





Abstract
Regulatory T (Tr) cells have been shown to arise in the periphery during induction of peripheral tolerance. However, the mechanism involved remains elusive. In the present study, we investigated the potential role of transforming growth factor (TGF)-β in the peripheral induction of regulatory phenotypes in the conventional CD4+CD25− T cells. Upon priming in the presence of TGF-β, there was greatly enhanced expression of CD25, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), and the natural Tr cell-specific transcription factor Foxp3 in naive CD4+CD25− T cells. The CD25+ cells that emerged later only in the TGF-β-treated culture failed to express CD69, so distinguishing this population from activated CD25+ effector cells. The TGF-β-treated T cells entered an anergic state following restimulation, as judged by enhanced induction of programmed death (PD)-1, as well as impaired responses in terms of proliferation and IL-2 production. Importantly, the TGF-β-costimulated CD4+CD25− T cells, prior to conversion to CD25+ cells, were able to suppress the proliferation of responder T cells via contact-dependent and interleukin-10-independent mechanisms. Taken together, these results demonstrate that the existence of TGF-β during early phase of priming is sufficient to induce CD4+CD25− Tr cells with anergic and immunoregulatory activities equivalent to thymus-derived CD4+CD25+ Tr cells, and these cells are programmed to be CD25+ cells under the prolonged resting conditions. Thus, our findings provide a novel mechanism by which TGF-β mediates infectious tolerance in the periphery.
Introduction
Immunological tolerance to self is essential to avoid autoimmunity and to maintain immune homeostasis. Several mechanisms of immune tolerance have been proposed. These include thymic deletion of autoreactive T cells, induction of peripheral anergy and active suppression of autoreactivity by regulatory T cells. CD4+CD25+ regulatory T (Tr) cells are a unique population of professional suppressor cells that constitute 5–10% of peripheral CD4+ T cells in both mice and humans (1–3). They are naturally anergic to stimulation via the TCR and inhibit the proliferation and function of CD4+CD25− and CD8+ T cells by a contact-dependent mechanism. Depletion of CD4+CD25+ Tr cells by neonatal thymectomy induces autoimmune disease of many organs that can be prevented by transfer of CD4+CD25+ T cells from normal naive mice, pointing to a crucial role of CD4+CD25+ Tr cells in maintaining peripheral tolerance (4–6). Unlike other Tr cell-associated molecules such as CD25, cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and glucocorticoid-induced tumor necrosis factor receptor (GITR), which are also expressed on activated T cells, the transcription factor forkhead box p3 (Foxp3) is Tr cell-specific (7–9). Foxp3-deficient mice lack Tr cells and develop massive inflammatory autoimmune disease. Conversely, conventional CD4+CD25− or CD8+ T cells ectopically expressing Foxp3 acquire a regulatory phenotype analogous to professional CD4+CD25+ Tr cells (7–9). Therefore, Foxp3 appears to be a master gene governing the development and function of CD4+CD25+ Tr cells.
Although the immunosuppressive ability of Tr cells is generally accepted, the underlying mechanisms remain unclear. In addition it is not known for certain where and how these cells develop. While CD4+CD25+ Tr cells are known to arise naturally from the thymus, there is also accumulating evidence that Tr phenotypes can be induced in the periphery in tolerizing conditions such as the induction of oral tolerance (10–12). The factors that promote this peripheral differentiation of Tr cells remain unidentified. Another debate focuses on the role of transforming growth factor (TGF)-β in the development and function of Tr cells. TGF-β, which is produced as a consequence of activation of regulatory cell populations such as CD25+ Tr and Th3 cells, is dispensable for the suppressive activity of natural Tr cells in vitro (13). On the other hand, TGF-β deficiency or blockade in vivo abolishes the suppressive activity of Tr cells (14,15), indicating that the development of some Tr cell capabilities is TGF-β dependent. These conflicting observations suggest the possibility that TGF-β is involved in the development and/or amplification of Tr activity via a mechanism other than direct suppression of responder T cells.
TGF-β is one of the key molecules contributing to peripheral tolerance. The finding that abrogation of TGF-β signaling in T cells alone results in spontaneous T cell differentiation and autoimmune disease identifies an essential role of TGF-β signaling in T cell homeostasis and the prevention of inflammatory autoimmunity (16,17). Several in vitro and in vivo investigations have demonstrated a link between TGF-β and regulatory cell populations. For example, TGF-β was able to amplify the functions of CD4+CD25+ Tr cells ex vivo by causing their expansion (18). In contrast, Th3 cells that are known to induce active suppression by producing TGF-β, could emerge upon stimulation of naive CD4+CD25− cells with exogenous TGF-β in vitro, and upon oral antigen administration which can induce large amounts of TGF-β (19,20). Nevertheless, the question of whether TGF-β can promote the formation of peripheral Tr cells in addition to Th3 cells needs to be clarified.
In the present study we demonstrated a novel mechanism by which TGF-β contributes to the amplification of Tr cell activity. We found that when naive CD4+CD25− T cells were costimulated with TGF-β they upregulated Tr cell-associated markers, such as CD25, CTLA-4 and Foxp3, and an anergy-associated molecule, PD-1. Surprisingly, the CD4+CD25− T cells, prior to conversion to CD25+ cells, acquired anergic and suppressive phenotypes similar to those of thymus-derived professional Tr cells, so revealing a mechanism by which TGF-β spreads a tolerant state from regulatory T cells to conventional T cells.
Methods
Reagents and antibodies
The mouse CD4+CD25+ regulatory T cell isolation kit was purchased from Miltenyi Biotec (Auburn, CA). Recombinant human TGF-β1 and murine IL-2 were from R&D systems (Minneapolis, MN) and PeproTech (London, UK), respectively. The following antibodies and reagents were purchased from BD Biosciences PharMingen (San Diego, CA); purified mAb for CD3 (145-2C11) and CD28 (37.51), anti-CD4–FITC (GK1.5), anti-IL-2–PE (JES6-5H4), anti-CD4–PerCP (RM4-5), anti-IL-10 (JES5-16E3), annexin V–FITC and Streptavidin–PE. Antibodies purchased from eBioscience (San Diego, CA) include biotinylated anti-PD-1 (J43), anti-CD69–FITC (H1.2F3), anti-CD152–PE (UC10-4B9), anti-PD-1 (J43) and hamster IgG.
Isolation of CD4+CD25− T cells and cell culture
Lymph node (LN) cells from C57BL/6 mice were gently minced in complete RPMI-1640 media supplemented with 10% FBS, and single cell suspensions were prepared by passage through a 100 μm nylon cell strainer (Becton Dickinson, Franklin Lakes, NJ). CD4+ T cells were enriched by depletion of cells expressing CD8α, CD11b, CD45R, CD49b and Ter-119 using the CD4+CD25+ regulatory T cell isolation kit. CD4+CD25− T cells were obtained by further depletion of cells expressing CD25. The purified CD4+CD25− T cell fraction contained <1.0% CD25+ cells.
Whole LN cells and derived fractions were stimulated with 5 μg/ml plate-bound anti-CD3 and 0.5 μg/ml soluble anti-CD28 antibodies in the presence or absence of 5 ng/ml TGF-β. After 48 h of stimulation, the cells were removed and rested in fresh media supplemented with 10 U/ml IL-2 for 48 or 96 h. In some experiments, they were extensively washed and restimulated with 1 μg/ml plate-bound anti-CD3 and 0.2 μg/ml soluble anti-CD28 antibodies.
Flow cytometry
To observe the expression of CD25, CD69 and PD-1 on the surface of CD4+ cells, cells were stained with appropriate combinations of mAb diluted in PBS containing 0.5% BSA and 0.1% sodium azide for 20 min at 4°C. For analysis of cell surface CD152 expression, cells were stained with anti-CD152–PE antibody for 2 h at 37°C (21). After washing, the labeled cells were fixed with 2% paraformaldehyde and analyzed using a FACSCaliber with CellQuest software (Becton Dickinson, San Jose, CA). Apoptotic cells were detected by double staining with anti-CD4–PerCP and annexin V–FITC.
To observe IL-2-producing cells by intracellular FACS assays, LN cells from C57BL/6 mice were stimulated and rested as described above. They were re-stimulated with 5 μg/ml anti-CD3 and 1 μg/ml anti-CD28 antibodies for a total of 6 h. For the last 4 h of re-stimulation, 10 μg/ml Brefeldin A (Sigma, St Louis, MO) was added to promote intracellular accumulation of IL-2, and the cells were then stained with anti-CD4–FITC and fixed with 2% paraformaldehyde. After washing, they were permeabilized in 0.5% saponin solution (Sigma) and stained with anti-IL-2–PE for 30 min. After washing, IL-2-expressing CD4+ cells were analyzed by flow cytometry.
RT–PCR
Total cellular RNA was extracted from the sorted cell populations with TRIzol reagent (Invitrogen, Carlsbad, CA). It was reverse transcribed with AMV reverse transcriptase XL (TaKaRa, Shiga, Japan) and random primers. PCR was performed using Taq DNA polymerase (TaKaRa) with Foxp3-specific primers. The relative quantity of Foxp3 cDNA was normalized by the amount of β2-microglobulin (β2-M) cDNA. For non-saturating amplification, reactions were conducted for 28 and 35 cycles for β2-M and Foxp3, respectively. The primer sequences were as follows; Foxp3: 5′-CTTCATGCATCAGCTCTCCAC-3′ and 5′-AGACTCCATTTGCCAGCAGTG-3′; β2-M: 5′-TGACCGGCTTGTATGCTATC-3′ and 5′-CAGTGTGAGCCAGGATATAG-3′.
Proliferation assay
LN cells from C57BL/6 mice were stimulated and rested as described above. Following this, 1.5 × 105 cells were re-stimulated with 1 μg/ml immobilized anti-CD3 and 0.2 μg/ml soluble anti-CD28 antibodies in 96-well plates for 45 h. After pulsing with [3H]thymidine (1 μCi/well; Amersham Biosciences, Buckinghamshire, UK) for the last 8 h of culture, the cells were harvested with a Cell Harvester (Skatron Instruments, Lier, Norway), and counted with a liquid scintillation counter (Wallac, Finland). All assays were conducted in triplicate.
Suppression assay
To determine the suppressive activity of TGF-β-converted Tr cells on responder cell proliferation, carboxy-fluorescein diacetate succinimidyl ester (CFSE)-labeled responder cells were co-cultured with TGF-β-exposed or non-exposed cells. In brief, freshly isolated LN cells from C57BL/6 mice were washed and resuspended at a concentration of 1.0 × 107 cells/ml in PBS. CFSE was added at a final concentration of 5 μM and incubated for 10 min at room temperature. The reaction was stopped by adding 500 μl of cold FBS and the cells were washed three times with PBS. CD4+CD25− cells were primarily stimulated for 48 h in the presence or absence of TGF-β, followed by resting phase for another 48 h. After extensive washing, they (1 × 105) were mixed with 1 × 106 CFSE-labeled responder cells at 500 μl/well in 48-well plates and stimulated with 1 μg/ml immobilized anti-CD3 and 0.2 μg/ml soluble anti-CD28 antibodies in the presence or absence of 10 μg/ml anti-PD-1 or control antibody. After 72 h of co-culture, the fluorescence intensity of live CFSE+ cells was determined by flow cytometry.
To determine the effect of IL-10 on the suppressive activity, 10 μg/ml neutralizing antibody to mouse IL-10 was added to 1 × 105 TGF-β-costimulated CD4+CD25− T cells cocultured with 1 × 106 CFSE-labeled responder cells in the presence of 2×105 mitomycin C-treated splenocytes from C57BL/6 mice and 0.1 μg/ml soluble anti-CD3 antibody.
In transwell experiments, 5.0 × 105 CFSE-labeled LN cells were stimulated with 0.5 μg/ml soluble anti-CD3 antibody and 2.0 × 106 mitomycin-C-treated splenocytes from C57BL/6 mice in the lower chamber of a transwell plate (0.4 μm pore size; Millipore, Bedford, MA) in the presence or absence of 5.0 × 105 TGF-β-costimulated CD25− cells in the same or in the upper chamber. The cells in the upper chamber were also stimulated with 0.5 μg/ml soluble anti-CD3 antibody and 2.0 × 106 mitomycin-C-treated splenocytes from C57BL/6 mice. After 72 h of culture, the cells in the lower chamber were harvested and the fluorescence intensity of live CFSE+ cells was analyzed by flow cytometry.
ELISA
Purified CD4+CD25− and CD4+CD25+ cells were stimulated with 5 μg/ml anti-CD3 and 0.5 μg/ml anti-CD28 antibodies for 48 h and supernatants were collected. CD4+CD25− cells stimulated in the presence or absence of TGF-β were rested for 48 h and restimulated with 5 μg/ml anti-CD3 and 0.5 μg/ml anti-CD28 antibodies. Supernatants were collected after 48 h. The concentrations of IL-10 in the supernatants were determined using R&D Duoset ELISA development system (R&D systems) according to the manufacturer's instructions.
Results
Late emergence of CD4+CD25+ T cells by TGF-β costimulation
The CD25 molecule serves as a marker of Tr cells. About 4% of the CD4+ T cells in naive mice expressed CD25, indicating the existence of Tr cells (Fig. 1A). To determine whether the presence of TGF-β during priming promotes the development of CD4+CD25+ Tr cells in vitro, naive T cells from LN were polyclonally stimulated with anti-CD3 and anti-CD28 antibodies for 48 h in the presence or absence of TGF-β and rested in fresh medium containing exogenous IL-2. As shown in Fig. 1(A), irrespective of the presence of TGF-β, cell surface expression of CD25 increased within 48 h of stimulation and returned to normal after a further 48 h in the resting phase. Whereas the majority of CD4+ T cells not exposed to TGF-β failed to express CD25 during the prolonged resting phase, ∼40% of the TGF-β costimulated CD4+ T cells expressed CD25 after the prolonged period of resting (Fig. 1A).
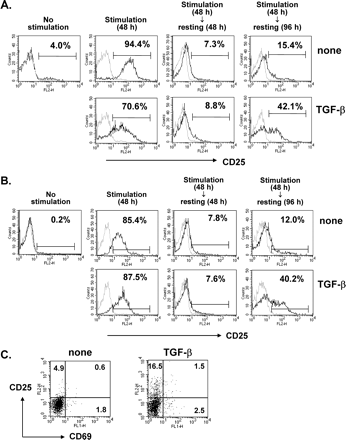
Late emergence of CD4+CD25+CD69− T cells upon TGF-β costimulation. Whole (A and C) or purified CD4+CD25− (B) LN cells from C57BL/6 mice were stimulated with anti-CD3 and anti-CD28 antibodies in the presence or absence of 5 ng/ml TGF-β for 48 h and rested in fresh medium supplemented with 10 U/ml IL-2 and 1 ng/ml TGF-β for 48 h or 96 h. (A and B) At the indicated times, they were harvested and stained with anti-CD4–FITC and anti-CD25–PE. CD4+ T cells were gated and histogram profiles of CD25 expression on the CD4+ T cells are displayed as black lines (gray lines, cells stained with isotype-matched control antibody). (C) Cells rested for 96 h were harvested and stained with anti-CD69–FITC, anti-CD25–PE, and anti-CD4–PerCP. CD4+ T cells were gated and the profiles of dual CD25 and CD69 expression on CD4+ T cells were determined by flow cytometry. A representative result of three independent experiments is shown.
To determine whether the late emerging CD4+CD25+ T cells were derived from CD4+CD25− cells or expanded from CD4+CD25+ cells, purified CD4+CD25− cells were assayed under the same conditions. The TGF-β-dependent biphasic profile of CD25 expression observed in the LN culture was very similar in the CD25+ cell-depleted culture, suggesting that the late emerging CD4+CD25+ cells were derived from CD4+CD25− cells (Fig. 1B). These cells did not express CD69, an activation marker, indicating that they differed from early-activated CD4+CD25+ effector cells (Fig. 1C).
TGF-β-costimulation induces Foxp3 expression in the CD4+CD25− T cells
CD4+CD25+ Tr cells have been shown to specifically express Foxp3, a transcription factor whose synthesis is sufficient to convert CD4+CD25− cells to CD4+CD25+ Tr cells (7–9). Therefore, we determined whether TGF-β costimulation induces Foxp3 in subsets of non-Tr cells, and found that the presence of TGF-β during polyclonal stimulation of LN T cells greatly enhanced the expression of Foxp3 transcripts (Fig. 2A). TGF-β inducibility of Foxp3 was most prominent in a subset of CD4+CD25− cells and, to a lesser extent, even in CD4− LN cells. The induced level of Foxp3 transcripts resulting from TGF-β costimulation of CD4+CD25− T cells was comparable to that in CD4+CD25+ T cells. The presence of TGF-β did not further increase the level of Foxp3 in the CD4+CD25+ T cells. Furthermore, the induced level of Foxp3 transcripts reached a peak after 48 h of costimulation and declined during the resting period (Fig. 2B), implying a requirement for both TGF-β and TCR stimulation for induction of Foxp3. Interestingly, restimulation of the rested cells that had been exposed to TGF-β with anti-CD3 plus anti-CD28 antibody in the absence of TGF-β restored Foxp3 induction to a level similar to that produced after the primary stimulation with TGF-β. This indicates that the CD4+CD25− T cells acquired a phenotype corresponding to Tr cells during the priming with TGF-β, and the presence of TGF-β only during the early phase of priming was sufficient to develop Foxp3-expressing cells.
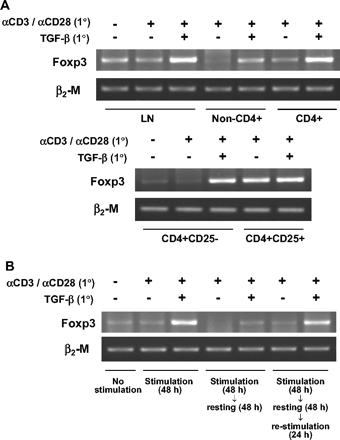
TGF-β-mediated Foxp3 induction in CD4+CD25− T cells. (A) LN cells from C57BL/6 mice were sorted into CD4+ and non-CD4+ T cells and the CD4+ T cells were further separated into CD25− and CD25+ cells. Whole LN cells and the purified cell populations were stimulated in the presence or absence of 5 ng/ml TGF-β for 48 h and assayed by RT and non-saturating PCR using Foxp3- or β2M-specific primers. (B) LN cells were stimulated, rested, and restimulated as described in Methods. At each of the indicated times, they were subjected to RT and non-saturating PCR. A representative of two independent experiments is shown.
Different kinetics of surface CTLA-4 and PD-1 expression in TGF-β-costimulated CD4+ T cells
CTLA-4 is known to be constitutively expressed on the surface of CD4+CD25+ Tr cells while transiently expressed on activated T cells. Less than 3% of CD4+ cells from naive mice expressed CTLA-4 on their cell surface. Surface CTLA-4 was upregulated upon polyclonal activation yet quickly downregulated thereafter. However, the kinetics of CTLA-4 expression was strikingly different when CD4+ T cells were costimulated with TGF-β. These cells contained significantly higher percentage of CTLA-4+ cells after resting for 96 h, compared with TGF-β-unexperienced cells (Fig. 3A).
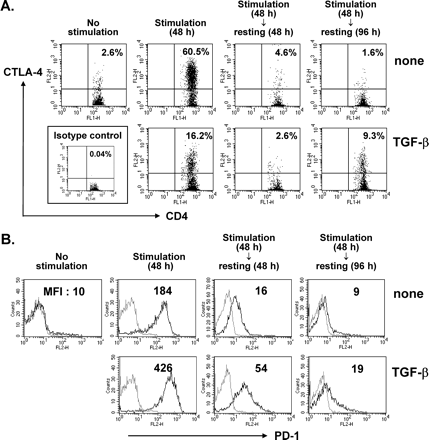
Upregulation of CTLA-4 and PD-1 on CD4+ T cells by TGF-β at different phases. LN cells were stimulated in the presence or absence of 5 ng/ml TGF-β for 48 h and rested for 48 h or 96 h. At each time point indicated, the cells were harvested and stained with anti-CD4–FITC and anti-CTLA-4–PE (A) or biotinylated anti-PD-1 plus Streptavidin–PE (B). CD4+ T cells were gated. The number represents the percentage of CD4+CTLA-4+ cells (A). Histogram profiles of PD-1 expression on the CD4+ T cells are displayed as black lines (gray lines, stained with isotype-matched control antibody; MFI, mean fluorescence intensity). A representative result of two (A) and three (B) independent experiments is shown.
In addition to CTLA-4, PD-1 is another costimulatory molecule which transduces negative and anergic signals to T cells (22,23). To test the possibility that the PD-1 pathway is associated with the mechanism of TGF-β-mediated generation of Tr cells, we analyzed the level of cell surface expression of PD-1 by FACS. As expected, cell surface expression increased transiently upon stimulation through the TCR and CD28 costimulatory pathways. Induction of PD-1 was increased by addition of TGF-β during the primary stimulation (Fig. 3B). Irrespective of the presence of TGF-β, the level of PD-1 returned to the basal level after the resting phase for 96 h, the time when late emerging CTLA-4 and CD25 were observed. Thus, along with the finding that the majority of purified natural CD4+CD25+ Tr cells did not express PD-1 constitutively on their cell surface (data not shown), this result suggests that TGF-β-mediated PD-1 induction during priming may not be directly involved in the acquisition of the phenotype of the CD4+CD25+ T cells.
Acquisition of anergic phenotypes by TGF-β-costimulated T cells
Thymus-derived CD4+CD25+ Tr cells have been shown to be naturally anergic to stimulation through their TCR (1–3). We therefore determined whether TGF-β costimulation could induce an anergic state by stimulating LN T cells with anti-CD3/anti-CD28 antibody in the presence or absence of TGF-β for 48 h, followed by a resting period of another 48 h. Upon polyclonal restimulation, the T cells that had been exposed to TGF-β exhibited a significant reduction in their proliferation, compared to cells not exposed to TGF-β (Fig. 4A). This hypo-responsiveness was fully reversed by adding exogenous IL-2 to the cells during restimulation, thus confirming that the TGF-β-exposed T cells were anergic. IL-2 production upon restimulation of the TGF-β-exposed cells was defective, consistent with their poor proliferative response: the number of IL-2-producing CD4+ T cells among the TGF-β-exposed cells was reduced by 90% relative to the non-exposed cells (Fig. 4B). TGF-β treatment did not reduce viability, as measured by annexin V staining, thus excluding the possibility that TGF-β-mediated hypo-responsiveness results from cell death rather than induction of anergy (Fig. 4C).
![TGF-β-mediated induction of T cell anergy. LN cells were stimulated in the presence or absence of 5 ng/ml TGF-β for 48 h and rested for 48 h. (A) After extensive washing, 1.5 × 105 cells were re-stimulated with 1 μg/ml anti-CD3 and 0.2 μg/ml anti-CD28 in the presence or absence of 200 U/ml IL-2 and in the absence of TGF-β for 45 h, followed by [3H]thymidine incorporation assays (*P < 0.01, Student's t-test). (B) 5.0 × 105 cells were re-stimulated with 5 μg/ml anti-CD3 and 1 μg/ml anti-CD28 antibodies for 6 h and stained with anti-CD4–FITC and anti-IL-2–PE, followed by FACS analyses. (C) To detect apoptotic cells after 48 h resting in fresh medium, cells were harvested and stained with anti-CD4–PerCP and annexin V–FITC. CD4+ T cells were gated and the percentage of annexin V-stained CD4+ T cells was determined by flow cytometry. A representative result of three independent experiments is shown.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/intimm/16/8/10.1093/intimm/dxh123/2/m_intimmdxh123f04_ht.gif?Expires=1717058485&Signature=0EuyWro7r9jtlpmxhYd2U3NAldZ0O39mRoh5JkMD9HvTL7M4uFVc3NglSV3BDaJ6h57ghKGHDt2WwUzrK6-yrVqprC4uqqTXGl7-sh6kF6v9dEIdWIIuisTwz1Wkw2Z4YtJtpb76J7Ppn4SqVdYT5g~DYzn0BEn~7bHzvVweP0xvEUBZU5zeT9o7BhjQU-VCNdMIGLM8s2DS1zEIq5W6Wm3RFM25i3FAq4XAdQLicsVjLTMf1kHrmdauAH0pUuOOhuvOrEN-W~-e~5SnqLQK--N5TaaGmfKormoiYMS4HuTi62L0iuWh79uteG8aJEFm8Jj-dUEpXdNfy~-E3wyTEg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TGF-β-mediated induction of T cell anergy. LN cells were stimulated in the presence or absence of 5 ng/ml TGF-β for 48 h and rested for 48 h. (A) After extensive washing, 1.5 × 105 cells were re-stimulated with 1 μg/ml anti-CD3 and 0.2 μg/ml anti-CD28 in the presence or absence of 200 U/ml IL-2 and in the absence of TGF-β for 45 h, followed by [3H]thymidine incorporation assays (*P < 0.01, Student's t-test). (B) 5.0 × 105 cells were re-stimulated with 5 μg/ml anti-CD3 and 1 μg/ml anti-CD28 antibodies for 6 h and stained with anti-CD4–FITC and anti-IL-2–PE, followed by FACS analyses. (C) To detect apoptotic cells after 48 h resting in fresh medium, cells were harvested and stained with anti-CD4–PerCP and annexin V–FITC. CD4+ T cells were gated and the percentage of annexin V-stained CD4+ T cells was determined by flow cytometry. A representative result of three independent experiments is shown.
Suppression of responder cell proliferation by TGF-β-experienced CD4+CD25− T cells
A functional hallmark of Tr cells is their ability to suppress the development and expansion of effector T cells. We therefore determined whether TGF-β-experienced CD4+CD25− T cells were able to suppress the proliferation of responder T cells. Naive CD4+ T cells or CD4+CD25− T cells were primed with TGF-β, rested for 48 h, and cocultured with CFSE-labeled responder cells. They were exposed simultaneously to polyclonal stimulation with anti-CD3 and anti-CD28 antibodies, followed by FACS analysis to determine the profile of responder cell division. As shown in Fig. 5(A), co-culture with the TGF-β-exposed CD4+CD25− T cells resulted in a major increase in the number of non-dividing responder cells, whereas control CD25− cells not exposed to TGF-β did not alter the proliferative profile of the responder cells. The suppressive activity of the TGF-β-experienced whole CD4+ cells was quite similar to that of the TGF-β-exposed CD25− cells, indicating that in our system the suppression of responder cell proliferation was largely attributed to CD4+CD25− T cells rather than natural CD4+CD25+ Tr cells.
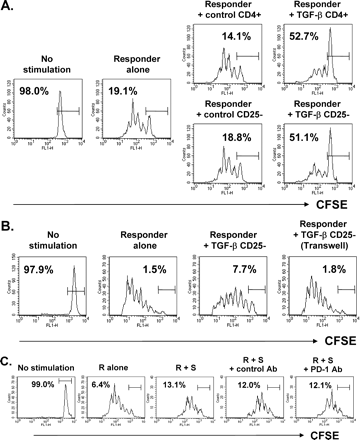
Suppression of responder cell proliferation by TGF-β-converted Tr cells. Purified CD4+ or CD4+CD25− T cells from C57BL/6 mice were stimulated in the presence (TGF-β CD4+ or TGF-β CD25−) or absence (control CD4+ or control CD25−) of 5 ng/ml TGF-β for 48 h and rested for 48 h. (A) After extensive washing, 1.0 × 105 cells were mixed with 1.0 × 106 CFSE-labeled LN cells from C57BL/6 mice and stimulated with immobilized anti-CD3 and soluble anti-CD28 antibodies. (B) CFSE-labeled LN cells were stimulated with soluble anti-CD3 antibody and mitomycin-C-treated splenocytes in the lower chamber of a transwell plate in the presence or absence of 5.0 × 105 TGF-β CD25− cells in the same or in the upper chamber. TGF-β CD25− cells placed in the upper chamber were also stimulated as the same as the lower chamber. (C) TGF-β-experienced CD4+CD25− T cells and CFSE-labeled responder cells were costimulated in the presence or absence of 10 μg/ml anti-PD-1 antibody or control IgG. The fluorescence intensity of live CFSE+ cells after 72 h of culture is displayed as a histogram. The percentages in the figure represent undivided CFSEhigh cells. These figures show a representative result of three independent experiments.
To assess whether TGF-β-exposed CD4+CD25− T cells exert their suppressive activity through a cell contact-dependent mechanism, responder and TGF-β-exposed CD25− cells were separated in transwell chambers and stimulated with soluble anti-CD3 antibody and APC. Separation of the two populations by the transwell partition abolished the suppressive action of the TGF-β-exposed CD25− T cells (Fig. 5B), demonstrating that they exert their suppressive action by cell contact. Neutralizing antibody to PD-1, which was previously used to block the PD-1 pathway (24), did not affect the suppressive activity of TGF-β-costimulated CD4+ T cells, implying that the PD-1 pathway may not be involved in the suppressive activity of Tr cells (Fig. 5C).
TGF-β-experienced CD4+CD25− T cells produced IL-10 yet exerted their suppressive activity via an IL-10-independent mechanism.
It remains controversial whether CD4+CD25+ Tr cells are able to produce IL-10 (7,25–27). To address this issue, purified CD4+CD25+ and CD4+CD25− T cells were activated and the culture supernatants were assayed to measure the amount of IL-10. CD25+ cells produced significantly more IL-10 than CD25− cells did, demonstrating IL-10-producing activity of naturally occurring CD25+ Tr cells in our system (Fig. 6A). Similarly, TGF-β-experienced CD4+CD25− cells produced more IL-10 than unexperienced cells upon restimulation. Although TGF-β-experienced CD4+CD25− cells could induce IL-10 production, their suppressive activity was totally independent of IL-10, since neutralizing antibody to IL-10 could not restore the proliferative response of T cells which were suppressed by TGF-β-converted Tr cells (Fig. 6B).
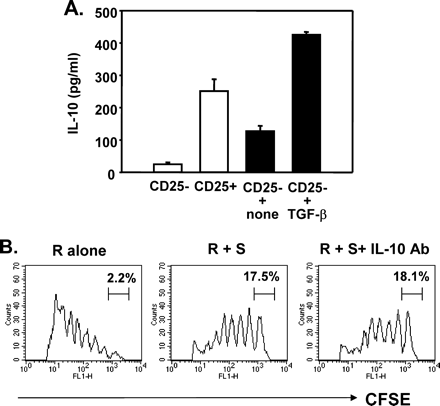
IL-10 production but IL-10-independent suppression by TGF-β-converted Tr cells. (A) Purified CD4+CD25− and CD4+CD25+ cells were polyclonally stimulated for 48 h (open bars). In separate experiments, purified CD4+CD25− cells were stimulated with or without TGF-β for 48 h and rested for 48 h, followed by restimulation for another 48 h (closed bars). The level of IL-10 in the supernatants was measured by quantitative ELISA. (B) CD4+CD25− T cells costimulated with TGF-β were rested for 48 h. These cells (S) were cocultured with CFSE-labeled responder cells (R) in the presence or absence of 10 μg/ml anti-IL-10 antibody. The fluorescence intensity of live CFSE+ cells after 72 h of culture is displayed as a histogram.
Discussion
TGF-β has been shown previously to regulate peripheral tolerance by a number of routes, including expansion of thymus-derived CD4+CD25+ Tr cells and promotion of the development of Th3 cells (18,19). In the present study, we demonstrated a novel action of TGF-β costimulation of naive CD4+CD25− T cells in the peripheral circulation as a result of which the T cells display potent immune regulatory activities analogous to those of thymus-derived CD4+CD25+ Tr cells. TGF-β-experienced CD4+ cells that retained a phenotype of CD25− exerted functionally anergic and suppressive activities. Importantly, these activities could take place in CD25+ Tr cell-depleted cultures, confirming that the Tr cells that emerged in response to TGF-β costimulation were derived from conventional CD4+CD25− cells, not from CD4+CD25+ thymic emigrants expanded by TGF-β, as reported by others (18). This thought is further supported by the observation that the suppressive activity of TGF-β-exposed CD4+ T cells which contained natural CD25+ Tr cells was similar to that of TGF-β-exposed CD4+CD25− cells in which natural CD25+ Tr cells were depleted. Our finding that the suppressive activity of the TGF-β-converted Tr cells was abolished by inhibition of contact with responder cells is critical for distinguishing TGF-β-converted Tr cells from Th3 and Tr1 cells whose activities are solely dependent on soluble TGF-β and IL-10, respectively, and hence are contact-independent. Although TGF-β-converted Tr cells could produce IL-10, they elicited their suppressive activity via an IL-10-independent mechanism. This observation exactly recapitulated previously established characteristics of natural Tr cells at least under in vitro culture conditions (5,25,26).
TGF-β costimulation potentiated the expression of CD25 and CTLA-4, Tr cell-associated markers, in a biphasic pattern, as observed by later emergence of CD25+CTLA-4+CD4+ cells after transiently expressing CD25, CTLA-4 and CD69 during early primary culture. This indicates that TGF-β is capable of programming CD4+CD25− T cells to express CD25 and these TGF-β-converted CD25+ T cells are distinct from activated effector T cells. Nevertheless, TGF-β-mediated acquisition of Tr phenotypes might not rely on the CD25 expression, since TGF-β-exposed CD4+CD25− T cells, prior to the conversion to CD25+ cells, effectively elicited their anergic and suppressive activities. This finding is inconsistent with the recent report by Chen et al. (28) demonstrating that, unlike TGF-β-converted CD25+ cells, TGF-β-treated cells that remained the CD25− phenotype lacked suppressive action to normal T cell proliferation. This discrepancy may largely be attributed to the difference of in vitro culture conditions. Our investigation is not the first one to report the existence of CD4+CD25− Tr cells in the periphery, as in many situations CD4+CD25− Tr cells are as effective as CD4+CD25+ Tr cells in regulating T cell homeostasis in vitro and in vivo. In a TCR-transgenic model of spontaneous diabetes, although CD4+CD25+ T cells played a suppressive role in the prevention of disease, so did CD4+CD25− T cells (29). In another model where mice express simultaneously a TCR transgene and its agonist ligand, both T cell subsets exhibited a similar potential to block naive T cell proliferation (30). Uraushihara et al. reported that CD4+CD25−GITR+ T cells as well as CD4+CD25+GITR+ T cells suppressed T cell proliferation and prevented murine inflammatory bowel disease (31). The CD4+CD25− Tr cells described in our study may not be identical subsets of Tr cells with those previously reported, since they induced Foxp3 expression and had a fate to be converted to CD25+ cells.
It has been proposed that Foxp3 expression is not only Tr cell-specific, but is also itself sufficient to convert conventional T cells into CD25+ Tr cells, since Foxp3 protein is specifically expressed by professional CD25+ Tr cells but not by other subsets of IL-10-producing regulatory T cells (32), and the induced expression of Foxp3 in non-Tr cells converts them into Tr cells (7–9). Therefore, the best evidence for the TGF-β-mediated transition of CD4+CD25− naive T cells to professional Tr cells consists in the observation that TGF-β costimulation with TCR induces CD4+CD25− T cells to express Foxp3. The ability of TGF-β to induce Foxp3 has also been observed in the non-CD4+ LN cell fraction that includes CD8+ T and B cells, though to a lesser extent. This result, together with the previous demonstration that conventional CD8+ T cells but not B cells can be converted to regulatory cells by ectopic expression of Foxp3 (9), suggests that TGF-β may be able to induce CD8+ T cells to retain a regulatory phenotype. The level of Foxp3 constitutively expressed by CD4+CD25+ Tr cells was not altered by the presence of TGF-β during priming, possibly indicating that the induction of Foxp3 takes place specifically in non-Tr cells, including CD4+CD25− and non-CD4+ T cells. Although the mechanism of Foxp3 action remains largely unknown, Foxp3 protein has been shown to translocate to the nucleus and serves as a transcriptional repressor inhibiting the transcription of the IL-2 gene (33). Consistent with this observation, we found that TGF-β costimulation with TCR resulted in impaired IL-2 production and cell proliferation, indicative of a state of anergy. However, we do not yet know what other target molecules there are whose transcription may be under the control of Foxp3 and that may collaborated with TGF-β in bringing about the lineage conversion.
An intriguing aspect of our results is that TGF-β costimulation of T cells led to an increase in the level of PD-1 protein on their surface. In vivo investigations have suggested that PD-1, an inhibitory costimulatory receptor induced on activated T, B and myeloid cells (22,34), plays an important role in the regulation of peripheral tolerance. For instance, PD-1 deficient mice develop lupus-like glomerulonephritis, arthritis, or autoimmune-dilated cardiomyopathy, depending on their genetic background (35,36), and PD-1 blockade accelerates the development of diabetes in NOD mice (24). Although the signaling pathway involved in this in vivo effect has not yet been established, it has been suggested that one outcome of PD-1 signaling in T cells may be the induction of an anergic state (22,23). Thus, the amplification of the PD-1 pathway by TGF-β costimulation may have contributed to the acquisition of anergy by the TGF-β-converted T cells. However, it is unlikely that the PD-1 pathway is directly associated with the suppressive activity of TGF-β-converted Tr cells, since PD-1 was not constitutively expressed on the surface of thymus-derived CD4+CD25+ cells and a PD-1 pathway blockade did not interfere with the suppression by TGF-β-converted Tr cells.
It has recently been proposed that peripheral tolerant states expand and spread because conventional T cells are converted to a regulatory phenotype. A well-established example of this ‘infectious’ tolerance is the generation of Th3 cells, a suppressor subset distinct from, but complementary to, CD25+ Tr cells, in the periphery by TGF-β and CD25+ Tr cells (19,37). Our finding that conventional CD4+CD25− T cells acquire phenotypes and functions of Tr cells, analogous to CD4+CD25+ Tr cells, by a TGF-β-mediated thymus-independent mechanism implies the existence of another pathway of infectious tolerance. Moreover, given the fact that professional Tr cells have remarkably potent suppressive activity on auto- and allo-immune responses, the ability to generate robust Tr cells ex vivo with TGF-β has immediate clinical relevance in, for example, the treatment of autoimmune diseases and the prevention of transplant rejection.
Transmitting editor: K. Yamamoto
We thank Drs Julian Gross and Woon Ki Paik for editing the manuscript. This work was supported by grants from the Korea Science and Engineering Foundation (R04-2002-000-20036-0 and R11-2002-098-01001-0 through the Rheumatism Research Center).
References
Sakaguchi, S., Fukuma, K., Kuribayashi, K. and Masuda, T.
Sakaguchi, S.
Shevach, E. M.
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. and Toda, M.
Asano, M., Toda, M., Sakaguchi, N. and Sakaguchi, S.
Takahashi, T., Kuniyasu, Y., Toda, M., Sakaguchi, N., Itoh, M., Iwata, M., Shimizu, J. and Sakaguchi, S.
Hori, S., Nomura, T. and Sakaguchi, S.
Fontenot, J. D., Gavin, M. A. and Rudensky, A. Y.
Khattri, R., Cox, T., Yasayko, S.-A. and Ramsdell, F.
Thorstenson, K. M. and Khoruts, A.
Hauet-Broere, F., Unger, W. W. J., Garssen, J., Hoijer, M. A., Kraal, G. and Samsom, J. N.
Zhang, X., Izikson, L., Liu, L. and Weiner, H. L.
Jonuleit, H., Schmitt, E., Stassen, M., Tuettenberg, A., Knop, J. and Enk, A. H.
Fowrie, F.
Fuss, I. J., Boirivant, M., Lacy, B. and Strober, W.
Gorelik, L. and Flavell, R. A.
Lucas, P. J., Kim, S.-J., Melby, S. J. and Gress, R. E.
Yamagiwa, S., Gray, J. D., Hashimoto, S. and Horwitz, D. A.
Zheng, S.G., Gray, J. D., Ohtsuka, K., Yamagiwa, S. and Horwitz, D. A.
Weiner, H. L.
Takahashi, T., Tagami, T., Yamazaki, S., Uede, T., Shimizu, J., Sakaguchi, N., Mak, T. W. and Sakaguchi, N.
Freeman, G. J., Long, A. J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., Fitz, L. J., Malenkovich, N., Okazaki, T., Byrne, M. C. et al.
Hatachi, S., Iwai, Y., Kawano, S., Morinobu, S., Kobayashi, M., Koshiba, M., Saura, R., Kurosaka, M., Honjo, T. and Kumagai, S.
Ansari, M. J., Salama, A. D., Chitnis, T., Smith, R. N., Yagita, H., Akiba, H., Yamazaki, T., Azuma, M., Iwai, H., Khoury, S. J. et al.
Dieckmann, D., Plottner, H., Berchtold, S., Berger, T. and Schuler, G.
Belkaid, Y., Piccirillo, C. A., Mendez, S., Shevach, E. M. and Sacks, D. L.
Horwitz, D. A., Zheng, S.G. and Gray, D.
Chen, W., Jin, W., Hardegen, N., Lei, K., Li, L., Marinos, N., McGrady, G. and Wahl, M.
Gonzalez, A., Andre-Schmutz, I., Carnaud, C., Mathis, D. and Benoist, C.
Apostolou, I., Sarukhan, A., Klein, L. and von Boehmer, H.
Uraushihara, K., Kanai, T., Ko, K., Totsuka, T., Makita, S., Iiyama, R., Nakamura, T. and Watanabe, M.
Vieira, P. L., Christensen, J. R., Minaee, S., O'Neill, E. J., Barrat, F. J., Boonstra, A., Barthlott, T., Stockinger, B., Wraith, D. C. and O'Garra, A.
Schubert, L. A., Jeffery, E., Zhang, Y., Ramsdell, F. and Ziegler, S. F.
Latchman, Y., Wood, C. R., Chernova, T., Chaudhary, D., Borde, M., Chernova, I., Iwai, Y., Long, A. J., Brown, J. A., Nunes, R. et al.
Nishimura, H., Nose, M., Hiai, H., Minato, N. and Honjo, T.
Nishimura, H., Okazaki, T., Tanaka, Y., Nakatani, K., Hara, M., Matsumori, A., Sadayama, S., Mizoguchi, A., Hiai, H., Minato, N. and Honjo, T.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}