Y., Fujikake, N., Ohno, K., Higashiyama, H., Popiel, H.A., Rahadian, J., Yamaguchi, M., Strittmatter, W.J., Burke, J.R. and Toda, T. (
Marsh, J.L., Walker, H., Theisen, H., Zhu, Y.Z., Fielder, T., Purcell, J. and Thompson, L.M. (
Kazemi-Esfarjani, P. and Benzer, S. (
Fernandez-Funez, P., Nino-Rosales, M.L., de Gouyon, B., She, W.C., Luchak, J.M., Martinez, P., Turiegano, E., Benito, J., Capovilla, M., Skinner, P.J. et al. (
Brachmann, C.B. and Cagan, R.L. (
Le Bourg, E. and Lints, F.A. (
Ganetzky, B. and Flanagan, J.R. (
Mangiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A., Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies, S.W. et al. (
Takeyama, K., Ito, S., Yamamoto, A., Tanimoto, H., Furutani, T., Kanuka, H., Miura, M., Tabata, T. and Kato, S. (
Steffan, J.S., Bodai, L., Pallos, J., Poelman, M., McCampbell, A., Apostol, B.L., Kazantsev, A., Schmidt, E., Zhu, Y.Z., Greenwald, M. et al. (
Jackson, G.R., Salecker, I., Dong, X., Yao, X., Arnheim, N., Faber, P.W., MacDonald, M.E. and Zipursky, S.L. (
Driscoll, M. and Gerstbrein, B. (
Lindquist, S., Krobitsch, S., Li, L. and Sondheimer, N. (
Meriin, A.B., Zhang, X., He, X., Newnam, G.P., Chernoff, Y.O. and Sherman, M.Y. (
DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P. and Aronin, N. (
Davies, S.W., Turmaine, M., Cozens, B.A., Difiglia, M., Sharp, A.H., Ross, C.A., Scherzinger, E., Wanker, E.E., Mangiarini, L. and Bates, G.P. (
Cha, J.H. (
Kazantsev, A., Preisinger, E., Dranovsky, A., Goldgaber, D. and Housman, D. (
Steffan, J.S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y.Z., Gohler, H., Wanker, E.E., Bates, G.P., Housman, D.E. and Thompson, L.M. (
Nucifora, F.C., Jr, Sasaki, M., Peters, M.F., Huang, H., Cooper, J.K., Yamada, M., Takahashi, H., Tsuji, S., Troncoso, J., Dawson, V.L. et al. (
Hughes, R.E., Lo, R.S., Davis, C., Strand, A.D., Neal, C.L., Olson, J.M. and Fields, S. (
McCampbell, A., Taye, A.A., Whitty, L., Penney, E., Steffan, J.S. and Fischbeck, K.H. (
Kazemi-Esfarjani, P. and Benzer, S. (
Taylor, J.P., Taye, A.A., Campbell, C., Kazemi-Esfarjani, P., Fischbeck, K.H. and Min, K.T. (
Hockly, E., Richon, V.M., Woodman, B., Smith, D.L., Zhou, X., Rosa, E., Sathasivam, K., Ghazi-Noori, S., Mahal, A., Lowden, P.A. et al. (
Wyttenbach, A., Carmichael, J., Swartz, J., Furlong, R.A., Narain, Y., Rankin, J. and Rubinsztein, D.C. (
Wyttenbach, A., Sauvageot, O., Carmichael, J., Diaz-Latoud, C., Arrigo, A.P. and Rubinsztein, D.C. (
Chai, Y., Koppenhafer, S.L., Shoesmith, S.J., Perez, M.K. and Paulson, H. (
Chai, Y., Koppenhafer, S.L., Bonini, N.M. and Paulson, H.L. (
Warrick, J.M., Chan, H.Y.E., Gray-Board, G.L., Chai, Y., Paulson, H.L. and Bonini, N.M. (
Auluck, P.K., Chan, H.Y., Trojanowski, J.Q., Lee, V.M. and Bonini, N.M. (
Cummings, C.J., Sun, Y., Opal, P., Antalffy, B., Mestril, R., Orr, H.T., Dillmann, W.H. and Zoghbi, H.Y. (
Sittler, A., Lurz, R., Lueder, G., Priller, J., Lehrach, H., Hayer-Hartl, M.K., Hartl, F.U. and Wanker, E.E. (
Heemskerk, J., Tobin, A.J. and Bain, L.J. (
Heiser, V., Scherzinger, E., Boeddrich, A., Nordhoff, E., Lurz, R., Schugardt, N., Lehrach, H. and Wanker, E.E. (
Apostol, B.L., Kazantsev, A., Raffioni, S., Illes, K., Pallos, J., Bodai, L., Slepko, N., Bear, J.E., Gertler, F.B., Hersch, S. et al. (
Kazantsev, A., Walker, H., Slepko, N., Bear, J.E., Preisinger, E., Steffan, J.S., Zhu, Y.-Z., Gertler, F.B., Housman, D.E., Marsh, J.L. et al. (
Chan, H.Y., Warrick, J.M., Gray-Board, G.L., Paulson, H.L. and Bonini, N.M. (
Chen, H.K., Fernandez-Funez, P., Acevedo, S.F., Lam, Y.C., Kaytor, M.D., Fernandez, M.H., Aitken, A., Skoulakis, E.M., Orr, H.T., Botas, J. et al. (
Higashiyama, H., Hirose, F., Yamaguchi, M., Inoue, Y.H., Fujikake, N., Matsukage, A. and Kakizuka, A. (
Kayed, R., Head, E., Thompson, J.L., McIntire, T.M., Milton, S.C., Cotman, C.W. and Glabe, C.G. (
Dedeoglu, A., Kubilus, J.K., Jeitner, T.M., Matson, S.A., Bogdanov, M., Kowall, N.W., Matson, W.R., Cooper, A.J.L., Ratan, R.R., Beal, M.F. et al. (
Karpuj, M.V., Becher, M.W., Springer, J.E., Chabas, D., Youssef, S., Pedotti, R., Mitchell, D. and Steinman, L. (
Sanchez, I., Mahlke, C. and Yuan, J. (
Feany, M.B. and Bender, W.W. (
Ferber, D. (
Jackson, G.R., Wiedau-Pazos, M., Sang, T.K., Wagle, N., Brown, C.A., Massachi, S. and Geschwind, D.H. (
Wittmann, C.W., Wszolek, M.F., Shulman, J.M., Salvaterra, P.M., Lewis, J., Hutton, M. and Feany, M.B. (

{kind=link}
{kind=link}
{kind=link}