




Abstract
Muir-Torre syndrome (MTS) is currently considered as a clinical variant of Lynch syndrome (LS). The clinical significance of the screening of patients with MTS-associated cutaneous tumors for the identification of LS has not yet been established. In addition, the prevalence and molecular characteristics of mismatch repair (MMR) protein deficiency in such tumors has scarcely been investigated in the Japanese population.
Immunohistochemistry (IHC) for MMR proteins (MLH1, MSH2, MSH6 and PMS2) was performed in formalin-fixed paraffin-embedded sections prepared from 16 sebaceous neoplasms (SNs) resected from 13 patients and 32 keratoacanthomas (KAs) resected from 31 patients at our institution between January 2005 and March 2014. Tumors showing MMR protein loss were further subjected to genetic analysis for detecting the presence of germline and/or somatic alterations of the MMR genes to identify the precise molecular mechanisms underlying the protein loss.
Among the 16 SNs resected from 13 patients, eight SNs resected from five patients (38.5%) showed loss of expression of MMR proteins (MLH1/PMS2 loss, one patient; MSH2/MSH6 loss, four patients). Genetic analyses showed a pathogenic germline MSH2 mutation in one patient, somatic hypermethylation of the MLH1 promoter region in one patient, and somatic alterations of MSH2 without detectable germline mutations of MSH2 in three patients. None of the KAs examined in the study showed any loss of MMR protein expression.
The efficacy of routine screening of cutaneous neoplasms known to be associated with MTS by IHC for MMR proteins to identify LS may be fairly limited. MMR protein loss as determined by IHC in SNs is not always diagnostic of LS, and appears, in most cases, to be a result of somatic inactivation of the MMR genes.
Introduction
Lynch syndrome (LS), previously termed hereditary non-polyposis colorectal cancer (CRC) (1), is a hereditary autosomal dominant disorder of cancer susceptibility that accounts for 0.5–5% of all cases of CRC (2), and is caused by germline mutations in one of the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2) (3), or 3′deletion of EPCAM, upstream of MSH2 (4). Individuals with LS are at an increased risk of developing CRC as well as other malignant tumors, including endometrial cancer (EC), ovarian cancer, gastric cancer, small intestinal cancer, bile duct cancer, pancreatic cancer, renal pelvic/ureteral cancer, brain tumor, as well as cutaneous tumors such as sebaceous neoplasms (SNs) and keratoacanthomas (KAs) (5).
Muir-Torre syndrome (MTS) is characterized by synchronous or metachronous occurrence of SNs and/or KAs, associated with at least one LS-related internal cancer, and is currently considered to be a clinical variant of LS. From a historical point of view, a British surgeon, Muir, was the first to report an association between multiple gastrointestinal tumors and facial KAs in 1967 (6). Then, in 1968, a US dermatologist, Torre, also reported the existence of an association between multiple sebaceous tumors and internal malignancies (7). In 1981, Lynch recognized the association of cutaneous lesions with what later came to be known as LS, and hypothesized that MTS was a subtype of the ‘cancer family syndrome,’ or LS (8). The diagnosis of MTS has traditionally been made based on the distinct clinical features; the diagnosis is made in patients who have a positive family history of MTS, with concurrent or sequential documentation of at least one SN plus a minimum of one visceral malignancy (9), or multiple KAs plus at least one visceral malignancy (10). Some patients with MTS have been definitely found to have germline mutations of MSH2 or MLH1 (11,12); some with MSH6 germline mutations have also been reported (13). In 2014, a clinical scoring system to predict MTS in patients with SNs was proposed by the Mayo Clinic (14).
IHC for analyzing MMR protein expression is becoming increasingly more popular than microsatellite instability (MSI) testing as a screening method for defective MMR (dMMR)-CRC (15), as well as other LS-associated tumors, including SNs/KAs (16–23). Both tests have been reported to have similar sensitivities (80–91%) and specificities (88–90.2%) (24) in CRC specimens. IHC offers the distinct advantage of cost-effectiveness and is also widely available in many institutions, as compared to MSI testing. One other additional advantage of IHC is that the MMR gene that is likely to be mutated can be pinpointed.
In western countries, universal tumor screening (UTS) of CRC specimens for the expression of MMR proteins by IHC and/or MSI testing has been proposed, and is actually widely undertaken to identify patients with LS among patients with CRC and EC; it is reported that 1–5% of CRCs and 1–5% of ECs are related to LS (25). The UTS approach for CRCs has a better sensitivity than clinical criteria for identifying patients with LS, and has the potential to be cost-effective if individuals and their relatives at risk can be identified and screened to reduce the morbidity and mortality (26). In line with the experience with CRC and EC, routine screening of MTS-associated tumors (SNs and/or KAs) for dMMR has also been proposed in order to identify patients with LS (18). However, until date, there is still a lack of sufficient information on the results of testing for germline mutations of the MMR genes in MTS patients with dMMR tumors, which makes the efficacy of the UTS approach difficult to evaluate. To the best of our knowledge, no comprehensive or systematic approach has been established until date to identify LS among patients with SNs or KAs in the Japanese population.
Recently, somatic mutations, in the absence of germline mutations, of the MMR genes, have been revealed as a novel mechanism for MMR protein deficiency in tumors in a subset of patients initially suspected as having LS (27). Such patients have been labeled as having ‘Lynch-like syndrome’ or as ‘Lynch syndrome mimics.’ Analysis for such mutations has been conducted in CRCs and ECs, but only a limited report (28) has documented the possible existence of such a condition among patients with MTS-associated cutaneous tumors. For example, biallelic germline inactivation of the MUTYH genes, which is known to be associated with MUTYH-associated polyposis, has been shown in SNs with loss of MSH2 as one of possible causes of somatic MMR gene inactivation in the absence of germline MMR gene mutations (29).
This single-institutional study was aimed at clarifying the significance of immunohistochemical screening of SN(s)/KA(s) for MMR proteins to identify cases of LS among patients with MTS-associated cutaneous neoplasms in a Japanese hospital-based population. In addition, the prevalence of MMR protein deficiency and the molecular mechanisms underlying MMR protein deficiency in such cutaneous tumors were comprehensively examined.
Patients and methods
Ethical considerations
This study was conducted with the approval of the Local Ethics Committee of Saitama Medical Center (No. 924, 925, 926 and 1355) and Saitama Medical University (No. 592 and 747). Among 13 patients with SNs and 31 patients with KAs, we offered genetic counseling to the patients with dMMR-SNs or -KAs prior to genetic testing for suspected LS. For the genetic testing of the MMR genes, informed consent was obtained from each patient.
Patients
The subjects of this study were consecutive patients who had undergone surgical resection for histologically proven SNs or KAs at Saitama Medical Center, Saitama Medical University, between January 2005 and March 2014. The patients were selected by searching the archives of the Department of Pathology at our institution. Data on the clinicopathological characteristics, as well as the personal and family history of the patients were extracted from the medical charts of the patients. Concerning the diagnosis of SNs, we referred to the report by Ponti and associates (16). KA is a distinctive, benign skin tumor that is characterized by initially rapid growth over several months followed by gradual involution; the most important microscopic feature is the characteristic features of the lesions on cross section, namely, overhanging edges, keratin-filled craters, and a hemispheric shape. The histologic diagnoses of the SNs were reevaluated by an expert pathologist (E.A.) with specialized knowledge of cutaneous tumors who was blinded to the MMR status of the tumors.
IHC for the MMR proteins
Formalin-fixed paraffin-embedded (FFPE) sections (4-μm-thick) were prepared from each resected specimen. The sections were deparaffinized and washed with water, and antigen retrieval was conducted, except for the case of PMS2, by autoclaving the slides in 0.01 M citrate buffer (pH 6.2) for 20 min; for the case of PMS2, the slides were immersed in 0.01 M EDTA (pH 8.0). The Leica IHC kit (NovoLink Polymer Detection System RE7150-K RE7150-CE, Leica Biosystems Newcastle Ltd) and the following mouse-monoclonal antibodies were used for the IHC: anti-hMLH1(clone ES05, Leica Biosystems Newcastle Ltd, Newcastle, UK, 1:150), anti-hMSH2 (clone 25D12, Leica Biosystems Newcastle Ltd, 1:150), anti-hMSH6 (clone PU29, Leica Biosystems Newcastle Ltd, 1:100), and anti-hPMS2 (clone M0R4G, Leica Biosystems Newcastle Ltd, 1:40).
Tumors that showed total absence of nuclear staining, with the adjacent normal tissue showing normal nuclear staining, were regarded as being ‘negative’ for MMR protein expression. Normal cutaneous cells adjacent to the tumor, lymphoid cells and stromal cells served as the internal positive controls. Given the biologic interactions of MLH1 with PMS2, and of MSH2 with MSH6, loss of one protein in the pair is often associated with the loss of the other.
All the staining results were evaluated by two investigators (K.K. and O.S.) under the supervision of a single pathologist (J.T.).
DNA and RNA isolation
Genomic DNA for genetic testing was extracted from the peripheral blood, and/or FFPE specimens were prepared from the resected tumors and corresponding normal cutaneous tissue, where necessary, using the QIA amp DNA blood Kit (Qiagen GmbH, Hilden, Germany) and/or QIA amp Tissue Kit (Qiagen), respectively, according to the manufacturer’s instructions. RNA was isolated from the peripheral blood cells treated or not treated with puromycin using the QIAamp RNA Blood Mini kit or RNeasy mini kit (Qiagen).
Detection of germline mutations, copy number variations and altered transcripts of the MMR genes
Germline sequence analysis of each exon and flanking regions of the MMR genes were performed by the Sanger sequencing method, as described previously (30). When no deleterious (pathogenic) mutations were detected by Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA) was performed for analyzing copy number alterations of the exons of MLH1, MSH2 and EPCAM, using the Salsa® MLPA® kit P-003 (MRC-Holland, Amsterdam, Netherlands).
The identified variants were assessed by the InSiGHT classification criteria (31), and variants categorized into Class 4 or Class 5 were considered to be deleterious (pathogenic). The germline alterations are described according to the recommendation of the Human Genome Variation Society (http://www.HGVS.org/varnomen).
When no germline mutations of the MMR genes were detected in the DNA samples, RNA sequencing was undertaken to search for structural alterations of the MMR genes. After synthesis of cDNAs with oligo-dT primer using the SuperScript First-strand Synthesis system (Thermo Fisher Scientific, Waltham, MA, USA), the coding DNA regions of the MMR genes were amplified by polymerase chain reaction (PCR) and subjected to library preparation using the Nextera XT Sample Prep kit (Illumina, Santa Clara, USA), according to the manufacturer’s protocol. The library was run on the MiSeq sequencer (Illumina). Otherwise, the prepared RNA was transcribed using the reverse-transcriptase ReverTraAce (TOYOBO), and the MMR (MLH1 or MSH2) cDNAs were amplified using the appropriate primer sets for each gene. Sanger sequencing analysis of the PCR products was performed.
Detection of germline mutations of MUTYH
In cases where no pathogenic MSH2 mutations were detected, analysis was also conducted for germline mutations of the MUTYH gene by Sanger sequencing using the appropriate primer sets, as described by López-Villar et al. (32).
Analysis for somatic hypermethylation of the MLH1 promoter region
To detect epigenetic inactivation of the MLH1 gene in case of loss of expression of MLH1/PMS2 protein in the absence of pathogenic germline mutations of MLH1, we evaluated FFPE specimens of the tumors for hypermethylation of the MLH1 promoter C-region. After laser microdissection of the tumor cells using Leica LMD 7000 (Leica Microsystems GmbH, Wetzlar, Germany), DNA was isolated from the collected tissue samples. Bisulfite conversion of the extracted DNA was performed using the innuCONVERT Bisulfite Basic Kit (AJ Innuscreen GmbH, Berlin, Germany). The promoter C-region of MLH1 was amplified, as described previously (30), and Sanger sequencing was performed. When only a cytosine residue signal was observed at the CpG sites in the MLH1 promoter C-region, the sample was classified as being ‘hypermethylated’, while when only a thymine residue signal was observed at the CpG sites, the sample was classified as being ‘unmethylated’.
Analysis for somatic alterations of the MSH2 gene in the tumor tissues
To analyze the molecular mechanisms underlying the MSH2/MSH6 protein loss in the tumors in the absence of pathogenic germline mutations of the MSH2 gene, full-sequence analysis and detection of copy number variations in the tumor cells were undertaken, as described previously (30).
Results
Patient characteristics
The clinicopathological features of the patients included in this study are summarized in Table 1. There were 16 SNs resected from 13 patients, including 12 patients with one SN each and one patient who underwent independent resections for four SNs during the specified period. The median age at diagnosis (age at the time of diagnosis of the first SN was considered for the analysis in the patient with multiple lesions) of the SNs was 70.5 years (range, 43–90 years). The male: female ratio was 7:6. The median maximal size of the tumor was 13 mm (range, 4–59 mm). The tumors in all the cases were located on the head/face. The histological diagnoses of the 16 SNs were as follows: sebaceous adenoma, two cases; sebaceoma (sebaceous epithelioma), three cases; sebaceous carcinoma, 11 cases. In addition, there were 32 KAs resected from 31 patients, including 31 patients with one KA each and one patient who underwent resection for an additional metachronous lesion during the specified period. The median age at diagnosis of the KA was 72 years (range, 42–98 years). The male: female ratio was 12:19. The median maximal size of the tumor was 9.5 mm (range, 3–45 mm). The tumor was located on the head/face in 27 cases (84.4%), in the neck in one case (3.1%), and at other sites in four cases (12.5%). Past history of malignant tumor was recorded in one (8%) patient with a SN, and four (12.5%) patients with KAs; these tumors included prostate cancer in one patient, breast cancer in two patients, colon cancer in one patient, primary peritoneal cancer in one patient, and malignant lymphoma in one patient. No clinically significant family history was recorded in the majority of cases.
Demographic and clinicopathological characteristics of patients with sebaceous neoplasms and keratoacanthomas
| Sebaceous neoplasms (N = 13) | Keratoacanthomas (N = 31) | |
|---|---|---|
| Age at diagnosisa (years) | 70.5 (43–90) | 72 (42–98) |
| Gender (Male: Female) | 7:6 | 12:19 |
| Number of lesions | 16 | 32 |
| Maximal tumor diameter* (mm) | 13 (4–59) | 9.5 (3–45) |
| Sites of lesions | ||
| Head/Face | 16 | 27 |
| Neck | 0 | 1 |
| Others | 0 | 4 |
| Histology | ||
| Sebaceous adenoma | 2 | — |
| Sebaceoma (sebaceous epithelioma) | 3 | — |
| Sebaceous carcinoma | 11 | — |
| Sebaceous neoplasms (N = 13) | Keratoacanthomas (N = 31) | |
|---|---|---|
| Age at diagnosisa (years) | 70.5 (43–90) | 72 (42–98) |
| Gender (Male: Female) | 7:6 | 12:19 |
| Number of lesions | 16 | 32 |
| Maximal tumor diameter* (mm) | 13 (4–59) | 9.5 (3–45) |
| Sites of lesions | ||
| Head/Face | 16 | 27 |
| Neck | 0 | 1 |
| Others | 0 | 4 |
| Histology | ||
| Sebaceous adenoma | 2 | — |
| Sebaceoma (sebaceous epithelioma) | 3 | — |
| Sebaceous carcinoma | 11 | — |
aMedian (range).
Demographic and clinicopathological characteristics of patients with sebaceous neoplasms and keratoacanthomas
| Sebaceous neoplasms (N = 13) | Keratoacanthomas (N = 31) | |
|---|---|---|
| Age at diagnosisa (years) | 70.5 (43–90) | 72 (42–98) |
| Gender (Male: Female) | 7:6 | 12:19 |
| Number of lesions | 16 | 32 |
| Maximal tumor diameter* (mm) | 13 (4–59) | 9.5 (3–45) |
| Sites of lesions | ||
| Head/Face | 16 | 27 |
| Neck | 0 | 1 |
| Others | 0 | 4 |
| Histology | ||
| Sebaceous adenoma | 2 | — |
| Sebaceoma (sebaceous epithelioma) | 3 | — |
| Sebaceous carcinoma | 11 | — |
| Sebaceous neoplasms (N = 13) | Keratoacanthomas (N = 31) | |
|---|---|---|
| Age at diagnosisa (years) | 70.5 (43–90) | 72 (42–98) |
| Gender (Male: Female) | 7:6 | 12:19 |
| Number of lesions | 16 | 32 |
| Maximal tumor diameter* (mm) | 13 (4–59) | 9.5 (3–45) |
| Sites of lesions | ||
| Head/Face | 16 | 27 |
| Neck | 0 | 1 |
| Others | 0 | 4 |
| Histology | ||
| Sebaceous adenoma | 2 | — |
| Sebaceoma (sebaceous epithelioma) | 3 | — |
| Sebaceous carcinoma | 11 | — |
aMedian (range).
IHC for the MMR proteins
The flowchart for the screening of patients for identification of LS is shown in Fig. 1. Of the 13 patients with 16 SNs enrolled in this study, five (38.5%) patients (including the patient with four SNs) showed loss of expression of the MMR proteins. While one of the patients showed loss of MLH1 and PMS2, the remaining four (including the patient with four SNs) showed loss of expression of both MSH2 and MSH6 (Fig. 2). On the other hand, none of the KAs showed loss of expression of any of the MMR proteins.
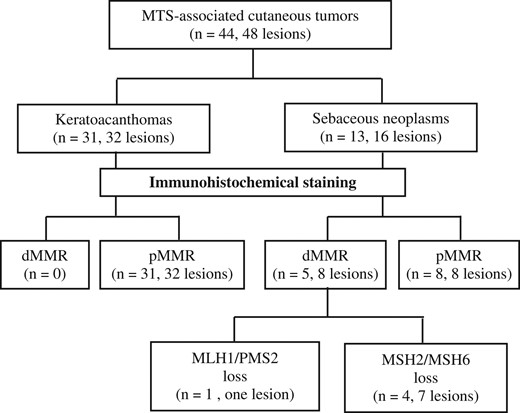
The flowchart for screening of patients with sebaceous neoplasms or keratoacanthomas for identifying LS using an immunohistochemical staining method. dMMR, defective MMR; pMMR, proficient MMR.
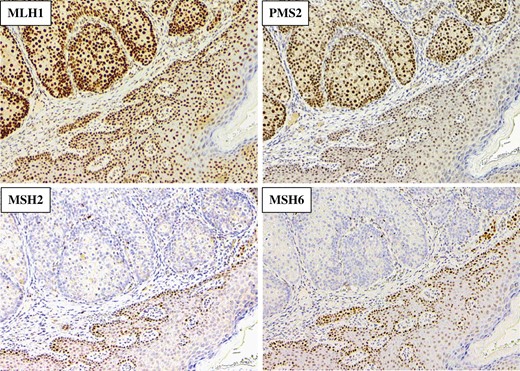
Representative immunohistochemical staining of MMR protein expression in the cancer tissue of the patient (Case 1) with loss of MSH2/MSH6. (magnification ×100).
Evaluations for germline and somatic alterations of the MMR genes
The results of evaluation for germline and somatic alterations of the MMR genes in the five patients with dMMR-SNs and the clinicopathological features of these patients are summarized in Table 2.
Tests for germline MMR mutations
Genetic testing of the MMR genes was carried out in all of the five patients who showed loss of MMR protein expression. Of these five patients, a pathogenic MSH2 mutation (NM_000 251.2(MSH2):c.942+3 A>T, p.Val 265_Gln314del) was identified in only one patient, a 67-year-old woman (Case 1). None of the remaining four patients were found to show any pathogenic or structural variations of the MMR genes using the MLPA method or RNA sequence.
Clinicopathological characteristics and evaluations of germline and somatic alterations of MMR genes in five SN patients with dMMR
| Case No. | Age | Gender | Site | Maximal diameter (mm) | Histology | Loss of MMR protein by IHC | Fulfillment of screening criteria | Germline and somatic alterations of MMR genes | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Revised Amsterdam | Revised Besethda | Germline mutation for MMRs and/or MUTYH | Class | Somatic alterations | Class | |||||||
| 1 | 67 | Female | Top of nose | 12 | SCA | MSH2/MSH6 | (-) | ○ | MSH2: c.942+3 A>T (p.Val265_Gln314del) | 5 | Not performed | – |
| 2 | 74 | Male | Left-sided forehead | 5 | SAD | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon3–4 duplication | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| 3 | 66 | Male | Right-sided nose | 30 | SEP | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon1 duplication | 5 |
| MSH2: c.2134 G > C(p.Val712Ile) | 3 | |||||||||||
| 4 | 43 | Male | Top of nose | 14 | SCA | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: c.1801C>T(p.Gln601*) | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| MSH2: c.1571 G>T(p.Arg524Leu) | 3 | |||||||||||
| MSH2: exon14–15 deletion | 5 | |||||||||||
| 51 | Left-sided spat | 4 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 52 | Left-sided nosal wing | 7 | SCA | MSH2/MSH6 | MSH2: c.2645 A>T(p.Lys882Ile) | 3 | ||||||
| 52 | Right-sided nosal wing | 12 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 5 | 66 | Male | Right-sided cheek | 12 | SEP | MLH1/PMS2 | (-) | (-) | Not detected (MLH1) | – | Hypermethylation of MLH1 promoter | – |
| Case No. | Age | Gender | Site | Maximal diameter (mm) | Histology | Loss of MMR protein by IHC | Fulfillment of screening criteria | Germline and somatic alterations of MMR genes | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Revised Amsterdam | Revised Besethda | Germline mutation for MMRs and/or MUTYH | Class | Somatic alterations | Class | |||||||
| 1 | 67 | Female | Top of nose | 12 | SCA | MSH2/MSH6 | (-) | ○ | MSH2: c.942+3 A>T (p.Val265_Gln314del) | 5 | Not performed | – |
| 2 | 74 | Male | Left-sided forehead | 5 | SAD | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon3–4 duplication | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| 3 | 66 | Male | Right-sided nose | 30 | SEP | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon1 duplication | 5 |
| MSH2: c.2134 G > C(p.Val712Ile) | 3 | |||||||||||
| 4 | 43 | Male | Top of nose | 14 | SCA | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: c.1801C>T(p.Gln601*) | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| MSH2: c.1571 G>T(p.Arg524Leu) | 3 | |||||||||||
| MSH2: exon14–15 deletion | 5 | |||||||||||
| 51 | Left-sided spat | 4 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 52 | Left-sided nosal wing | 7 | SCA | MSH2/MSH6 | MSH2: c.2645 A>T(p.Lys882Ile) | 3 | ||||||
| 52 | Right-sided nosal wing | 12 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 5 | 66 | Male | Right-sided cheek | 12 | SEP | MLH1/PMS2 | (-) | (-) | Not detected (MLH1) | – | Hypermethylation of MLH1 promoter | – |
SCA, sebaceous carcinoma; SAD, sebaceous adenoma; SEP, sebaceous epithelioma.
Clinicopathological characteristics and evaluations of germline and somatic alterations of MMR genes in five SN patients with dMMR
| Case No. | Age | Gender | Site | Maximal diameter (mm) | Histology | Loss of MMR protein by IHC | Fulfillment of screening criteria | Germline and somatic alterations of MMR genes | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Revised Amsterdam | Revised Besethda | Germline mutation for MMRs and/or MUTYH | Class | Somatic alterations | Class | |||||||
| 1 | 67 | Female | Top of nose | 12 | SCA | MSH2/MSH6 | (-) | ○ | MSH2: c.942+3 A>T (p.Val265_Gln314del) | 5 | Not performed | – |
| 2 | 74 | Male | Left-sided forehead | 5 | SAD | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon3–4 duplication | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| 3 | 66 | Male | Right-sided nose | 30 | SEP | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon1 duplication | 5 |
| MSH2: c.2134 G > C(p.Val712Ile) | 3 | |||||||||||
| 4 | 43 | Male | Top of nose | 14 | SCA | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: c.1801C>T(p.Gln601*) | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| MSH2: c.1571 G>T(p.Arg524Leu) | 3 | |||||||||||
| MSH2: exon14–15 deletion | 5 | |||||||||||
| 51 | Left-sided spat | 4 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 52 | Left-sided nosal wing | 7 | SCA | MSH2/MSH6 | MSH2: c.2645 A>T(p.Lys882Ile) | 3 | ||||||
| 52 | Right-sided nosal wing | 12 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 5 | 66 | Male | Right-sided cheek | 12 | SEP | MLH1/PMS2 | (-) | (-) | Not detected (MLH1) | – | Hypermethylation of MLH1 promoter | – |
| Case No. | Age | Gender | Site | Maximal diameter (mm) | Histology | Loss of MMR protein by IHC | Fulfillment of screening criteria | Germline and somatic alterations of MMR genes | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Revised Amsterdam | Revised Besethda | Germline mutation for MMRs and/or MUTYH | Class | Somatic alterations | Class | |||||||
| 1 | 67 | Female | Top of nose | 12 | SCA | MSH2/MSH6 | (-) | ○ | MSH2: c.942+3 A>T (p.Val265_Gln314del) | 5 | Not performed | – |
| 2 | 74 | Male | Left-sided forehead | 5 | SAD | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon3–4 duplication | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| 3 | 66 | Male | Right-sided nose | 30 | SEP | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: Exon1 duplication | 5 |
| MSH2: c.2134 G > C(p.Val712Ile) | 3 | |||||||||||
| 4 | 43 | Male | Top of nose | 14 | SCA | MSH2/MSH6 | (-) | (-) | Not detected (MSH2, MUTYH) | – | MSH2: c.1801C>T(p.Gln601*) | 5 |
| MSH2: c.2038 C>T(p.Arg680*) | 5 | |||||||||||
| MSH2: c.1571 G>T(p.Arg524Leu) | 3 | |||||||||||
| MSH2: exon14–15 deletion | 5 | |||||||||||
| 51 | Left-sided spat | 4 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 52 | Left-sided nosal wing | 7 | SCA | MSH2/MSH6 | MSH2: c.2645 A>T(p.Lys882Ile) | 3 | ||||||
| 52 | Right-sided nosal wing | 12 | SCA | MSH2/MSH6 | MSH2: c.1571 G>T(p.Arg524Leu) | 3 | ||||||
| 5 | 66 | Male | Right-sided cheek | 12 | SEP | MLH1/PMS2 | (-) | (-) | Not detected (MLH1) | – | Hypermethylation of MLH1 promoter | – |
SCA, sebaceous carcinoma; SAD, sebaceous adenoma; SEP, sebaceous epithelioma.
Tests for germline MUTYH mutations
In three of the four patients (Case 2, Case 3 and Case 4) who showed loss of MSH2/MSH6 expression in the absence of germline mutations of MSH2, additional analysis was performed for MUTYH mutations. The results revealed no mutations, either biallelic or monoallelic, of the MUTYH gene in any of these three patients.
MLH1 promoter methylation
Analysis of the methylation status of the promoter region of MLH1 in the patient who showed loss of MLH1/PMS2 in the tumor (Case 5) demonstrated ‘hypermethylated’ of the promoter region (Supplementary Fig. 1).
Analysis for somatic alterations of MSH2
Analysis for somatic alterations involved in the loss MMR protein expression was conducted in the three patients who showed MSH2/MSH6 loss without detectable pathogenic germline MSH2 mutations (including two patients with one tumor each [Case 2 and Case 3] and one patient with four tumors [Case 4]). Sanger sequencing and MLPA demonstrated duplication of exons 3–4 and one pathogenic variant (c.2038 C>T (p.Arg680*), Class 5) in the MSH2 gene in the single tumor from Case 2, duplication of exon 1 and one variant of uncertain significance (VUS) (c.2134 G > C (p.Val712Ile), Class 3) in the single tumor from Case 3, and two pathogenic variants (Class 5), one VUS (Class 3), and deletion of exons 14–15 in the largest tumor from Case 4; the other three tumors from Case 4 showed one VUS (Class 3) each (Table 2).
Characteristics of the patient (Case 1) identified as having LS and her relatives
The pedigree of the patient (Case 1) with the pathogenic germline MSH2 mutation is shown in Fig. 3. The proband (III-1) had undergone resection for ascending colon cancer at the age of 56 years; she developed locally advanced bile duct cancer at the age of 68 years and died of this cancer without undergoing surgery. She fulfilled the revised Bethesda Guidelines, but not the revised Amsterdam Criteria for suspected LS (Table 2). The Mayo MTS risk score of the patient was 2. Four affected family members (II-9, II-10, II-11 and III-2) had LS-associated tumors. After identification of the pathogenic mutation in the MSH2 gene in this case, we carried out genetic testing of her relatives (III-2, III-3, IV-1, 2 and 3). Only her younger brother (III-2) was identified as having the same MSH2 mutation as that in the patient. He had multiple (synchronous and metachronous) bilateral renal pelvic cancers and urinary bladder cancer, all of which were treated by curative surgery, and also early colon cancer, which was treated by endoscopic resection. The patient’s mother (II-9) and aunt (II-11) had brain tumors. Another aunt (II-10) had CRC. These individuals (II-9, 10 and 11) could not undergo genetic testing, since they had already died by the time the genetic diagnosis was made in the proband.
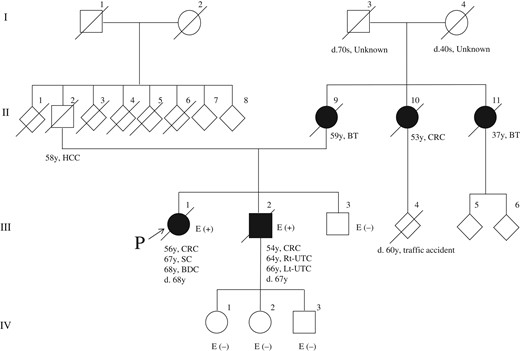
The pedigree of the patient (Case 1). BT, brain tumor; CRC, colorectal cancer; SC, sebaceous cancer; UTC, urothelial cancer; BDC, bile duct cancer; HCC, hepatocellular carcinoma; E, MSH2 genetic testing.
Discussion
Although this was a single-institutional study with a small sample size, it nonetheless seems to provide useful information about MTS-associated cutaneous neoplasms. We showed that: (1) the percentage of patients showing dMMR among the 13 patients with SNs was 38.5% (5/13); (2) only one of the patients who fulfilled the revised Bethesda Guidelines (Case 1) showed a pathogenic germline MMR gene mutation (NM_000 251.2(MSH2):c.942+3 A>T, p.Val 265_Gln314del); (3) hypermethylation of the promoter region of MLH1 was identified in the patient who showed loss of MLH1/PMS2 (Case 5); (4) all of the three patients with cancer showing deficiency of the MSH2/MSH6 in the absence of pathogenic germline mutations of MSH2 (Case 2, Case 3 and Case 4) showed at least two somatic events causing inactivation of MSH2, which may be consistent with the definition of Lynch-like syndrome, although we could not determine whether these mutations were present in the same allele or not (5) none of the patients with KAs examined in this study showed dMMR in the tumors. We shall discuss the aforementioned results, focusing separately on SNs and KAs.
Since none of the KAs examined in this study showed any loss of MMR proteins, it was concluded that screening of KAs is not useful for identifying LS. However, according to the revised Bethesda Guidelines (33) published in 2004, in CRC patients with KAs, analysis of the CRC specimen for MSI is recommended (currently, IHC for MMR protein analysis is used more often than MSI testing), to exclude the possibility of LS. On the other hand, to the best of our knowledge, there have been no reports of UTS by IHC for MMR protein analysis of KAs, even though KAs are very common in patients with MTS (up to 20%) (34). Ponti et al. (16) conducted selective screening of early-onset KA(s) and multiple (synchronous/metachronous) KAs, and found dMMR in only 2 of the 93 KAs examined. Of these two cases, one had a single KA and the other had multiple KAs. Occasionally, cases of LS with a solitary KA at the first presentation have been reported (35). Thus, we screened all consecutive patients with KAs treated by surgical resection, all of whom, except one, had solitary lesions.
Based on studies (36,37) demonstrating a high incidence of loss of MSH2 or MLH1 expression in MTS-associated cutaneous neoplasms, the results of universal or selective screening of SNs associated or not associated with KAs have been reported (Supplementary Table 1). The incidence of loss of MMR expression in the SNs varied widely from 8% to 66%, presumably depending on the method of sample selection, location of the SNs, number of anti-MMR protein antibodies used, and the histology of the SNs examined. In recently published studies (17–21,23), including the present study, four antibodies (against MLH1, MSH2, MSH6 and PMS2) were used in order not to overlook SNs with defective MSH6 or PMS2, as in the case of screening of CRCs and ECs. Among the studies that have reported the results of screening by IHC for MMR expression, genetic testing for identifying LS has been performed in only five studies, including the present study (16,18,19,22), with the reported rate of detection of pathogenic germline mutations among patients with MMR protein loss varying from 20% to 50%. Notably, dMMR-SNs are not diagnostic of LS, and up to 50% of dMMR-SNs may possibly be categorized under Lynch-like syndrome.
In the present study, no germline mutations of MSH2 were identified in three out of the four patients who showed loss of MSH2/MSH6 in the SNs. The possibility of a technical limitation causing failure of detection of large rearrangements such as inversion (38) cannot be excluded. However, this may not have been the case, since they were detected to harbor at least two somatic events causing inactivation of MSH2 in the cancer tissue, and should thus be categorized under the so-called ‘Lynch-like syndrome,’ which may strictly be defined as being characterized by biallelic inactivation of the MMR gene(s) in the absence of germline MMR gene mutations, as documented in CRCs (39). To the best of our knowledge, Joly et al. (28) were the first to describe ‘Lynch-like syndrome’ among patients with SNs. They detected somatic heterozygous alterations or multiple mutations concurrently with biallelic inactivation in seven out of 12 patients with dMMR-SNs not harboring germline MMR gene mutations, suggesting that somatic rather than germline mutations are more often responsible for a Muir-Torre syndrome-like (or Lynch-like syndrome) condition.
Recently, a clinical scoring system, called the ‘Mayo MTS risk score,’ has been proposed for identifying patients with SNs who are at risk for MTS (14). The scoring is based on four items, including the age (younger than 60 years assigned a score of 1), number of SNs (two or more assigned as score of 2), personal history of any LS-associated cancer (presence assigned a score of 1), and family history of any LS-associated cancer (presence assigned a score of 1). Absence of the respective risk factors is assigned a score of 0. They recommend evaluation for LS in SN patients with a score of two or higher. Although we did not calculate the Mayo MTS risk score in all the SN patients in the present study, the scores in Case 1 and Case 4 among the five patients with dMMR-SNs were 2 and 3, respectively, whereas the score in the remaining three patients with dMMR-SNs (Case 2, Case 3 and Case 5) was 0. Case 1 was definitively diagnosed as having MTS, with the c.942+3 A>T mutation in the MSH2 gene. Since this genotype appears to be more frequent among the pathogenic mutations of MSH2 in MTS patients, it is possibly associated with an increased risk for the MTS phenotype (14). The tumors in Case 4, who had multiple SNs, did not harbor any germline mutations of MSH2, despite the Mayo MTS risk score of 3. Therefore, the involvement of some constitutional genetic alterations other than in the MMR genes in the pathogenesis of dMMR was strongly suspected. The Mayo MTS risk score may be useful as a screening tool for MTS in a large number of SN patients, similar to the revised Amsterdam Criteria and the revised Bethesda Guidelines. However, it may not be possible to evaluate the score accurately due to the difficulty in obtaining a precise family history of LS-associated cancer owing to the trend toward nuclear families in Japan.
We carried out analysis of the MUTYH for germline mutations in three patients suspected as having ‘Lynch-like syndrome,’ but did not identify either monoallelic or biallelic MUTYH gene mutations in any of the three patients. Our study apparently had some limitations in detecting alterations of the MUTYH gene: since the amounts of DNA extracted from the FFPE specimens were not sufficient, we could not perform the MLPA test for detecting copy number variance of MUTYH. It is known that the highly variable phenotype of MUTYH-associated polyposis, which is a hereditary autosomal recessive disease, can overlap with the LS phenotype. Biallelic MUTYH mutations impair the base excision repair process and can result in somatic mutational inactivation of both MMR-alleles mimicking LS by being associated with MMR-deficient tumors (40). Morak et al. (40) analyzed the MUTYH gene in 85 ‘unresolved’ patients with tumors showing MMR loss by IHC in the absence of detectable MMR germline mutations. Biallelic MUTYH germline mutations were detected in one patient (1.2%) each with CRC, urothelial cancer and sebaceous cancer. They demonstrated two somatic G>T transversion mutations specific to MUTYH-associated polyposis in the MSH2 gene in the sebaceous cancer resulting in MMR deficiency and a loss of MSH2/MSH6 expression by IHC. Further collection of data is required to establish the association between germline MUTYH mutations and somatic MMR deficiency.
In the present study, one patient (Case 5) of sebaceous epithelioma (sebaceoma) with MLH1/PMS2 loss showed hypermethylation of the MLH1 gene promoter region. This epigenetic event is well known to be involved in the serrated neoplasia pathway in sporadic MMR-deficient CRC, and the sebaceous neoplasm was also considered to develop via an epigenetic pathway, as in some CRCs. However, since the validity of methylation analysis has not yet been established for triaging patients with SNs towards undergoing genetic testing for MLH1 germline mutations, we performed genetic testing before the methylation analysis.
In conclusion, in this single-institutional study, we examined, for the first time (even though the sample size in our study was relatively small), the significance of immunohistochemical screening of patients with SN(s)/KA(s) for MMR proteins to identify LS patients in a Japanese hospital-based population. The efficacy of routine screening of patients with MTS-associated cutaneous neoplasms, especially of SNs, may be extremely limited, since dMMR in SNs is not diagnostic of LS, but mostly suggests somatic inactivation of the MMR genes. In other words, we should note that there might be a significant proportion of patients with the so-called ‘Lynch-like syndrome’ along with those with epigenetic inactivation of MLH1 through promoter region hypermethylation among unselected patients with MTS-associated cutaneous neoplasms. Finally, it would appear from the results of this study that UTS for KAs cannot be recommended, although further investigation in a larger patient series is warranted.
Supplementary data
Supplementary data are available at Japanese Journal of Clinical Oncology online.
Acknowledgements
We thank Dr Shin-ichi Ansai, Director, Department of Dermatology, Nippon Medical School Musashi Kosugi Hospital for useful comments. We also thank Dr Tetsuo Tachikawa, Department of Cancer Prevention and Molecular Genetics, Saitama Prefectural Cancer Center, and Mrs Nao Kamae for genetic counseling and support of preparing the manuscript.
Funding
This study was supported in part by a grant supported by the Program for an Integrated Database of Clinical and Genomic Information from Japan Agency for Medical Research and Development (AMED) under Grant number JP18kk0205004.
Conflict of interest statement
None declared.
References
Author notes
Kouki Kuwabara and Okihide Suzuki Contributed equally.

{kind=link}
{kind=link}
{kind=link}