






Abstract
Helicobacter pylori colonizes the human stomach for decades unless pharmacologically eradicated. We hypothesized that this flagellated pathogen escapes immune clearance, in part, by avoiding detection by the flagellin receptor Toll-like receptor 5 (TLR5). In contrast to other gram-negative microbes, H. pylori did not release flagellin. Furthermore, recombinant H. pylori flagellin (FlaA) was significantly less potent (1000-fold) than Salmonella typhimurium flagellin in activating TLR5-mediated interleukin (IL)-8 secretion. TLR5 can mediate flagellin-induced IL-8 secretion via p38 mitogen-activated protein kinase signaling; however, compared with potent induction by S. typhimurium flagellin, H. pylori FlaA-dependent p38 activation was substantially attenuated. In addition, disruption of H. pylori flaA decreased motility but had no effect on H. pylori-induced IL-8 secretion, which indicates that H. pylori flagellin plays no role in activating epithelial orchestration of inflammation. We conclude that H. pylori evades TLR5-mediated detection, which may contribute to its long-term persistence in individual hosts.
Approximately 50% of the world's population is colonized by Helicobacter pylori, and, although the majority of these infections are asymptomatic, long-term interactions between H. pylori and humans significantly increase the risk for peptic ulcer disease and distal gastric adenocarcinoma [1,2]. Regardless of clinical outincome, the vast majority of colonized persons never eliminate H. pylori, unless a targeted antibiotic regimen is employed; thus, a signature feature of H. pylori-induced gastritis is its capacity to persist for decades. This is in marked contrast to inflammatory reactions induced by other gram-negative enteric pathogens, such as Salmonella and Escherichia coli species, which either resolve within weeks or progress to eliminate the host.
H. pylori likely uses a variety of mechanisms to avoid immune clearance. One mechanism through which H. pylori may limit the host inflammatory response is in duction of the production of the anti-inflammatory cytokine interleukin (IL)-10 [3,4]. H. pylori arginase also attenuates nitric oxide-mediated bacterial killing in vitro [5]. Another level of host defense that may be circumvented by H. pylori is innate immunity. Toll-like receptors (TLRs) are an evolutionarily conserved family of eukaryotic receptors that function in innate immunity via recognition of somewhat invariant regions in bacterial molecules, which are termed microbe-associated molecular patterns 6–9]. TLR4 and TLR5 efficiently recognize picomolar concentrations of lipopolysaccharide (LPS) and flagellin, respectively, that are released by a wide variety of gram-negative organisms, including Salmonella typhimurium, E. coli, and Pseudomonas aeruginosa8–11], although the potency of these microbial constituents varies, depending on the type and activation state of the targeted host cell [12,13]. The means by which H. pylori may evade innate immune clearance involves avoidance of at least 1 of these pathways, because H. pylori LPS is >100-fold less potent than E. coli LPS for activating TLR4-mediated proinflammatory gene expression [14,15].
To facilitate locomotion within gastric mucus and to counteract peristalsis, H. pylori possesses 5 or 6 polar flagella consisting of 2 structural subunits: a major 53-kD FlaA and a minor 54-kD FlaB [16,17]. The genes encoding these 2 flagellins are located at distant sites on the H. pylori chromosome and are transcriptionally regulated by different promoters [16]. The essential role for both flaA and flaB in the establishment of persistent colonization has been demonstrated by Eaton et al. [18], who showed that aflagellate H. pylori strains only transiently colonized gnotobiotic piglets; however, the ability of H. pylori flagellins to regulate other host responses, such as induction of inflammation, remains undefined.
Because H. pylori is a flagellated organism [16] and TLR5 is expressed in primary and transformed gastric epithelial cells [15], we aimed to determine whether H. pylori could activate TLR5-mediated innate immunity in vitro and, if not, how such immune evasion may be regulated. We report that, although gastric epithelial cells can functionally respond to S. typhimurium flagellin monomers, the primary flagellar structural component of H. pylori (FlaA) is not released and, in addition, is inherently much less proinflammatory than flagellin from S. typhimurium (FliC), which indicates that H. pylori harbors 2 distinct mechanisms for avoiding TLR5-mediated immunity.
METHODS
Cells and reagents. AGS human gastric epithelial cells (ATCC CRL 1739) were grown in RPMI 1640 medium (Gibco BRL) supplemented with 10% fetal bovine serum (FBS) and 20 µg/ mL gentamicin in an atmosphere of 5% CO2 at 37°C. Coculture experiments, in which AGS cells were incubated with viable H. pylori, were performed in antibiotic-free medium containing 10% FBS. Model human intestinal epithelia were prepared by culturing the colonic cell line T84 on collagen-coated permeable supports, as described elsewhere [19]. Flagellin was purified, as described elsewhere [20,21].
H. pylori wild-type (wt) and isogenic mutant strains. In the present study, H. pylori strain 60190 (ATCC 49503) was the parental wt strain, and an isogenic flaA mutant was generated in strain 60190 by insertional mutagenesis [22]. In brief, the 5′region of flaA was amplified from H. pylori strain 60190 by use of the following primers: 5′-TGGGCAAACTACGGAATCTC-3′ and 5′-AACATTTTCATCGCTGATTCG-3′. The resultant polymerase chain reaction (PCR) product was cloned into pGem-T Easy (Promega), and the resultant plasmid was subjected to inverse PCR by use of the following primers: 5′-CGGGATCCTAACGCGCCTGTAGCGATAC-3′ and 5′-CGGGATCCGGCTCTACTAACTACGGAAG-3′, which include BamHI sites for the insertion of aphA from pUC4K [23]. H. pylori isogenic flaA mutants were generated by natural transformation and allelic exchange and were selected on brucella agar with kanamycin (25 µg/mL), as described elsewhere [22].
H. pylori culture and IL-8 quantitation. H. pylori were grown in Brucella broth with 10% FBS for 48 h, harvested by centrifugation at 2000 g, resuspended in antibiotic-free RPMI 1640 medium with 10% FBS, and added to cells in 6-well plates at bacteria:cell concentrations ranging from 1:1 to 100:1 (1 × 106 to 1 × 108H. pylori: 1 × 106 AGS cells/well). Broth culture supernatants from H. pylori cultures containing FBS were concentrated 30-fold by ultrafiltration (Amicon), using a molecular weight cutoff of 30 kDa. Protein concentrations were determined by use of a Micro-BCA assay (Pierce). For quantitation of IL-8, AGS cell monolayers in 6-well plates were cocultured with or without H. pylori or flagellin in triplicate, and supernatants were centrifuged at 15,000 g and used for ELISA, as described elsewhere [24]. IL-8 secretion in polarized T84 cells was measured by ELISA, as described elsewhere [25].
Preparation of recombinant flagellins. The complete coding region of S. typhimurium and H. pylori flagellins (fliC and flaA) were PCR amplified from S. typhimurium strain SL3201 and H. pylori strain 60190 genomic DNA, respectively, by use of the following primers: fliC, 5′-GAATTCATGGCACAAGTCATTAATACAA-3′ and 5′-TCTAGATTAACGCAGTAAAGAGAGGACG-3′; flaA, 5′-ATAGGATCCATGGCTTTTCAGGTCAATACA-3′ and 5′-GCCAGATCTAGTTAAAAGCCTTAAGATATT-3′. The resulting PCR products were then digested with BamHI and BglII and ligated into pTrcHisA (Invitrogen), which had been restricted with the same enzymes. After sequence verification of the plasmids, E. coli were transfected and induced 18 h later with 1 mmol/L isopropyl-β-D-thiogalactopyranoside for 4 h, cell lysates were collected, and flagellins were purified, as described elsewhere [21].
Generation of constitutive TLR5-expressing cells. MDCK cells were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% FBS. For stable transfections, pEF6/V5-TLR5 (a plasmid encoding V5-tagged TLR5; provided by Jongdae Lee, Scripps Institute, La Jolla, CA) was linearized by ScaI. Five micrograms of linearized plasmid DNA was added to MDCK cells in 100-mm culture dishes that were ∼30% confluent by use of Superfectin (Qiagen) as transfection reagent, as described elsewhere [26]. After 24 h, transfected cells were passed at low density and were cultured in DMEM supplemented with 5–10 mg/mL Blasticidin (Invitrogen) until the appearance of colonies, which then were transferred to a new culture container and propagated. Expression of TLR5 was confirmed by Western blot, using V5 antibody (Invitrogen), as described elsewhere [26].
Immunoblot analysis. The presence of flagellin in H. pylori-filtered supernatants or whole-cell sonicates was assayed by SDS-PAGE immunoblotting (50-µg protein/sample), using a polyclonal antibody raised against a preparation of S. typhimurium flagellin (Becton Dickinson) diluted 1:2000. Phosphop38 levels were measured in MDCK cells stably expressing TLR5 or AGS cells, by use of Western blot (25-µg protein/sample), as described elsewhere [26].
Statistical analysis. Results are expressed as mean ± SD. The Mann-Whitney U test was used for statistical analyses of intergroup comparisons. P ≤ .05 was considered to be significant.
Results
Gastric epithelial cells functionally responsive to flagellin. We tested whether AGS gastric epithelial cells, which express TLR5 [15], could functionally respond to flagellin. As shown in figure 1, AGS cells secreted copious amounts of IL-8 in response to nanomolar concentrations of purified S. typhimurium flagellin. Pretreatment of flagellin with proteinase K (60 min at 37°C, which was followed by 10 min at 100°C, to inactivate the proteinase K) abolished this response (figure 1), and there was no decrease in AGS cell viability after treatment with proteinase K alone (data not shown). These results demonstrate that the ability to induce IL-8 secretion was attributable to flagellin, rather than a nonproteinaceous contaminant, such as LPS. Therefore, TLR5 expressed by AGS cells permits activation of proinflammatory gene expression after detection of flagellin.
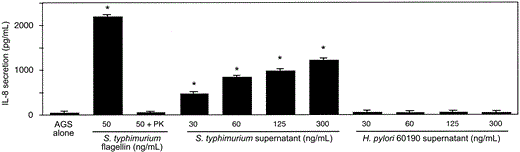
Secretion of interleukin (IL)-8 by AGS cells after incubation with Salmonella typhimurium flagellin and bacteria-free S. typhimurium supernatants, but not Helicobacter pylori-conditioned medium. AGS cells were incubated with or without S. typhimurium flagellin in the absence or presence of proteinase K (PK; 1 µg/µL) or varying protein concentrations of S. typhimurium or H. pylori strain 60190 filtrate. IL-8 production in coculture supernatants was quantified by ELISA. Lowest S. typhimurium supernatant concentration tested was 30 ng/mL, which represents a 1:10,000 dilution of a S. typhimurium filtrate. Data represent at least 3 independent experiments performed in triplicate and are expressed as picograms per milliliter. Bars, SD. * P < .05, vs. AGS cells in medium alone.
Secretion of IL-8 in AGS cells after incubation with bacteria-free S. typhimurium- but not H. pylori-conditioned medium. Many flagellated microbial species release flagellin into culture medium, and we observed that medium conditioned by S. typhimurium induced IL-8 secretion from AGS cells, even when added at dilutions of up to 1:10,000 (figure 1). In contrast, medium conditioned by H. pylori in a similar manner did not contain detectable IL-8-inducing bioactivity, even when added at higher concentrations (figure 1). This suggests either that H. pylori does not release flagellin into its medium or that its flagellin is not recognized by TLR5.
Absence of H. pylori flagellin in conditioned medium. To discern whether H. pylori encoded a noninflammatory flagellin or simply did not release flagellin monomers, we next investigated whether H. pylori-conditioned medium contained flagellin. SDS-PAGE immunoblotting was performed on H. pylori whole-cell sonicates and supernatants. When H. pylori was subjected to intense sonication, flagellin was detected in the medium (figure 2A), which suggests that flagellin may be released under certain circumstances (i.e., bacterial lysis). However, flagellin was not detectable in H. pylori-conditioned medium (figure 2A), with a minimum threshold of detection being 20 ng/mL (as determined by use of the recombinant flagellin described below). These results indicate that this organism releases flagellin much less readily than other gram-negative microbes, such as S. typhimurium, which suggests that H. pylori may evade TLR5-mediated immunity by failing to shed flagellin.
![Absence of Helicobacter pylori FlaA in conditioned medium and lack of its effect for induction of interleukin (IL)-8 secretion in AGS gastric epithelial cells. A, Bacterial whole-cell sonicates (lanes 1 and 2) and supernatants (filter-concentrated [lanes 5 and 6] and unconcentrated [lanes 7 and 8]) from broth-grown H. pylori wild-type (wt) strain 60190 (lanes 1, 5, and 7) and its isogenic flaA-mutant derivative (lanes 2, 6, and 8) were analyzed by use of Western blot, using a polyclonal antibody raised against Salmonella typhimurium flagellin. Purified S. typhimurium flagellin (lane 3) and commercially available H. pylori FlaA (Austral Biologicals; lane 4) were used as positive controls for antibody recognition. Molecular mass of native H. pylori FlaA is ∼54 kDa, which is glycosylated within the H. pylori flagellar filament by neuA/flmD, a bicistronic operon [56]. Recombinant FlaA (Austral Biologicals) was produced in genetically engineered E. coli, and its molecular weight is ∼51 kDa. It is likely that E.coli lack the ability to glycosylate recombinant FlaA, which leads to the production of a lower molecular-weight protein than that for native H.pylori FlaA. Experiments were performed at least twice; arrow, H. pylori FlaA. B, AGS cells were incubated with or without H. pylori wt strain 60190 (solid symbols) or an isogenic 60190 flaA-mutant (open symbols) at varying cell:bacteria concentrations (1:1, 1:10, and 1:100) for 3, 6, and 24 h, and IL-8 production in coculture supernatants was quantified by ELISA. Data represent at least 3 independent experiments performed in triplicate and are expressed as mean levels of IL-8 release relative to AGS control cells that were incubated in tissue-culture medium without H. pylori. Bars, SD.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/189/10/10.1086/386289/2/m_189-10-1914-fig002.gif?Expires=1716383595&Signature=i5fFh7nrq6iHpcwTZ~0mnOxUbjYIatnV6UL99DCVhRnHIR9yC1MPYV~Dhno26Ci~4Q47yW7Zz~y1crFC4LA-tR0vnNPxjMh0fJxa9LaYI4xcqhow-v-CP1zOFAqo9tca7VPH5kr79vqWui8tpdGBjgz50fNXGURGsfn6Mrgp7TU-7Q00yD2g1ZHkVInKKvGUIIC~T6lukJUvS5Vq-PbhkRVQ34WW4ssPYppxNmRalF57dTO9SuL8woGzR~alswAWwVbj3FPKljuudju1xdJVRArxBexIUblQ3iZhCZ6URUTVoykuLUQuGlw6Ni3rUh6lptIrQaMmNBrtP-UZ3becPg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Absence of Helicobacter pylori FlaA in conditioned medium and lack of its effect for induction of interleukin (IL)-8 secretion in AGS gastric epithelial cells. A, Bacterial whole-cell sonicates (lanes 1 and 2) and supernatants (filter-concentrated [lanes 5 and 6] and unconcentrated [lanes 7 and 8]) from broth-grown H. pylori wild-type (wt) strain 60190 (lanes 1, 5, and 7) and its isogenic flaA-mutant derivative (lanes 2, 6, and 8) were analyzed by use of Western blot, using a polyclonal antibody raised against Salmonella typhimurium flagellin. Purified S. typhimurium flagellin (lane 3) and commercially available H. pylori FlaA (Austral Biologicals; lane 4) were used as positive controls for antibody recognition. Molecular mass of native H. pylori FlaA is ∼54 kDa, which is glycosylated within the H. pylori flagellar filament by neuA/flmD, a bicistronic operon [56]. Recombinant FlaA (Austral Biologicals) was produced in genetically engineered E. coli, and its molecular weight is ∼51 kDa. It is likely that E.coli lack the ability to glycosylate recombinant FlaA, which leads to the production of a lower molecular-weight protein than that for native H.pylori FlaA. Experiments were performed at least twice; arrow, H. pylori FlaA. B, AGS cells were incubated with or without H. pylori wt strain 60190 (solid symbols) or an isogenic 60190 flaA-mutant (open symbols) at varying cell:bacteria concentrations (1:1, 1:10, and 1:100) for 3, 6, and 24 h, and IL-8 production in coculture supernatants was quantified by ELISA. Data represent at least 3 independent experiments performed in triplicate and are expressed as mean levels of IL-8 release relative to AGS control cells that were incubated in tissue-culture medium without H. pylori. Bars, SD.
Induction of IL-8 secretion in AGS cells without the requirement of H. pylori flagellin. If, in contrast to S. typhimurium, H. pylori evades TLR5-mediated innate immunity, one would predict that disruption of H. pylori flagellin synthesis would have no effect on IL-8 production induced by this organism. To test this prediction, the gene encoding the major flagellin structural subunit (flaA) in H. pylori strain 60190 was isogenically inactivated, and IL-8 release from AGS cells was quantified by ELISA. Successful disruption of flaA was confirmed by immunoblotting whole-cell sonicates harvested from H. pylori wt and flaA-mutant strains (figure 2A) and by demonstrating loss of motility of the flaA-derivative in soft agar (data not shown). Inactivation of flaA did not result in an impaired ability to induce IL-8 secretion, compared with the wt strain, at any concentration or time point tested (figure 2B), which indicates that, indeed, flagellin does not play a significant role in mediating epithelial proinflammatory gene expression induced by this organism.
Weak activation of TLR5 by purified H. pylori FlaA. Although flagellin may not be readily shed by H. pylori, it may be released under certain conditions, such as bacterial autolysis, which is analogous to the TLR9 ligand CpG DNA [27]. Therefore, we next investigated whether H. pylori flagellin could activate TLR5-mediated proinflammatory gene expression when given direct access to this receptor. Recombinant epitope-tagged H. pylori flagellin (FlaA) and S. typhimurium flagellin (FliC) were added to T84 intestinal epithelial cells that express endogenous TLR5 [6], and IL-8-inducing bioactivities were compared (figure 3A). S. typhimurium FliC, similar to flagellin purified from native S. typhimurium, tpotently activated IL-8 secretion, with half maximal concentration of ∼0.5 ng/mL and a maximal IL-8 secretion response of ∼6 ng/106 epithelial cells. In contrast, the EC50 for recombinant H. pylori FlaA was ∼3 logs higher, in conjunction with a 4-fold decrease in maximal induction of IL-8 secretion at saturating concentrations (figure 3A).
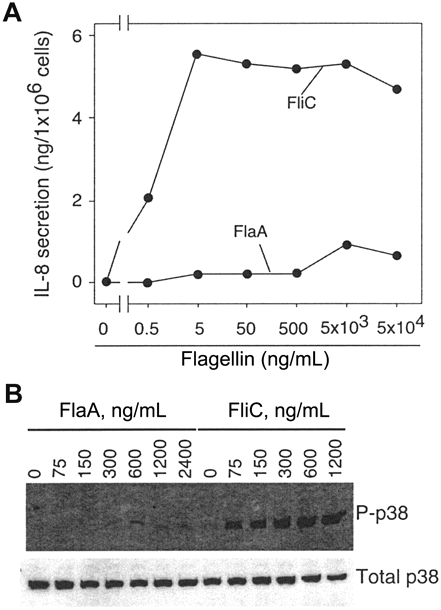
Induction of an attenuated Toll-like receptor 5 (TLR5)-mediated interleukin (IL)-8 response by purified Helicobacter pylori FlaA. A, Purified recombinant epitope-tagged FlaA and FliC from H. pylori and Salmonella typhimurium, respectively, were added at increasing concentrations to T84 intestinal epithelial cells that express endogenous TLR5 and IL-8 production in coculture supernatants was quantified by ELISA. Data are expressed as nanograms of IL-8 secretion/ 1 × 106 epithelial cells. B, MDCK cells engineered to stably express TLR5 were treated with indicated concentrations of H. pylori FlaA or S. typhimurium FliC and phosphorylated p38 (P-p38) was detected 30 min later by immunoblotting, using a phosphospecific anti-p38 antibody. A control Western blot, which used an antibody that recognizes total p38, is also shown.
To confirm that the differential potency of FliC and FlaA for induction of IL-8 secretion resulted from a differential ability to activate TLR5, we also examined activation of p38 mitogenactivated protein kinase (MAPK), an early signaling event that we have recently shown to result directly from FliC-mediated activation of TLR5 [26]. In response to low concentrations of S.typhimurium FliC, p38 was strongly activated in MDCK cells engineered to stably express TLR5; however, H. pylori FlaA only very weakly activated p38 (figure 3B), which indicates that the relative inability of FlaA to induce IL-8 secretion corresponds to an inability to activate TLR5.
Failure of purified H. pylori flagellin to induce IL-8 secretion or activate p38 MAPK in gastric epithelial cells. Colonization of H. pylori is specific for gastric epithelium; therefore, we sought to determine whether the ability of H. pylori flagellin to evade TLR5-mediated pathways in T84 or MDCK cells could be extended to gastric cells. Recombinant H. pylori FlaA and S. typhimurium FliC were added to AGS cells at varying concentrations, and IL-8-inducing and p38-activating properties were compared (figure 4). Similar to our observations for T84 and MDCK cells, S. typhimurium FliC potently stimulated IL-8 secretion and p38 activation, whereas the response to H. pylori FlaA was significantly attenuated (figure 4).
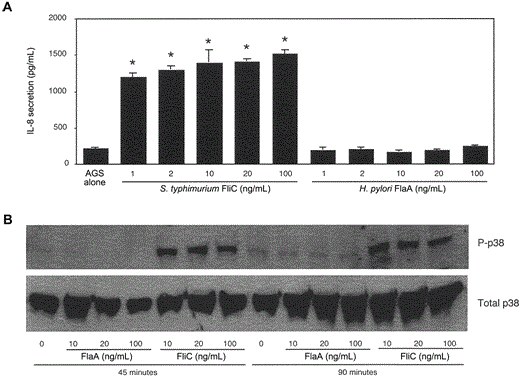
Failure of purified Helicobacter pylori FlaA to stimulate interleukin (IL)-8 secretion or p38 activation in gastric epithelial cells. A, Purified recombinant FliC and FlaA from Salmonella typhimurium and H. pylori, respectively, were added at increasing concentrations to AGS gastric epithelial cells that express endogenous Toll-like receptor 5 (TLR5), and IL-8 production in coculture supernatants was quantified by ELISA. Data represent at least 3 independent experiments that were performed in triplicate and are expressed as picograms per milliliter. Bars, SD. * P < .05, vs. AGS cells in medium alone. B, AGS cells were treated with indicated concentrations of H. pylori FlaA or S. typhimurium FliC, and phosphorylated p38 (P-p38) was detected 45 and 90 min later by immunoblotting, using a phosphospecific anti-p38 antibody. A control Western blot, which used an antibody that recognizes total p38, is also shown.
Therefore, H. pylori not only fails to release FlaA, but its flagellin is a much less potent activator of TLR5 than flagellin derived from other gram-negative microbes.
Discussion
The mucosal immune response to H. pylori is insufficient to eliminate this pathogen from the gastric niche [28], and, as a result, microbial persistence can lead to diseases, such as peptic ulceration and distal gastric cancer. The mechanisms underlying such immune evasion are not clearly delineated, because H. pylori can induce both an innate inflammatory and an adaptive immune response [1]. We have now demonstrated that H. pylori can evade TLR5-mediated innate immunity by expressing a flagellin that is required for the establishment and maintenance of gastric colonization, but that is inherently less proinflammatory than that derived from other gram-negative enteric microbes [20,6,7,10,11,18,20,29].
Adherence of H. pylori to gastric epithelial cells is required for the induction of IL-8 secretion, and one microbial constituent that regulates secretion of this chemokine is the cag pathogenicity island, a 27-gene locus that is present in 60%– 70% of US strains [30–33]. Several cag genes encode products that possess homology to components of type IV secretion systems that export proteins, and befitting these homologies, the product of the terminal gene in the island (CagA) is trans-located into host epithelial cells after bacterial attachment, where it undergoes Src-dependent tyrosine phosphorylation and activates a eukaryotic phosphatase (SHP-2), which leads to dephosphorylation of host cell proteins and cellular morphological changes 34–43]. A CagA-independent consequence of cag island-mediated H. pylori-epithelial cell contact is induction of IL-8 secretion, which is regulated by activation of the transcription factor NF-κB and MAPKs by certain cag genes (e.g., cagE, but not cagA) [44–51]. Although IL-8 release has been linked to certain cag genes, the specific molecule that triggers epithelial proinflammatory gene expression remains undefined. Flagellin is a major determinant of IL-8 secretion that results from S. typhimurium or E. coli colonization of cultured intestinal epithelia [6,7]; therefore, we investigated whether H. pylori flagellin might play a similar role. However, our findings indicate that IL-8 production is not regulated by H. pylori flagellin, and, because contact-mediated responses appear to be inherently less robust than those mediated by TLR-mediated detection of soluble products [52], these observations are consistent with the chronicity of H. pylori colonization.
The absence of flagellin monomers in H. pylori supernatants is consistent with previous proteome analyses of secreted H. pylori proteins [53] and with ultrastructural studies, which demonstrated that the flagellum from this organism is cloaked by a sheath that is an extension of the bacterial outer membrane [54]. Although this sheath was originally postulated to protect the acid-labile flagellar structure from gastric contents [54], our results suggest that it also contributes to the ability of H. pylori to evade TLR5-mediated immunity by preventing the release of a potentially immunogenic, albeit less proinflammatory, protein.
It is apparent that the regulation of chronic inflammation by H. pylori is governed by levels of host-bacteria equilibria that are not found during cellular interactions with acute enteric pathogens. Our observation that, unlike other enteric bacteria, the primary flagellar structural component of H. pylori does not trigger TLR5-mediated immunity may underlie the failure of the host to rapidly clear this pathogen. Although innate immunity and TLRs, in particular, are often thought of as playing an early role in the establishment of infection, TLR4-deficient mice only exhibited a decreased ability to control physiologic levels of M. tuberculosis months after colonization [55]. Therefore, it is possible that failure of the adaptive immune system to clear H. pylori may reflect inadequate innate immune recognition.
In conclusion, although gastric epithelial cells can functionally respond to S. typhimurium flagellin monomers, the primary flagellar structural component of H. pylori is not released and is substantially less proinflammatory than flagellin from S. typhimurium, which indicates that H. pylori harbors 2 distinct mechanisms for avoiding TLR5-mediated immunity. Such evasion of TLR4-and TLR5-mediated immunity may contribute to the ability of H. pylori to persist and cause disease within the gastric niche for the virtual lifetime of its cognate host.
Presented in part: Digestive Diseases Week, Orlando, 17–22 May 2003 (abstract W907).
Financial support: National Institutes of Health (grants DK-02792 and DK-061417 to ATG; grants DK-58587 and CA-77955 to RMP); Medical Research Service of the Department of Veterans Affairs (to R.M.P.); Crohn's and Colitis Foundation of America (to A.T.G.).
References
Author notes
A.T.G. and Y.Y. contributed equally to this work.


{kind=link}
{kind=link}
{kind=link}
{kind=link}