





Abstract
Clostridioides difficile infection (CDI) remains a high burden worldwide. DAV131A, a novel adsorbent, reduces residual gut antimicrobial levels, reducing CDI risk in animal models.
We used a validated human gut model to investigate the efficacy of DAV131A in preventing moxifloxacin-induced CDI.
C. difficile (CD) spores were inoculated into two models populated with pooled human faeces. Moxifloxacin was instilled (43 mg/L, once daily, 7 days) alongside DAV131A (5 g in 18 mL PBS, three times daily, 14 days, Model A), or PBS (18 mL, three times daily, 14 days, Model B). Selected gut microbiota populations, CD total counts, spore counts, cytotoxin titre and antimicrobial concentrations (HPLC) were monitored daily. We monitored for reduced susceptibility of CD to moxifloxacin. Growth of CD in faecal filtrate and medium in the presence/absence of DAV131A, or in medium pre-treated with DAV131A, was also investigated.
DAV131A instillation reduced active moxifloxacin levels to below the limit of detection (50 ng/mL), and prevented microbiota disruption, excepting Bacteroides fragilis group populations, which declined by ∼3 log10 cfu/mL. DAV131A delayed onset of simulated CDI by ∼2 weeks, but did not prevent CD germination and toxin production. DAV131A prevented emergence of reduced susceptibility of CD to moxifloxacin. In batch culture, DAV131A had minor effects on CD vegetative growth, but significantly reduced toxin/spores (P < 0.005).
DAV131A reduced moxifloxacin-induced microbiota disruption and emergence of antibiotic-resistant CD. Delayed onset of CD germination and toxin production indicates further investigations are warranted to understand the clinical benefits of DAV131A in CDI prevention.
Introduction
Clostridioides (Clostridium) difficile1 infection (CDI) is a leading cause of antibiotic-associated diarrhoea2 and a major worldwide burden.3,4 Treatment options are limited and are associated with high recurrence rates (∼20%).5,6 Many antimicrobial classes can induce CDI,7–9 notably fluoroquinolones.10,11 This is likely due to disruption of normal gut microbiota, thus reducing colonization resistance.12 DAV132 is a non-specific adsorbent formulated to irreversibly capture antibiotics in the late ileum, caecum and colon of humans before they can significantly alter the microbiota. It is particularly efficacious in binding fluoroquinolones, e.g. levofloxacin or moxifloxacin, and protects the gut microbiota from antibiotic-mediated disruption, without affecting plasma antimicrobial levels.13 Thus, co-administration of DAV132 alongside fluoroquinolones may reduce their propensity to induce CDI. Indeed, it has been demonstrated in animal models that DAV131A, the rodent-adapted version of DAV132, can prevent moxifloxacin-mediated microbiota disruption in hamsters and confer protection from lethal CDI.14,15
The in vitro gut model has been used to investigate the propensity of multiple antibiotics to induce CDI,16–20 and results correlate well with clinical data. For example, fluoroquinolones and cephalosporins induce simulated CDI in the model, whereas piperacillin/tazobactam does not.16–20 Here we have used this in vitro gut model system to investigate the effects of DAV131A instillation on moxifloxacin-mediated gut microbiota disruption, C. difficile growth and toxin production, and whether emergence of reduced susceptibility to fluoroquinolone occurs in C. difficile. The test item used here was DAV131A, which is the non-formulated adsorbent of the DAV132 product, to be used for in vitro studies.
Materials and methods
In vitro gut model
C. difficile strains
The PCR ribotype 027 strain used in this experiment (027 210) was isolated during an outbreak of CDI at the Maine Medical Centre (Portland, MA, USA) and was kindly supplied by Dr Rob Owens.
Gut model
The model consisted of three chemostat vessels, pH controlled (vessel 1, pH 5.5 ± 0.2; vessel 2, pH 6.2 ± 0.2; vessel 3, pH 6.8 ± 0.2) and arranged in a weir cascade system. Vessel 1 was top fed with a complex growth medium,18 and all vessels were sparged with nitrogen to maintain an anaerobic atmosphere. The model was inoculated with a pooled faecal slurry (10% in pre-reduced PBS). Faeces was from elderly volunteers (>60 years) (n = 3–5) with no history of antimicrobial therapy (last 3 months), and was screened for C. difficile (by culture on selective agar). Only faecal samples confirmed as C. difficile negative were used to create the faecal slurry.
Gut model experimental design
Two gut models were then run simultaneously as outlined in Figure 1.
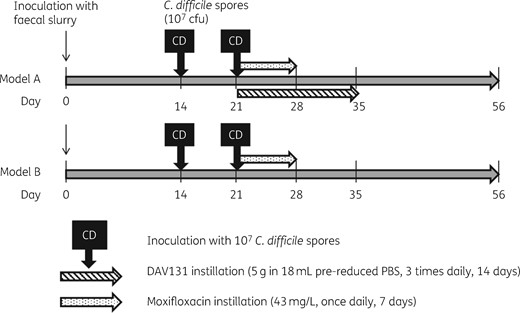
Gut model experimental design.
The models were set up and primed with pooled faecal slurry, then left for 2 weeks to reach steady-state, before addition of an aliquot of C. difficile spores (∼107 cfu). One week later, moxifloxacin instillation commenced (43 mg/L, once daily, 7 days). Another aliquot of C. difficile spores (∼107 cfu) was added with the first moxifloxacin dose. Model B was instilled with moxifloxacin only, and Model A was instilled with moxifloxacin and DAV131A (5 g in 18 mL pre-reduced PBS, three times daily, 14 days). Eighteen millilitres of pre-reduced PBS diluent (three times daily, 14 days) was added to Model B to keep the flow rates of the two models comparable.
Bacterial enumeration
Gut microbiota populations and C. difficile total viable and spore counts were enumerated by culture on solid media. Colonies were identified to genus level on the basis of colony morphology, Gram reaction, microscopic appearance and/or MALDI-TOF identification on selective and non-selective agars as follows: fastidious anaerobe agar supplemented with 5% horse blood (total anaerobes); Beerens agar [42.5 g/L Columbia agar, 5 g/L agar technical, 0.5 g/L cysteine HCl, 5 g/L glucose, 5 mL propionic acid, adjusted to pH 5 (bifidobacteria)]; Bacteroides bile aesculin agar supplemented with 5 mg/L haemin, 10 μL/L vitamin K, 7.5 mg/L vancomycin, 1 mg/L penicillin G, 75 mg/L kanamycin and 10 mg/L colistin (Bacteroides fragilis group); LAMVAB agar [20 g/L agar technical, 52.2 g/L MRS broth, 0.5 g/L cysteine HCl, 20 mg/L vancomycin, adjusted to pH 5 (lactobacilli)]; nutrient agar (total facultative anaerobes); MacConkey’s agar No. 3 (lactose-fermenting Enterobacteriaceae); kanamycin aesculin azide agar supplemented with 10 mg/L nalidixic acid, 10 mg/L aztreonam and 20 mg/L kanamycin (enterococci); alcohol shock and Brazier’s CCEYL agar supplemented with 2% lysed horse blood, 5 mg/L lysozyme, 250 mg/L cycloserine and 8 mg/L cefoxitin (C. difficile spores); Brazier’s CCEYL agar as above and supplemented with 2 mg/L moxifloxacin (C. difficile total viable counts).
Cytotoxin testing
The presence of C. difficile cytotoxins was determined by Vero cell cytotoxicity assay (CA).19 Gut model fluid (1 mL) was centrifuged at 16 000 g, 4°C for 15 min. Supernatants were then serially diluted 1:10 in sterile PBS to 10−6. Twenty microlitres of each dilution was added to Vero cell monolayers and a further 20 μL of Clostridium sordellii antitoxin (diluted 1:10 in sterile distilled water) placed into the corresponding antitoxin row. Monolayers were examined after 24 and 48 h incubation at 37°C in 5% CO2, with a positive result indicated by the presence of cell rounding with concurrent neutralization of effect by C. sordellii antitoxin. Cytotoxin titres (relative units, RU) were an arbitrary log10 scale and the cytotoxin titre reported in the highest dilution with >70% cell rounding, i.e. 100 = 1 RU, 10−1 = 2 RU, 10−2 = 3 RU.
Measurement of antimicrobial concentrations by HPLC
Samples (1 mL) from all vessels of each gut model were centrifuged (16 000 g, 10 min) and the supernatants sterilized by filtration through 0.22 μm syringe filters, resulting in elimination of charcoal and any particulate material from the medium, before being stored at −20°C for measurement of antimicrobial concentrations. This was achieved by HPLC coupled with fluorescence detection, and was performed by AmatsiAvogadro (Fontenilles, France). Samples were spiked with 2.5 mg/L enrofloxacin used as an internal standard, and extracted by solubilization with 4% phosphoric acid followed by loading onto a solid-phase cation exchange sorbent (Oasis MCX 60 mg 3 cc cartridges, Waters) that was successively washed with 2% formic acid and methanol, dried and finally eluted with 5% ammonia in methanol. Dried samples were reconstituted with 0.1% formic acid in 90:10 water:acetonitrile, and separated by HPLC onto a Kinetex PhenylHexyl 100 × 3 mm 0.26 μm (Phenomenex) column that was eluted with a gradient from 90:10 to 30:70 of mobile phases, respectively, consisting of 20 mM ammonium formate and 0.1 M formic acid in acetonitrile. Fluorometric detection of the eluted products (excitation at 290 nm, emission at 500 nm) made it possible to reach a lower limit of detection of 50 ng/mL for moxifloxacin. Non-interference of the matrix with the assay was ensured by the fact that control as well as calibration samples with known amounts of moxifloxacin made in matrix or buffer gave similar results in the assay.
Emergence of reduced susceptibility
The emergence of C. difficile populations showing reduced susceptibility to moxifloxacin was monitored on antibiotic-containing agar plates as described previously.16 Brazier’s CCEYL agar containing 32 or 64 mg/L moxifloxacin, as well as the usual supplements, was used in addition to normal agars to enumerate C. difficile total viable counts (TVCs) and spores. The MIC of moxifloxacin for the C. difficile strain used here was 32 mg/L.
C. difficile growth and toxin production in batch culture
Three clinical isolates submitted to the C. difficile ribotyping network (CDRN) in 2013 were selected for batch culture growth experiments. Isolates were chosen to represent the epidemic ribotypes 027, 001 and 078. The growth of each strain was investigated in both brain heart infusion (BHI) broth and faecal filtrate prepared from faeces provided by healthy volunteers aged >60 years (the at-risk population for CDI). A 10% (w/v) faecal slurry was prepared in pre-reduced PBS. Faecal slurry was centrifuged and filtered through 0.22 μm filters to remove all viable organisms.
Each medium was treated in four different ways: A, control (sterile broth); B, spun control (sterile broth centrifuged and filtered through 0.22 μm filters before use); C, DAV131A exposed (sterile broth preincubated with 0.05 g/mL of DAV131 for 2 h before use); and D, spun DAV131A exposed (sterile broth preincubated with 0.05 g/mL of DAV131A for 2 h then centrifuged and filtered through 0.22 μm filters before use to eliminate DAV131A from the resulting broth).
Broths were pre-reduced overnight and either incubated with 0.05 g/mL of DAV131A (C, DAV131A exposed; D, spun DAV131A exposed only) for 2 h, or not (A, control; B, spun control); B and D were centrifuged (16 000 g, 15 min) and decanted into new tubes. All media were then filter sterilized and inoculated with C. difficile as follows: C. difficile was grown on CCEYL agar for 48 h, and the growth was suspended in pre-reduced saline to ∼0.5 McFarland; 200 μL of the C. difficile suspension was added to all broths, and incubated anaerobically at 37°C. Samples were taken for C. difficile enumeration (TVCs and spore counts) and toxin quantification at 48 h. The supernatant from the A broths after 48 h growth was used as a toxin-positive control. This supernatant was then incubated anaerobically with 0.05 g/mL DAV131A for 2 h, filtered and assayed for toxin (E, DAV131A-exposed supernatant).
C. difficile TVCs and spore counts were enumerated (in triplicate) as described above. Toxin levels were assayed using a cell CA as described above (in duplicate). Experiments were repeated in biological duplicate for each different ribotype. Statistical significance was determined using a paired t-test using Stata/IC 13.1 software.
Ethics
The collection/use of faecal donations from healthy adult volunteers following informed consent was approved by the Leeds Institute of Health Sciences, Leeds Institute of Genetics, Health and Therapeutics and Leeds Institute of Molecular Medicine, University of Leeds joint ethics committee (reference HSLTLM/12/061).
Results
In vitro gut model
Antimicrobial concentrations
In Model A, the instillation of DAV131A prevented the detection of any moxifloxacin throughout the gut model experiment in vessels 1 and 3 (vessel 3 data shown in Figure 2a). Moxifloxacin was detected (0.3 mg/L) only on a single day (day 21) in vessel 2 of Model A (data not shown). Concentrations of moxifloxacin in Model B detected by HPLC peaked at ∼120 mg/L in vessel 1, ∼90 mg/L in vessel 2 (data not shown) and ∼100 mg/L in vessel 3 (vessel 3 data shown in Figure 2b).
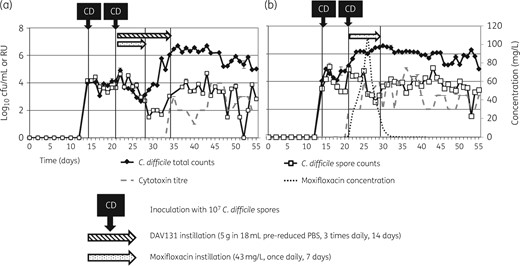
Mean (±SE) C. difficile total viable counts (log10 cfu/mL), spore counts (log10 cfu/mL), cytotoxin titre (RU) and moxifloxacin concentration (mg/L) in vessel 3 of (a) Model A (instilled with DAV131A and moxifloxacin) and (b) Model B (instilled with moxifloxacin alone).
Gut microbiota populations
Without the addition of DAV131A (Model B), moxifloxacin instillation caused substantial disruption to microbiota populations (Figure 3b and d). Decreases were observed in populations of lactose fermenters and B. fragilis group (∼6 log10 cfu/mL), bifidobacteria (∼4 log10 cfu/mL) and lactobacilli (∼3 log10 cfu/mL). All populations recovered to steady-state levels ∼10 days after the end of moxifloxacin infusion. In Model A, the effects of moxifloxacin instillation on the gut microbiota were greatly reduced (Figure 3a and c), with only a small, temporary decline in Bacteroides spp. counts observed (∼3 log10 cfu/mL).
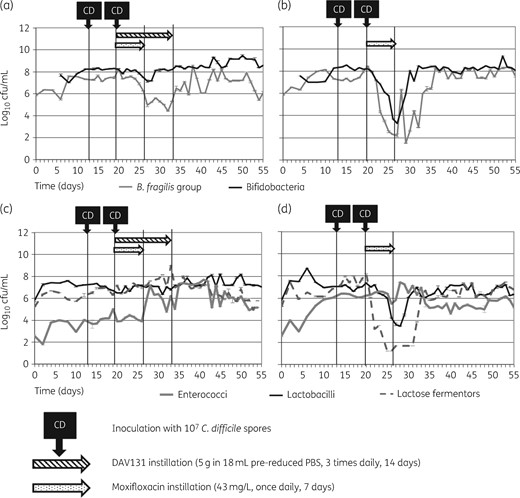
Mean (±SE) viable counts of selected microbiota populations from vessel 3 of the gut models. (a) Obligate anaerobes in Model A (instilled with DAV131A and moxifloxacin); (b) obligate anaerobes in Model B (instilled with moxifloxacin alone); (c) facultative anaerobes in Model A; (d) facultative anaerobes in Model B.
C. difficile total and spore counts and toxin titres
In the absence of DAV131A (Model B), moxifloxacin instillation caused rapid (1–2 days into instillation) germination and proliferation of C. difficile. Toxin production was also very rapid (1–2 days into instillation). In Model A, germination was delayed by ∼7 days in comparison with Model B, occurring 1 day after the end of moxifloxacin instillation. Toxin production was not detected until 7 days after the end of moxifloxacin instillation, when DAV131A instillation also ceased.
Emergence of reduced susceptibility to moxifloxacin
In both models, C. difficile counts on agar containing 32 mg/L moxifloxacin were comparable to those on agar containing 2 mg/L moxifloxacin (moxifloxacin MIC of strain 027 210 = 32 mg/L) (Figure 4). In Model A, no C. difficile was isolated on 64 mg/L agar (Figure 4a); however, in Model B, following moxifloxacin instillation there was an increase in C. difficile isolated on agar containing 64 mg/L moxifloxacin (Figure 4b), reaching ∼3 log10 cfu/mL.
![Mean (±SE) C. difficile total viable counts (log10 cfu/mL), isolated on breakpoint agar [moxifloxacin (MXF) 32 and 64 mg/L] from vessel 3 of (a) Model A (instilled with DAV131A and moxifloxacin) and (b) Model B (instilled with moxifloxacin alone).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jac/75/6/10.1093_jac_dkaa062/1/m_dkaa062f4.jpeg?Expires=1716428261&Signature=Loa1PDg8C3kNU6BPR2waPstwj9X0ey~Gr4tKQnds8TjY~R7oBlsIOAvnZ-tFO3XImXNfdk9cz-F07GhkLeHgYxqqOmY8q9-oP3mG4Oy~oMjdqk6zxogeaQ1IrB6PMKr~6RuDWY1~RX8Jrf16vdn7eQNzb~iKJv4ePh558aiyFWVfH6v2yYCtefDLmGH1mlkSC2NHSLb3DGPmnAUrKBRnt9yiBlzNCCBYX4MHxQGeGfnWl3GXX6N6WWdjZW6-w~WEGBZZb~-z8zsWrUXtG5cArqbjMjSKR6lngOu3xDiddyfqaOEDkLRlHyI5M7XKzbdSpZII3qJV2o9NevDeid7fyQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Mean (±SE) C. difficile total viable counts (log10 cfu/mL), isolated on breakpoint agar [moxifloxacin (MXF) 32 and 64 mg/L] from vessel 3 of (a) Model A (instilled with DAV131A and moxifloxacin) and (b) Model B (instilled with moxifloxacin alone).
C. difficile growth and toxin production in batch culture
Although variation was observed between growth characteristics of different C. difficile strains (data not shown), pooled data are presented here to indicate the overall effects of DAV131A on C. difficile growth and toxin production. As expected, no differences were observed in C. difficile growth or toxin production between control samples and spun control samples in either BHI or faecal filtrate (Figure 5).
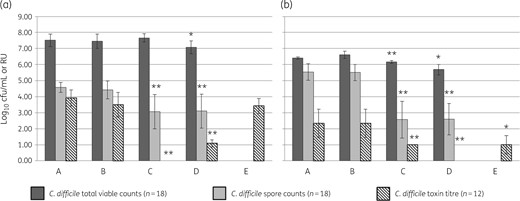
Mean (±SE) C. difficile TVC (log10 cfu/mL), spore count (log10 cfu/mL) and cytotoxin titre (RU) of three different C. difficile strains (of PCR ribotype 027, 001, 078) grown in (a) BHI and (b) faecal filtrate. A, control (sterile broth); B, spun control (sterile broth centrifuged and filtered through 0.22 μm filters before use); C, DAV131A exposed (sterile broth exposed to 0.05 g/mL of DAV131A for 2 h before use); D, spun DAV131A exposed (sterile broth exposed to 0.05 g/mL of DAV131A for 2 h then centrifuged and filtered through 0.22 μm filters before use); E, DAV131A-exposed supernatant (toxin-positive supernatant from control broths incubated anaerobically with 0.05 g/mL DAV131A for 2 h, centrifuged and filtered through 0.22 μm filters before use). Significant differences from the control (A) samples are indicated by ** (P < 0.005) or * (P < 0.05). Each strain was assayed in biological duplicate. TVCs were enumerated in technical triplicate (n = 18), and toxin in technical duplicate (n = 12).
Inclusion of DAV131A in the BHI media had no effect on TVCs (P = 0.38), but significantly reduced spore counts (P < 0.005) and toxin levels (P < 0.005). Inclusion of DAV131A in BHI followed by centrifugation and filtration before inoculation of C. difficile affected total viable counts to some extent (P = 0.03), and significantly reduced spore counts (P < 0.005) and toxin levels (P < 0.005). Incubation of BHI toxin-positive supernatant with DAV131A followed by centrifugation decreased toxin detection slightly (mean amount of decrease 3.93 to 3.42 RU; not significant, P = 0.16).
Preincubation of DAV131A in faecal filtrate, followed or not by centrifugation and filtration before C. difficile inoculation, significantly reduced total viable and spore counts and toxin levels (P < 0.005 in all cases). Incubation of faecal filtrate toxin-positive supernatant with DAV131A followed by centrifugation decreased mean toxin detection in the supernatant from 2.33 to 1 RU (P = 0.007).
Discussion
This study investigated the effects of a novel non-specific absorbent, DAV131A, on moxifloxacin-induced simulated CDI in an in vitro gut model. We have previously demonstrated that moxifloxacin administration instilled at 43 mg/L, once daily for 7 days, to reflect a standard clinical dosing regimen and achieve faecal antibiotic levels,21 induces simulated CDI in our in vitro model system.16 This observation is consistent with clinical data showing that fluoroquinolone administration is a risk factor for CDI.10,11
We show here that DAV131A instillation prevented detection of active moxifloxacin for the duration of the gut model experiment, indicating that DAV131A successfully adsorbed and inactivated substantial quantities of moxifloxacin. This is reflected in the fact that the majority of changes in gut microbiota populations observed following moxifloxacin instillation were not seen in the presence of DAV131A. These data are consistent with the recently reported clinical trial where DAV132, the targeted-release product for humans containing the same adsorbent as DAV131A, reduced exposure of the intestinal microbiota to moxifloxacin by ∼99%, and largely preserved the richness and composition of the microbiota seen in healthy volunteers.13
However, despite the presence of DAV131A, some microbiota disruption was observed, specifically a ∼3 log10 decline in B. fragilis group populations. This was substantially less than the ∼6 log10 cfu/mL decrease observed in the absence of DAV131A, but indicates that some active, but undetected moxifloxacin [i.e. below the limit of detection (LOD) of the HPLC method used here, 50 ng/mL], may still be present despite DAV131A instillation. The detection of 0.3 mg/L of moxifloxacin in vessel 2 on day 21 supports the suggestion that some level of active moxifloxacin is persisting. Whilst the MIC of moxifloxacin for B. fragilis ATCC 25285 is 0.25 mg/L, it is expected that the range of B. fragilis group species within the gut microbiota would have a range of MIC values, and so it is possible that moxifloxacin concentrations could be supra-MIC for some Bacteroides spp. populations in the model, but still below the HPLC assay LOD as discussed above. Alternatively, this minimal disruption could be due to the presence of DAV131A. As a non-specific adsorbent, DAV131A will sequester other components of the microbiota milieu, which may affect the growth of certain populations. This work has demonstrated that DAV131A appears to have minimal effects on cultivable microbiota, supporting the findings of clinical studies.13
Instillation of DAV131A delayed the onset of C. difficile spore germination by ∼1 week and C. difficile toxin production by ∼2 weeks. In the absence of DAV131A, toxin production occurred simultaneously with germination, very soon after moxifloxacin instillation commenced. This is similar to previously reported observations following moxifloxacin instillation in the gut model.16 However, with the co-administration of DAV131A, germination was not observed until ∼1 week after moxifloxacin instillation ended, and toxin production was delayed until ∼5 days after germination was observed. DAV131A was instilled for a further 7 days after the end of moxifloxacin instillation. Germination occurred during DAV131A instillation, but interestingly toxin detection was delayed until after DAV131A instillation ceased. This may represent delayed toxin production, or the fact that DAV131A adsorbed toxin while it was being instilled, so preventing subsequent toxin detection in the CA.
In order to facilitate instillation of DAV131A into the model, an increased fluid volume (54 mL/day) was required. This is a notably higher fluid instillation than used in previous gut model experiments and will have increased the flow rate of the system. It is possible that the increased flow rate may have some effects on the growth/behaviour of microbiota populations, including C. difficile. However, the instillation of pre-reduced PBS in the non-DAV131A-exposed model ensured that the flow rate of the two model systems was identical, allowing the effects of DAV131A exposure to be examined.
Batch culture experiments were utilized to try to further elucidate the mechanisms by which DAV131A might affect toxin detection. Data indicate that although DAV131A appears to sequester some toxin (from a toxin-positive culture supernatant), this did not reduce detected toxin to the same extent as when DAV131A was either included in the growth medium or simply used to treat the medium before C. difficile inoculation, suggesting that DAV131A may be affecting toxin production and/or detection. Since DAV131A is a non-specific adsorbent, the fact that inclusion of DAV131A in the growth medium, or simple pre-treatment of the medium by DAV131A similarly reduced sporulation and toxin production/detection, suggests it is likely acting by adsorbing medium components, thereby altering the environmental conditions in which C. difficile is growing. Many nutritional and environmental factors have been reported to affect toxin production, including temperature,22 bicarbonate concentration,23 sub-inhibitory antimicrobial concentrations,24,25 short-chain fatty acids,26 amino acid concentrations,23 and glucose or other rapidly metabolized carbon sources.27 Spo0A is the master regulator of sporulation in Clostridioides (and Bacillus) species and has been reported to play a role in toxin mediation,28–31 again linking sporulation and toxin production. In a complex, multispecies gut environment (such as the gut model or host gut), these nutritional and environmental factors are mediated by members of the microbiota communities. As these communities are altered by antibiotic exposure, this may affect resistance to colonization, in particular to C. difficile, and predispose to CDI, as has been demonstrated for Clostridium scindens-mediated bile acid metabolism.32 This suggests a potential 2-fold mechanism of action by which DAV132 administration in humans may help to prevent antibiotic-induced CDI: in addition to preventing antibiotic-induced changes to the microbiota leading to loss of colonization resistance, DAV132 could sequester key nutrients and germinants in the colonic environment, thereby reducing C. difficile germination and toxin production by the few C. difficile that could develop, notwithstanding these unfavourable conditions.
Notably, DAV131A instillation prevented the emergence of C. difficile with elevated resistance to moxifloxacin. In the absence of DAV131A, instillation of moxifloxacin caused a population (∼3 log10 cfu/mL) of C. difficile with moxifloxacin MIC >64 mg/L to emerge and persist. This was not observed following DAV131A co-administration, consistent with the considerable lowering of antibiotic selective pressure by DAV131A. In humans, such a mechanism might reduce the emergence of highly fluoroquinolone-resistant strains of C. difficile consequential to moxifloxacin administration.33
In the gut model, DAV131A successfully reduced detectable active moxifloxacin levels, substantially reduced moxifloxacin-induced deleterious effects on gut microbiota populations, prevented moxifloxacin-induced emergence of C. difficile with reduced susceptibility to moxifloxacin, and delayed, but did not totally prevent, the onset of simulated CDI. DAV131A successfully prevented moxifloxacin-induced CDI in hamsters.15 Both the hamster model and the human gut model have been shown to correlate with clinical use of CDI therapeutics. The gut model includes a human colonic microbiome, but does not simulate a humoral or cell-mediated immune response (beyond that present in the faecal samples used to prime the system). It is therefore possible that the observed delay in germination and toxin production caused by DAV131A instillation in the gut model, in conjunction with an effective (anti-toxin antibody) immune response in immunocompetent hosts, may prevent the actual development of CDI, and/or allow greater recovery of the gut microbiota, thereby improving colonization resistance to CD.
The C. difficile 027 strain used in the experiments described here is a highly virulent epidemic strain, whereas the strain used in the hamster model was non-epidemic. It is possible that the differences in outcomes between the two studies may be strain/ribotype specific. It should also be noted that the inoculum of C. difficile spores (107 cfu/mL) reflects levels of spores in the faeces of an infected patient, and is likely to be significantly higher than the exposure level of an at-risk patient in a healthcare setting. Thus, the model may have provided a very stringent test of the capacity of DAV131A to adsorb moxifloxacin and to prevent its deleterious effects. In this context, DAV131A was at least partially protective.
Conclusions
These gut model results complement hamster data in indicating that DAV131A may provide some protection against moxifloxacin-induced CDI. Whilst instillation of DAV131A did not prevent the onset of simulated CDI in this experiment, it caused an ∼2 week delay. It also substantially protected the gut microbiota examined in this study from the deleterious effects of moxifloxacin, and prevented emergence of C. difficile populations displaying reduced susceptibility to moxifloxacin. These results confirm clinical findings indicating that DAV132 has potential clinical benefit in humans, in reducing antibiotic-induced disruption of the gut microbiota.13 Whether DAV132 may confer a clinical benefit in prevention of CDI remains to be shown, but these results indicate that further investigation is warranted.
Acknowledgements
We thank Sakina Sayah-Jeanne for her contribution to this study, and also Violaine Augustin for the HPLC determination of moxifloxacin concentrations.
Funding
This study was initiated and financially supported by Da Volterra.
Transparency declarations
C.M. was an employee of Da Volterra at the time of the study; A.A. and J. de Gunzburg are consultants and shareholders of Da Volterra. C.H.C., G.S.C. and M.H.W. have received grant support from Da Volterra. M.H.W. has received: consulting fees from Actelion, Astellas, bioMerieux, Da Volterra, Merck, Meridian, Pfizer, Sanofi-Pasteur, Seres, Singulex, Summit, Synthetic Biologics, Valneva and Vaxxilon; lecture fees from Alere, Astellas, Merck, Pfizer & Singulex; and grant support from Actelion, Alere, Astellas, bioMerieux, Merck, MicroPharm, Morphochem AG, MotifBio, Paratek, Sanofi-Pasteur, Seres, Summit & Tetraphase.
References
Author notes
Present address: Vetoquinol, 37, rue de la Victoire, Paris, France.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}