





Abstract
The modification of adenosine to inosine at the first position of transfer RNA (tRNA) anticodons (I34) is widespread among bacteria and eukaryotes. In bacteria, the modification is found in tRNAArg and is catalyzed by tRNA adenosine deaminase A, a homodimeric enzyme. In eukaryotes, I34 is introduced in up to eight different tRNAs by the heterodimeric adenosine deaminase acting on tRNA. This substrate expansion significantly influenced the evolution of eukaryotic genomes in terms of codon usage and tRNA gene composition. However, the selective advantages driving this process remain unclear. Here, we have studied the evolution of I34, tRNA adenosine deaminase A, adenosine deaminase acting on tRNA, and their relevant codons in a large set of bacterial and eukaryotic species. We show that a functional expansion of I34 to tRNAs other than tRNAArg also occurred within bacteria, in a process likely initiated by the emergence of unmodified A34-containing tRNAs. In eukaryotes, we report on a large variability in the use of I34 in protists, in contrast to a more uniform presence in fungi, plans, and animals. Our data support that the eukaryotic expansion of I34-tRNAs was driven by the improvement brought by these tRNAs to the synthesis of proteins highly enriched in certain amino acids.
Introduction
Transfer RNAs (tRNAs) are central components of the translation machinery whose function is to physically link codon information to amino acid identity. The genomic composition of tRNA genes is extremely different between archaea, bacteria, and eukarya, and tRNA gene copy number is a phylogenetic character that closely reconstitutes the canonical phylogenetic tree (Novoa et al. 2012). The selection forces that drove the evolution of tRNA populations are unclear and likely connected to a large number of variables. An important factor influencing tRNA function is posttranscriptional modifications, which are extremely abundant in tRNAs and mostly kingdom specific (Machnicka et al. 2014).
tRNAs carrying the modified nucleotide inosine at the first position of the tRNA anticodon (position 34, wobble position) (I34) are strongly enriched in eukaryotes. I34 is one of the few essential posttranscriptional modifications on tRNAs and results from the deamination of adenosine (A34) (Gerber and Keller 1999; Wolf et al. 2002; Tsutsumi et al. 2007; Zhou et al. 2014; Torres et al. 2015). Modifications in the anticodon stem-loop modulate codon–anticodon interactions (Phizicky and Hopper 2010; El Yacoubi et al. 2012; Towns and Begley 2012; Jackman and Alfonzo 2013), and I34 allows anticodons to pair with codons ending in cytidine (C), uridine (U), and adenosine (A) (Crick 1966; Torres et al. 2014). As a result, I34 modifies the tRNA pool available to certain codons, and accounting for this effect reveals an otherwise hidden correlation between codon usage and tRNA gene copy number in mammals (Novoa et al. 2012). It has been proposed that I34 improves translation fidelity and efficiency (Schaub and Keller 2002; Novoa et al. 2012), especially for genes highly enriched in codons translated by I34-tRNAs (Rafels-Ybern et al. 2015; Rafels-Ybern et al. 2018).
In bacteria, I34 modifications are catalyzed by the homodimeric enzyme tRNA adenosine deaminase A (TadA) and have historically been considered exclusive to (Wolf et al. 2002). In eukaryotes, the same modification is catalyzed by the heterodimeric enzyme adenosine deaminase acting on tRNA (ADAT), which deaminates tRNAs coding for the amino acids T, A, P, S, L, I, V, and R (TAPSLIVR) (i.e., , , , , , , , and , respectively) (Gerber and Keller 1999) (table 1). I34 is absent in archaeal tRNAs (Marck and Grosjean 2002).
tRNAs Modified with Inosine at Position 34 (I34-tRNAs) Described in Bacteria and Eukarya, and the Codons They Recognize.
| Bacteria | |
| I34-tRNAs | Codons recognized |
| CGU, CGC, CGA | |
| a | CUU, CUC, CUA |
| Eukarya | |
| I34-tRNAs | Codons recognized |
| ACU, ACC, ACA | |
| GCU, GCC, GCA | |
| CCU, CCC, CCA | |
| UCU, UCC, UCA | |
| CUU, CUC, CUA | |
| AUU, AUC, AUA | |
| GUU, GUC, GUA | |
| CGU, CGC, CGA | |
| Bacteria | |
| I34-tRNAs | Codons recognized |
| CGU, CGC, CGA | |
| a | CUU, CUC, CUA |
| Eukarya | |
| I34-tRNAs | Codons recognized |
| ACU, ACC, ACA | |
| GCU, GCC, GCA | |
| CCU, CCC, CCA | |
| UCU, UCC, UCA | |
| CUU, CUC, CUA | |
| AUU, AUC, AUA | |
| GUU, GUC, GUA | |
| CGU, CGC, CGA | |
Described in this work.
tRNAs Modified with Inosine at Position 34 (I34-tRNAs) Described in Bacteria and Eukarya, and the Codons They Recognize.
| Bacteria | |
| I34-tRNAs | Codons recognized |
| CGU, CGC, CGA | |
| a | CUU, CUC, CUA |
| Eukarya | |
| I34-tRNAs | Codons recognized |
| ACU, ACC, ACA | |
| GCU, GCC, GCA | |
| CCU, CCC, CCA | |
| UCU, UCC, UCA | |
| CUU, CUC, CUA | |
| AUU, AUC, AUA | |
| GUU, GUC, GUA | |
| CGU, CGC, CGA | |
| Bacteria | |
| I34-tRNAs | Codons recognized |
| CGU, CGC, CGA | |
| a | CUU, CUC, CUA |
| Eukarya | |
| I34-tRNAs | Codons recognized |
| ACU, ACC, ACA | |
| GCU, GCC, GCA | |
| CCU, CCC, CCA | |
| UCU, UCC, UCA | |
| CUU, CUC, CUA | |
| AUU, AUC, AUA | |
| GUU, GUC, GUA | |
| CGU, CGC, CGA | |
Described in this work.
Sequence analyses, phylogenetic studies, and structural comparisons indicate that TadA evolved from ancestral cytidine deaminases (Gerber and Keller 2001). A recent computational analysis has concluded that the evolution of TadA from cytidine deaminases (CDAs) took place before the emergence of extant bacterial phyla (Diwan and Agashe 2018). In turn, the two ADAT subunits (ADAT2 and ADAT3) evolved from TadA through gene duplication and divergence (Gerber and Keller 1999), possibly in an ancestral eukaryotic genome. This evolution was likely followed by the increase in the number of tRNAs modified to I34 (Auxilien et al. 1996; Gerber and Keller 1999; Elias and Huang 2005; Rubio et al. 2007; Torres et al. 2014, 2015; Zhou et al. 2014), although the order of these events remains unclear.
TadA and ADAT have been shown to be essential in Escherichia coli, Trypanosoma brucei, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Arabidopsis thaliana, Fusarium graminearum, and Homo sapiens (Gerber and Keller 1999; Wolf et al. 2002; Rubio et al. 2007; Tsutsumi et al. 2007; Zhou et al. 2014; Torres et al. 2015; Liu et al. 2016), but the nature of the selection forces driving the evolution from TadA to ADAT remains an open question. Given the effect of I34 upon the pairing ability of anticodons, it stands to reason that the main selection parameter driving this process should be linked to the role of I34 in translation elongation. However, ADAT has been shown to play additional specialized functions in Trypanosoma brucei such as C-to-U deamination of DNA (Gaston et al. 2007) that could also have influenced the evolution of the enzyme.
We have shown that genes coding for TAPSLIVR-rich proteins are more abundant in eukaryotic genomes than in bacterial ones. Moreover, eukaryotic genes, but not bacterial ones, coding for such proteins display a preference for codons cognate to I34-containing tRNAs (Rafels-Ybern et al. 2015,, 2018). Thus, a potential selective advantage conferred by ADAT to eukaryotes is the facilitation of the synthesis of TAPSLIVR-rich protein regions (stretches). To understand the functional parameters that drove the evolution of ADAT from TadA, we have performed a comprehensive analysis of the evolution of the I34-associated molecular machinery. This includes the characterization of the phylogenetic distribution of A34-containing tRNAs (A34-tRNAs), the phylogenetic analyses of TadA and ADAT, a genomic analysis of TAPSLIVR-codon usage in prokaryotes and eukaryotes, and the experimental characterization of the substrate specificities of TadA and ADAT in relevant species.
First, we characterized the distribution of genes coding for A34-tRNAs across all major bacterial and eukaryotic groups. This analysis provided a first approximation to the distribution of I34 in the phylogenetic tree. In agreement with a recent report (Diwan and Agashe 2018), we found that several bacterial groups lack A34-tRNAs completely. These groups include two classes considered ancestral in nature, and several species that have undergone secondary genome reductions. On the other hand, we found that some unrelated species of bacteria have expanded the diversity of A34-tRNAs in their genomes. We experimentally determined that the genome of the firmicute Oenococcus oeni has an expanded repertoire of A34-tRNAs that includes tRNAArg, tRNALeu, tRNASer, and tRNAThr. We showed that all four genes are transcriptionally active, but only tRNAArg and tRNALeu are modified to I34 under standard culture conditions. Thus, I34 tRNA expansion is not exclusive to eukaryotes, and it can begin with the emergence of unmodified A34-tRNAs.
We also found that, in eukaryotes, a division can be established between protists, which display large variability in their genomic contents of A34-tRNA genes, and fungi, plants, and animals, which remain uniform in the same regard. We report that, despite the variability seen in protists, the tRNAome of the protozoan Tetrahymena thermophila contains the same set of I34-containing tRNAs than metazoans, showing that a fully functional ADAT most likely evolved early in eukaryotic evolution.
We then developed a pipeline to identify genes coding for bona fide TadA or ADAT, and we used it to determine the phylogenetic distribution of TadA and ADAT genes in bacteria and eukaryotes, respectively. Our findings indicate that all bacterial groups that lack A34-tRNAs also lack TadA. On the other hand, we found no evidence for lateral gene transfer of the tadA gene among species with increased diversity of A34-tRNAs, nor the existence of obvious sequence modifications that may explain the changes in substrate specificity in bacterial enzymes that possibly deaminate more than one tRNA.
Finally, we characterized the proteome composition of bacterial and eukaryotic groups in terms of TAPSLIVR-rich protein abundance and codon composition of the respective genes. We found that the abundance of TAPSLIVR-rich proteins gradually increases from bacteria to protists, and to multicellular eukaryotes. This increase is mirrored by a growing preference for codons that require I34-modified tRNAs. In summary, we have dissected the evolutionary and functional transition from TadA to ADAT and the changes linked to this evolution in terms of tRNA gene composition, I34-modified tRNAome, and codon usage. We propose that the role played by I34-tRNAs in the synthesis of TAPSLIVR-rich proteins was a selection force that drove the evolution of the system.
Results
Analysis of the Phylogenetic Distribution of A34-tRNAs
As a first step to determine the distribution of I34-tRNAs, we selected 956 bacterial and 150 eukaryotic completely sequenced genomes to represent all major phylogenetic phyla of both kingdoms (see Materials and Methods). Using tRNAscan-SE software (Lowe and Eddy 1997), we systematically searched each genome to identify tRNA genes cognate to TAPSLIVR with A at the wobble position (A34-tRNAs). These A34-tRNAs are potential substrates for TadA or ADAT. We defined “A34-tRNA diversity” as the number of different tRNA isoacceptors present in a genome (fig. 1). For example, the H. sapiens genome contains A34-tRNAs cognate for the amino acids TAPSLIVR so its A34-tRNA diversity is 8. We then used a consensus phylogenetic tree (Letunic 2015) to map the distribution of A34-tRNA diversity scores (fig. 1).
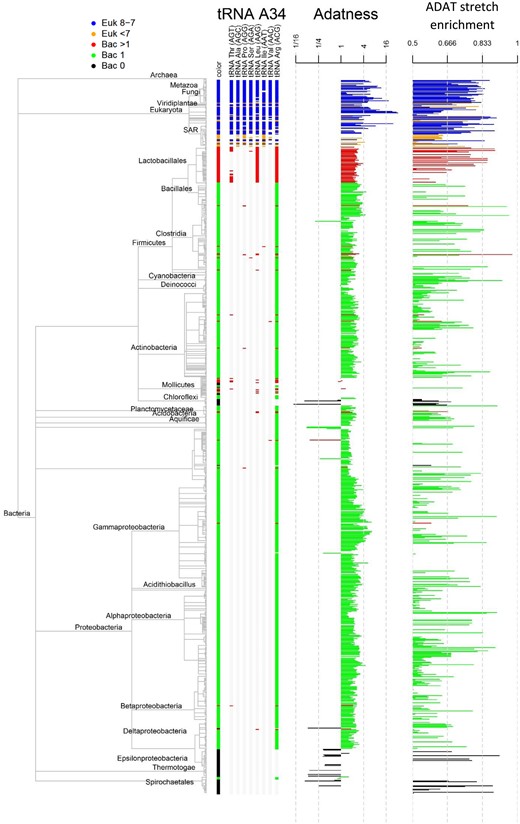
Standard phylogeny based on NCBI taxonomy (NCBI). Each final node corresponds to a species that is colored according to its tRNA diversity (legend on the top left corner). For each species, we depict its identified A34-tRNA genes (tRNA ANN), the Adatness value for ADAT2 or TadA (Adatness), and its ADAT-stretch enrichment score.
As expected, in the vast majority of bacterial phyla, A34 was only found in tRNAArg (A34-tRNA diversity = 1), consistent with the notion that TadA only deaminates this tRNA. In agreement with previous reports (Weisburg et al. 1989; Yokobori et al. 2013; Grosjean et al. 2014; Diwan and Agashe 2018), we found several species belonging to the orders of Tenericutes, ε-Proteobacteria, Chloroflexi, Spirochaetes, and Thermotogae that do not contain detectable genes coding for A34-tRNAs. Mollicutes present a massive secondary genomic reduction that drove the disappearance of and TadA (Grosjean et al. 2014). However, we confirmed a widespread lack of A34-tRNAs in bacterial groups, including Thermotogae, which do not display genome reduction and are considered deeply rooted in the bacterial phylogenetic tree (fig. 1 and table 2). These observations are compatible with an ancestral absence of A34-tRNAs in some bacteria and a secondary loss of these tRNAs in Tenericutes (Yokobori et al. 2013; Grosjean et al. 2014) (fig. 1).
Representative Species of A34-tRNA Diversity Groups.
| Organism | Class | Domain | A34 Diversity | ANN Anticodon | GNN Anticodon | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | ||||
| agu | agc | agg | aga | aag | aau | aac | acg | ggu | ggc | ggg | gga | gag | gau | gac | gcg | ||||
| Homo sapiens | Metazoa | Eukarya | 8–7 | 10 | 34 | 10 | 10 | 11 | 15 | 11 | 7 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 |
| Arabidopsis thaliana | Plantae | Eukarya | 10 | 16 | 16 | 37 | 12 | 17 | 15 | 11 | 1 | 0 | 0 | 3 | 1 | 0 | 2 | 0 | |
| Saccharomyces cerevisiae | Fungi | Eukarya | 11 | 11 | 2 | 11 | 0 | 13 | 14 | 7 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | |
| Acanthamoeba castellanii | Protist | Eukarya | 19 | 22 | 11 | 8 | 21 | 10 | 13 | 16 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Tetrahymena thermophila | SAR | Eukarya | 22 | 32 | 20 | 20 | 19 | 28 | 21 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Blastocystis hominis | SAR | Eukarya | 6 | 9 | 6 | 5 | 3 | 7 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 | |
| Phytophthora pinifolia | SAR | Eukarya | <7 | 2 | 6 | 1 | 1 | 1 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| Nannochloropsis gaditana | SAR | Eukarya | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | |
| Stramenopiles sp. | SAR | Eukarya | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Oenococcus oeni | Firmicutes | Bacteria | >1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Lactobacillus casei | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 1 | 1 | 0 | 1 | 0 | 3 | 0 | 0 | |
| Mycoplasma bovis | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Escherichia coli | Gammaprot. | Bacteria | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 2 | 2 | 1 | 1 | 1 | 4 | 2 | 0 |
| Aquifex aeolicus | Aquificae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 0 | |
| Staphylococcus aureus | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 2 | 1 | 0 | 0 | |
| Mycoplasma pneumoniae | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 1 |
| Spirochaeta thermophila | Spirochaetes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Chloroflexus aggregans | Chloroflexi | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Thermotoga maritima | Thermotogae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Organism | Class | Domain | A34 Diversity | ANN Anticodon | GNN Anticodon | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | ||||
| agu | agc | agg | aga | aag | aau | aac | acg | ggu | ggc | ggg | gga | gag | gau | gac | gcg | ||||
| Homo sapiens | Metazoa | Eukarya | 8–7 | 10 | 34 | 10 | 10 | 11 | 15 | 11 | 7 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 |
| Arabidopsis thaliana | Plantae | Eukarya | 10 | 16 | 16 | 37 | 12 | 17 | 15 | 11 | 1 | 0 | 0 | 3 | 1 | 0 | 2 | 0 | |
| Saccharomyces cerevisiae | Fungi | Eukarya | 11 | 11 | 2 | 11 | 0 | 13 | 14 | 7 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | |
| Acanthamoeba castellanii | Protist | Eukarya | 19 | 22 | 11 | 8 | 21 | 10 | 13 | 16 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Tetrahymena thermophila | SAR | Eukarya | 22 | 32 | 20 | 20 | 19 | 28 | 21 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Blastocystis hominis | SAR | Eukarya | 6 | 9 | 6 | 5 | 3 | 7 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 | |
| Phytophthora pinifolia | SAR | Eukarya | <7 | 2 | 6 | 1 | 1 | 1 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| Nannochloropsis gaditana | SAR | Eukarya | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | |
| Stramenopiles sp. | SAR | Eukarya | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Oenococcus oeni | Firmicutes | Bacteria | >1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Lactobacillus casei | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 1 | 1 | 0 | 1 | 0 | 3 | 0 | 0 | |
| Mycoplasma bovis | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Escherichia coli | Gammaprot. | Bacteria | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 2 | 2 | 1 | 1 | 1 | 4 | 2 | 0 |
| Aquifex aeolicus | Aquificae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 0 | |
| Staphylococcus aureus | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 2 | 1 | 0 | 0 | |
| Mycoplasma pneumoniae | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 1 |
| Spirochaeta thermophila | Spirochaetes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Chloroflexus aggregans | Chloroflexi | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Thermotoga maritima | Thermotogae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
Note.—For each species’ genome, the respective gene copy numbers of A34- and G34-tRNA isoacceptors for TAPSLIVR amino acids are shown.
Representative Species of A34-tRNA Diversity Groups.
| Organism | Class | Domain | A34 Diversity | ANN Anticodon | GNN Anticodon | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | ||||
| agu | agc | agg | aga | aag | aau | aac | acg | ggu | ggc | ggg | gga | gag | gau | gac | gcg | ||||
| Homo sapiens | Metazoa | Eukarya | 8–7 | 10 | 34 | 10 | 10 | 11 | 15 | 11 | 7 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 |
| Arabidopsis thaliana | Plantae | Eukarya | 10 | 16 | 16 | 37 | 12 | 17 | 15 | 11 | 1 | 0 | 0 | 3 | 1 | 0 | 2 | 0 | |
| Saccharomyces cerevisiae | Fungi | Eukarya | 11 | 11 | 2 | 11 | 0 | 13 | 14 | 7 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | |
| Acanthamoeba castellanii | Protist | Eukarya | 19 | 22 | 11 | 8 | 21 | 10 | 13 | 16 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Tetrahymena thermophila | SAR | Eukarya | 22 | 32 | 20 | 20 | 19 | 28 | 21 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Blastocystis hominis | SAR | Eukarya | 6 | 9 | 6 | 5 | 3 | 7 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 | |
| Phytophthora pinifolia | SAR | Eukarya | <7 | 2 | 6 | 1 | 1 | 1 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| Nannochloropsis gaditana | SAR | Eukarya | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | |
| Stramenopiles sp. | SAR | Eukarya | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Oenococcus oeni | Firmicutes | Bacteria | >1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Lactobacillus casei | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 1 | 1 | 0 | 1 | 0 | 3 | 0 | 0 | |
| Mycoplasma bovis | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Escherichia coli | Gammaprot. | Bacteria | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 2 | 2 | 1 | 1 | 1 | 4 | 2 | 0 |
| Aquifex aeolicus | Aquificae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 0 | |
| Staphylococcus aureus | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 2 | 1 | 0 | 0 | |
| Mycoplasma pneumoniae | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 1 |
| Spirochaeta thermophila | Spirochaetes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Chloroflexus aggregans | Chloroflexi | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Thermotoga maritima | Thermotogae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Organism | Class | Domain | A34 Diversity | ANN Anticodon | GNN Anticodon | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | Thr | Ala | Pro | Ser | Leu | Ile | Val | Arg | ||||
| agu | agc | agg | aga | aag | aau | aac | acg | ggu | ggc | ggg | gga | gag | gau | gac | gcg | ||||
| Homo sapiens | Metazoa | Eukarya | 8–7 | 10 | 34 | 10 | 10 | 11 | 15 | 11 | 7 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 |
| Arabidopsis thaliana | Plantae | Eukarya | 10 | 16 | 16 | 37 | 12 | 17 | 15 | 11 | 1 | 0 | 0 | 3 | 1 | 0 | 2 | 0 | |
| Saccharomyces cerevisiae | Fungi | Eukarya | 11 | 11 | 2 | 11 | 0 | 13 | 14 | 7 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | |
| Acanthamoeba castellanii | Protist | Eukarya | 19 | 22 | 11 | 8 | 21 | 10 | 13 | 16 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Tetrahymena thermophila | SAR | Eukarya | 22 | 32 | 20 | 20 | 19 | 28 | 21 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Blastocystis hominis | SAR | Eukarya | 6 | 9 | 6 | 5 | 3 | 7 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 1 | 4 | 0 | |
| Phytophthora pinifolia | SAR | Eukarya | <7 | 2 | 6 | 1 | 1 | 1 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| Nannochloropsis gaditana | SAR | Eukarya | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 0 | |
| Stramenopiles sp. | SAR | Eukarya | 2 | 0 | 0 | 1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Oenococcus oeni | Firmicutes | Bacteria | >1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Lactobacillus casei | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 1 | 1 | 0 | 1 | 0 | 3 | 0 | 0 | |
| Mycoplasma bovis | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| Escherichia coli | Gammaprot. | Bacteria | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 2 | 2 | 1 | 1 | 1 | 4 | 2 | 0 |
| Aquifex aeolicus | Aquificae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 0 | |
| Staphylococcus aureus | Firmicutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 2 | 1 | 0 | 0 | |
| Mycoplasma pneumoniae | Tenericutes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 1 |
| Spirochaeta thermophila | Spirochaetes | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Chloroflexus aggregans | Chloroflexi | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| Thermotoga maritima | Thermotogae | Bacteria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
Note.—For each species’ genome, the respective gene copy numbers of A34- and G34-tRNA isoacceptors for TAPSLIVR amino acids are shown.
Unexpectedly, we also detected bacterial species harboring A34-tRNAs cognate for amino acids other than arginine (A34-tRNA diversity > 1). For example, we found a gene coding for A34-tRNAIle in the genome of the Tenericute Mycoplasma bovis, and A34-tRNAs cognate for isoleucine, serine, and threonine in the Firmicute O. oeni (table 2). At first sight, the disperse distribution of bacterial species with increased diversity of A34-tRNAs may suggest that this increase took place independently in different bacterial orders.
The distribution of A34-tRNA genes among eukaryotic species also displayed significant heterogeneity. We first divided eukaryotes into four major groups: metazoa, fungi, plantae, and the rest of eukarya (hereinafter “Protists”) and found that the vast majority contain A34-tRNAs coding for TAPSLIVR (A34-tRNA diversity = 8). However, we noticed that the A34-tRNA gene diversity in Protists was extremely variable among the Stramenopiles, Alveolata, and Rizharia (SAR) group (fig. 1). For example, we identified A34-tRNA genes cognate for only four amino acid (Ala, Ser, Arg, and Ile) in the stramenopile alga Nannochloropsis gaditana, whereas we detected a full complement of A34-tRNAs cognate for TAPSLIVR in the ciliate T. thermophila (table 2). For this reason, we decided to analyze the group SAR as an additional set of eukaryotic species, in parallel with the canonical division of protists, plants, fungi, and metazoans.
Experimental Analysis of A34-Deamination in O. oeni and T. thermophila
Our computational analysis revealed bacterial species with A34-tRNA diversity higher than 1, whereas eukaryotic organisms in the SAR group displayed high heterogeneity with an A34-tRNA diversity between 0 and 8. Given the possibility that these results may be due to artifacts caused by the inability of tRNAscan-SE to correctly identify the tRNAs of interest, we set to experimentally determine the modification status of the A34-tRNAs found in the bacteria O. oeni and the SAR T. thermophila.
First, we investigated whether the four genes coding for A34-tRNAs found in O. oeni were transcribed and modified to I34. This would shed light on the potential for bacterial organisms to synthesize I34 in tRNAs other than , and on the order of events required for this substrate expansion. We purified total RNA from O. oeni and sequenced its tRNAs to determine the presence of A34 or I34 in tRNAs cognate for TAPSLIVR. I34 is read as G by reverse transcriptase, resulting in a substitution that can be detected by DNA sequencing. This analysis revealed that all four O. oeni A34-tRNAs are transcribed and showed that and are modified to I34 (supplementary fig. S1A, Supplementary Material online).
In order to unequivocally determine the presence of inosine at position 34 in O. oeni and , we used “splinted ligation-based inosine detection” (SL-ID) (Torres et al. 2018). First, total RNA from O. oeni was treated with Endonuclease V, which cleaves tRNAs after inosine. This treatment cleaves all tRNAs with I34 generating a 5′-half molecule. Next, bridge oligonucleotides were designed to specifically anneal to the cleaved 5′-halves of , , , or (supplementary table S1, Supplementary Material online) and to a 32P-labeled ligation oligonucleotide. The 5′-halves and the ligation oligonucleotide can then be ligated, resulting in RNA:DNA chimeras of approximately 49 nucleotides in length. In the absence of tRNA cleavage by Endonuclease V, no productive ligation is observed (Torres et al. 2018). Using this technique, we confirmed the presence of I34 in O. oeni and , and the lack of this modification in O. oeni and (fig. 2). This demonstrates that some bacteria have increased their repertoire of I34-tRNAs and suggests that this process of expansion begins with the emergence of A34-tRNA genes that are initially not modified to I34.
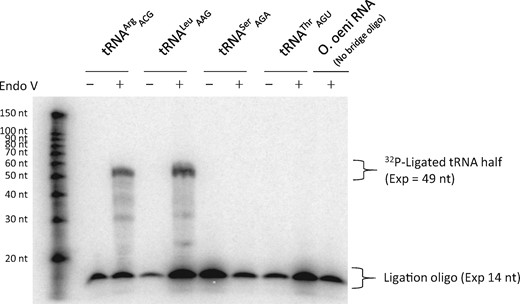
Detection of I34 in Oenococcus oeni by SL-ID using bridge oligonucleotides against , , , and . Productive I34-dependent ligation is observed for and . A reaction containing O. oeni total RNA without a bridge oligo to capture tRNA sequences was used as a negative control.
We then explored whether similar situations of coexisting I34- and A34-tRNAs take place in the unicellular eukaryote T. thermophila, which could represent an intermediate stage in the process of increasing A34-tRNA diversity from bacteria to eukaryotes. We purified and sequenced total RNA from T. thermophila. This initial analysis indicated the presence of I34 in T. thermophila tRNAs cognate for Thr, Ser, Leu, Ile, Val, and Arg (supplementary fig. S1B, Supplementary Material online). Sequencing results for the tRNAs cognate for Ala and Pro were inconclusive, but further experiments using SL-ID showed these tRNAs to also contain I34 (supplementary fig. S1C, Supplementary Material online). Thus, full modification of all A34-tRNAs for TAPSLIVR exists among SAR species. Although we cannot rule out that other species in this group contain unmodified A34-tRNAs, these results suggest that the full substrate expansion from TadA to ADAT took place early in the evolution of eukaryotic organisms.
Analysis of the Genomic Enrichment in A34-tRNA Genes
Our initial analysis confirmed that A34-tRNA diversity increases gradually from bacteria to metazoans (fig. 1). Genomic analysis indicated that deep-rooted bacterial phyla might lack A34-tRNAs completely, whereas other bacterial species may contain A34-tRNAs cognate for several amino acids. Among eukaryotes, some unicellular species of the SAR group contain a limited number of genes for A34-tRNAs, whereas others like T. thermophila host A34-tRNA genes cognate for TAPSLIVR and modify them all to I34. We have previously shown that the number of genes coding for A34-tRNAs (A34-tRNA gene copy number) dramatically increases relative to other isoacceptor tRNAs in eukaryotic genomes, a process that coincided with the increase of A34-tRNA diversity and the emergence of ADAT in these species (Novoa et al. 2012).
We asked whether the gradual variation in A34-tRNA diversity that we identified in bacteria and SAR is linked to an increase in the abundance of A34-tRNA genes relative to their respective isoacceptors. To answer this question, we quantified the total set of tRNA genes cognate for TAPSLIVR in each of the genomes of our data set, and we calculated the ratio of A34-tRNA genes to C34-, U34-, and G34-tRNA gene isoacceptors of the same 4-box family (A34-tRNA ratio) (note that Ser, Arg, an Leu have two extra tRNA isoacceptors that were also considered). Figure 3 shows a boxplot analysis where the values of A34-tRNA ratios are clustered by phyla. As expected, A34-tRNA ratios are minimal in the bacterial phyla where A34-tRNA diversity is 0. Among the rest of bacterial groups, the A34-tRNA ratios increase minimally from phyla that mostly contain species with to species that contain genes for more than one type of A34-tRNA isoacceptor. Thus, it does not appear that the increase in A34-tRNA diversity leads to a significant increase in A34-tRNA genes ratio in bacteria (fig. 3). The modest increase detected is due to the appearance of new A34-tRNAs cognate for amino acids other than arginine, rather than a relative decrease of the rest of isoacceptors (fig. 3).
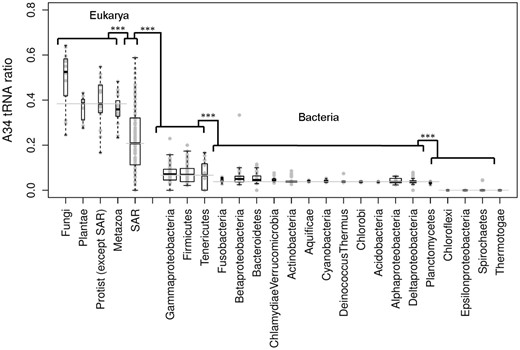
Boxplot analysis of A34-tRNA ratios for eukaryotic groups, the eukaryotic superphylum SAR, and different bacterial phyla. The y axis represents the fraction of A34-tRNAs among all tRNAs cognate for TAPSLIVR amino acids. The width of each boxplot is proportional to the number of organisms analyzed. The horizontal lines represent the median values for the sets they highlight. ***P value < 0.01.
As mentioned, the genomes of fungi, plants, Protists, and metazoans display a sharp increase in A34-tRNA ratios coinciding with the expansion in A34-tRNA diversity. Among Protists, SAR species display significant internal variability in A34-tRNA ratios with a median value below the other eukaryotic phyla. Thus, some extant SAR species may represent an intermediate step in the evolution of I34-tRNAs from bacteria to metazoans (fig. 3) or, alternatively, may have lost these tRNAs secondarily. As a control, we explored whether the increase in A34-tRNA ratios was linked to general increases in genes coding for all isoacceptor tRNAs coding for TAPSLIVR and found that the ratio of TAPSLIVR isoacceptor tRNAs remains constant across all species tested (supplementary fig. S2, Supplementary Material online).
Identification of TadA and ADAT Genes, and Phylogenetic Analysis
Based on sequence and structure comparisons, it has been proposed that TadA evolved from the superfamily of CDAs (Gerber and Keller 1999), a relationship reflected in the conservation of important structural and functional motifs. ADAT evolved through duplication of a TadA gene, and it retains a detectable sequence identity with CDA. We wondered whether the variation in A34-tRNA diversity among different bacterial and eukaryotic phyla would be reflected in the evolution of TadA and ADAT, respectively. To answer this question, we set up to analyze the evolution of the two enzymes using molecular phylogenetics.
A required initial step for the phylogenetic analyses of TadA and ADAT is the reliable identification of the genes coding for these enzymes, and the exclusion of CDA sequences in the library of genes used for the analysis. To this end, we developed a new approach designed to identify, for each available bacterial or eukaryotic genome, the sequence more likely to correspond to TadA, ADAT2, or ADAT3, and obtain a measure of its divergence from CDAs (“Adatness”; supplementary fig. S3, Supplementary Material online).
First, we implemented a protein–protein search (BlastP) using the Blast+ software (Camacho et al. 2009) using a set of bona fide TadA, ADAT2, or ADAT3 protein sequences to search the nonredundant (NR) protein sequences database. For each species, the hit with the best “BlastP e-value” (e-value[TadA/ADAT]) was retained as the gene more likely to encode for TadA or ADAT. Next, a second BlastP search was performed using the sequence retained from each species to blast a database of bona fide CDA superfamily sequences (including cytosine deaminases, cytidine deaminases, deoxycytidine deaminases, adenosine deaminases, C to U RNA deaminases, and guanine deaminases). The best alignment found was selected (e-value[CDA]) (supplementary fig. S4, Supplementary Material online). The Adatness score was calculated as the ratio between the exponents of e-value(TadA/ADAT) and e-value(CDA). Sequences with an Adatness score higher than 1 were accepted as a bona fide TadA/ADAT (see also Materials and Methods).
The application of this search strategy revealed that those species whose genome contains one or more genes coding for A34-tRNAs also contain genes that are more similar to adenosine deaminases than to cytidine deaminases. In contrast, in bacterial species lacking A34-tRNA genes, only genes with higher sequence similarity for CDA than for TadA could be detected. This indicates that all bacterial species lacking A34-tRNAs also lack TadA (fig. 1 and supplementary fig. S4, Supplementary Material online).
The data sets of bacterial TadA, eukaryotic ADAT2, and ADAT3 sequences were aligned using conserved structural and sequence motifs as references (supplementary fig. S5, Supplementary Material online), and used to construct molecular phylogenies with the maximum likelihood (ML) method. The phylogenetic analysis of TadA sequences revealed that species with A34-tRNA diversity higher than 1 do not form a monophyletic group (fig. 4). This strongly suggests that increases in A34-tRNA diversity are not linked to the lateral gene transfer of genes coding for TadA variants of broad substrate specificity. This conclusion was supported by the analysis of a restricted set of TadA sequences from Firmicute and Tenericute bacteria that contain one or more A34-tRNA genes. For example, O. oeni TadA (a species with four different A34-tRNAs) is highly similar to and monophyletic with enzymes of species that only contain . Thus, TadA can increase its substrate range without major sequence or structural rearrangements, and the increase in A34-tRNA genes likely took place independently in different bacterial phyla (fig. 4).
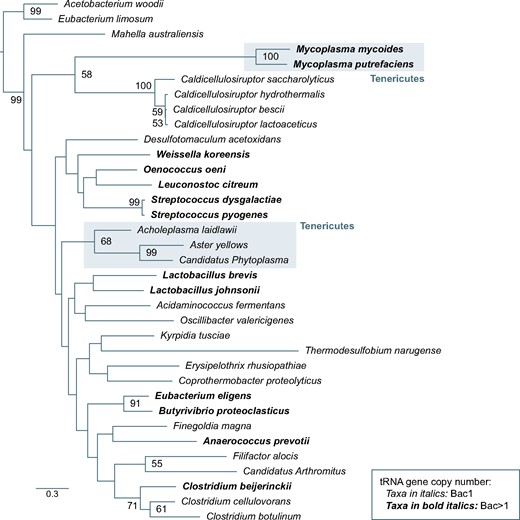
ML phylogenetic tree based on TadA amino acid sequences from species belonging to the Tenericutes (shaded boxes) and Firmicutes phyla. Species in bold denote organisms whose A34-tRNA diversity is >1. Nodal supports (100-replicate ML bootstrap) are indicated when higher than 50.
The phylogenetic analyses of ADAT2 are generally consistent with the canonical distribution of eukaryotic species (fig. 5). On the other hand, ADAT3-based phylogenies fail to recapitulate a canonical eukaryotic tree, likely due to the poor sequence conservation among ADAT3 sequences (fig. 5). However, all SAR species whose genomes contain reduced numbers of A34-tRNA genes also contain genes coding for ADAT2 and ADAT3 that are monophyletic with sequences from species with full complements of A34-tRNAs. This is compatible with the gene duplication that gave rise to ADAT preceding the expansion of A34 to all TAPSLIVR isoacceptor tRNA genes.
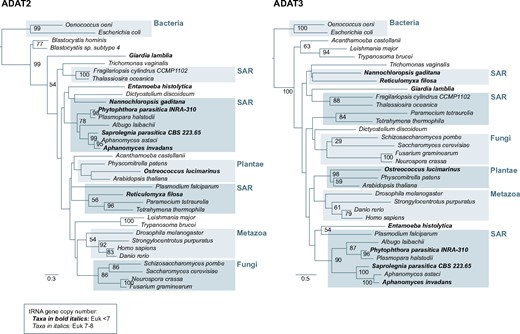
ML phylogenetic tree based on ADAT2 and ADAT3 amino acid sequences. Species belonging to each major eukaryotic group are boxed and labeled accordingly. Species in bold denote organisms whose A34-tRNA diversity is <7. Nodal supports (100-replicate ML bootstrap) are indicated when higher than 50. TadA from the bacteria Escherichia coli and Oenococcus oeni are used as outgroups.
TAPSLIVR-Rich Protein Abundance and I34-Sensitive Codon Usage
We have shown that eukaryotic genes coding for TAPSLIVR-rich proteins predominantly use codons recognized by I34-tRNAs, whereas such codon usage bias is absent in bacteria (Rafels-Ybern et al. 2018). We proposed that this codon preference in eukaryotes is due to a functional advantage provided by I34-tRNAs, which allowed eukaryotic cells to more efficiently translate TAPSLIVR-rich stretches. This resulted in an increase in abundance and length of such proteins in eukaryotic proteomes (Rafels-Ybern et al. 2018).
Using algorithms previously described (Rafels-Ybern et al. 2015), we compared the proteomes of all the species analyzed in this work in terms of abundance, length, and codon composition of TAPSLIVR-rich proteins (fig. 1 and table 3). This analysis revealed that eukaryotic proteomes contain higher numbers of TAPSLIVR-rich proteins, which also tend to be longer in these species. After normalizing for average total CDS contents, an 8-fold difference in abundance of TAPSLIVR-rich proteins was found between eukaryotes and bacteria (table 3).
Quantification of the Length, Abundance, and Composition of TAPSLIVR Stretches by Phylogenetic Groups.
| Eukarya | Bacteria | |||||||
|---|---|---|---|---|---|---|---|---|
| Fungi | Metazoa | Plantae | Protist | SAR | Black | Green | Red | |
| Num. organisms | 13 | 16 | 9 | 15 | 36 | 86 | 794 | 76 |
| With stretches | 13 (100%) | 16 (100%) | 9 (100%) | 15 (100%) | 36 (100%) | 22 (26%) | 466 (59%) | 33 (43%) |
| Num. stretch | 1,131 | 2,733 | 2,208 | 3,174 | 6,489 | 417 | 6,194 | 295 |
| Mean stretch size | 117 ± 61 | 124 ± 68 | 115 ± 48 | 120 ± 64 | 116 ± 51 | 114 ± 43 | 106 ± 40 | 110 ± 48 |
| Mean stretch enrich. | 0.761 ± 0.10 | 0.762 ± 0.12 | 0.619 ± 0.16 | 0.7 ± 0.15 | 0.63 ± 0.15 | 0.619 ± 0.11 | 0.541 ± 0.11 | 0.576 ± 0.14 |
| Mean CDS enrich. | 0.706 ± 0.04 | 0.692 ± 0.03 | 0.603 ± 0.07 | 0.667 ± 0.08 | NA | 0.686 ± 0.05 | 0.630 ± 0.08 | 0.712 ± 0.06 |
| Num. CDS | 1.25E+05 | 4.10E+05 | 2.40E+05 | 2.26E+05 | 6.29E+05 | 1.83E+05 | 2.93E+06 | 1.76E+05 |
| Stretch/CDS | 0.0090 | 0.0067 | 0.0092 | 0.0140 | 0.0103 | 0.0023 | 0.0021 | 0.0017 |
| Stretch/organisms | 87 | 171 | 245 | 212 | 180 | 5 | 8 | 4 |
| Eukarya | Bacteria | |||||||
|---|---|---|---|---|---|---|---|---|
| Fungi | Metazoa | Plantae | Protist | SAR | Black | Green | Red | |
| Num. organisms | 13 | 16 | 9 | 15 | 36 | 86 | 794 | 76 |
| With stretches | 13 (100%) | 16 (100%) | 9 (100%) | 15 (100%) | 36 (100%) | 22 (26%) | 466 (59%) | 33 (43%) |
| Num. stretch | 1,131 | 2,733 | 2,208 | 3,174 | 6,489 | 417 | 6,194 | 295 |
| Mean stretch size | 117 ± 61 | 124 ± 68 | 115 ± 48 | 120 ± 64 | 116 ± 51 | 114 ± 43 | 106 ± 40 | 110 ± 48 |
| Mean stretch enrich. | 0.761 ± 0.10 | 0.762 ± 0.12 | 0.619 ± 0.16 | 0.7 ± 0.15 | 0.63 ± 0.15 | 0.619 ± 0.11 | 0.541 ± 0.11 | 0.576 ± 0.14 |
| Mean CDS enrich. | 0.706 ± 0.04 | 0.692 ± 0.03 | 0.603 ± 0.07 | 0.667 ± 0.08 | NA | 0.686 ± 0.05 | 0.630 ± 0.08 | 0.712 ± 0.06 |
| Num. CDS | 1.25E+05 | 4.10E+05 | 2.40E+05 | 2.26E+05 | 6.29E+05 | 1.83E+05 | 2.93E+06 | 1.76E+05 |
| Stretch/CDS | 0.0090 | 0.0067 | 0.0092 | 0.0140 | 0.0103 | 0.0023 | 0.0021 | 0.0017 |
| Stretch/organisms | 87 | 171 | 245 | 212 | 180 | 5 | 8 | 4 |
Note.—Num. organisms = total number of species analyzed, with stretches = number of species in each group containing TAPSLIVR stretches, num. stretch = total number of TAPSLIVR stretches found in each group, mean stretch size = mean value of the total TAPSLIVR stretch lengths measured for each group, mean stretch enrich. = mean value of the percentage of ADAT-dependent codons in the set of TAPSLIVR stretches measured for each group, mean CDS enrich. = mean value of the percentage of ADAT-dependent codons in genes with TAPSLIVR stretches measured for each group, num. CDS = number of genes containing TAPSLIVR stretches measured for each group, stretch/CDS = mean value of the “number of TAPSLIVR stretches/number of CDS” ratio for each group, and stretch/organisms = mean value of TAPSLIVR stretches per organism in each group.
Quantification of the Length, Abundance, and Composition of TAPSLIVR Stretches by Phylogenetic Groups.
| Eukarya | Bacteria | |||||||
|---|---|---|---|---|---|---|---|---|
| Fungi | Metazoa | Plantae | Protist | SAR | Black | Green | Red | |
| Num. organisms | 13 | 16 | 9 | 15 | 36 | 86 | 794 | 76 |
| With stretches | 13 (100%) | 16 (100%) | 9 (100%) | 15 (100%) | 36 (100%) | 22 (26%) | 466 (59%) | 33 (43%) |
| Num. stretch | 1,131 | 2,733 | 2,208 | 3,174 | 6,489 | 417 | 6,194 | 295 |
| Mean stretch size | 117 ± 61 | 124 ± 68 | 115 ± 48 | 120 ± 64 | 116 ± 51 | 114 ± 43 | 106 ± 40 | 110 ± 48 |
| Mean stretch enrich. | 0.761 ± 0.10 | 0.762 ± 0.12 | 0.619 ± 0.16 | 0.7 ± 0.15 | 0.63 ± 0.15 | 0.619 ± 0.11 | 0.541 ± 0.11 | 0.576 ± 0.14 |
| Mean CDS enrich. | 0.706 ± 0.04 | 0.692 ± 0.03 | 0.603 ± 0.07 | 0.667 ± 0.08 | NA | 0.686 ± 0.05 | 0.630 ± 0.08 | 0.712 ± 0.06 |
| Num. CDS | 1.25E+05 | 4.10E+05 | 2.40E+05 | 2.26E+05 | 6.29E+05 | 1.83E+05 | 2.93E+06 | 1.76E+05 |
| Stretch/CDS | 0.0090 | 0.0067 | 0.0092 | 0.0140 | 0.0103 | 0.0023 | 0.0021 | 0.0017 |
| Stretch/organisms | 87 | 171 | 245 | 212 | 180 | 5 | 8 | 4 |
| Eukarya | Bacteria | |||||||
|---|---|---|---|---|---|---|---|---|
| Fungi | Metazoa | Plantae | Protist | SAR | Black | Green | Red | |
| Num. organisms | 13 | 16 | 9 | 15 | 36 | 86 | 794 | 76 |
| With stretches | 13 (100%) | 16 (100%) | 9 (100%) | 15 (100%) | 36 (100%) | 22 (26%) | 466 (59%) | 33 (43%) |
| Num. stretch | 1,131 | 2,733 | 2,208 | 3,174 | 6,489 | 417 | 6,194 | 295 |
| Mean stretch size | 117 ± 61 | 124 ± 68 | 115 ± 48 | 120 ± 64 | 116 ± 51 | 114 ± 43 | 106 ± 40 | 110 ± 48 |
| Mean stretch enrich. | 0.761 ± 0.10 | 0.762 ± 0.12 | 0.619 ± 0.16 | 0.7 ± 0.15 | 0.63 ± 0.15 | 0.619 ± 0.11 | 0.541 ± 0.11 | 0.576 ± 0.14 |
| Mean CDS enrich. | 0.706 ± 0.04 | 0.692 ± 0.03 | 0.603 ± 0.07 | 0.667 ± 0.08 | NA | 0.686 ± 0.05 | 0.630 ± 0.08 | 0.712 ± 0.06 |
| Num. CDS | 1.25E+05 | 4.10E+05 | 2.40E+05 | 2.26E+05 | 6.29E+05 | 1.83E+05 | 2.93E+06 | 1.76E+05 |
| Stretch/CDS | 0.0090 | 0.0067 | 0.0092 | 0.0140 | 0.0103 | 0.0023 | 0.0021 | 0.0017 |
| Stretch/organisms | 87 | 171 | 245 | 212 | 180 | 5 | 8 | 4 |
Note.—Num. organisms = total number of species analyzed, with stretches = number of species in each group containing TAPSLIVR stretches, num. stretch = total number of TAPSLIVR stretches found in each group, mean stretch size = mean value of the total TAPSLIVR stretch lengths measured for each group, mean stretch enrich. = mean value of the percentage of ADAT-dependent codons in the set of TAPSLIVR stretches measured for each group, mean CDS enrich. = mean value of the percentage of ADAT-dependent codons in genes with TAPSLIVR stretches measured for each group, num. CDS = number of genes containing TAPSLIVR stretches measured for each group, stretch/CDS = mean value of the “number of TAPSLIVR stretches/number of CDS” ratio for each group, and stretch/organisms = mean value of TAPSLIVR stretches per organism in each group.
In general, the enrichment in TAPSLIVR-rich proteins correlated well with A34-tRNA diversity and Adatness score (fig. 1). For example, ADAT stretches are consistently abundant in species with A34-tRNA diversity higher than 7 and Adatness scores higher than 1. In contrast, bacterial species with A34-tRNA diversity of 1 and Adatness higher than 1 have variable contents of TAPSLIVR-rich proteins. Finally, bacterial species with A34-tRNA diversity of 0 and low Adatness scores mostly lack TAPSLIVR-rich proteins (e.g., Thermotogae).
Based on these observations, we divided our species data set in five groups: eukaryotes with A34-tRNA diversity equal or higher than 7, eukaryotes with A34-tRNA diversity lower than 7, bacterial species with A34-tRNA diversity higher than 1, bacterial species with A34-tRNA diversity equal to 1, and bacterial species with A34-tRNA diversity equal to 0. We then compared the abundance of TAPSLIVR-rich proteins, and the preference for ADAT codons in their respective genes, among these five groups.
This comparison revealed a gradual increase in codon bias for ADAT codons from bacteria with A34-tRNA diversity of 1 to eukaryotes (fig. 6). Interestingly, we detected a small number of stretches in bacteria lacking A34-tRNAs that were accompanied by a significant increase in I34-dependent codon bias. However, the majority of species in this set (74%) completely lack TAPSLIVR stretches in their proteomes (fig. 1 and table 3). Thus, TAPSLIVR stretches in bacteria with A34-diversity equal to 0 are rare and only present in a restricted group of species. A clear preference for I34-dependent codons is only detectable in eukaryotes with A34-tRNA diversity higher than 7 (fig. 6 and supplementary fig. S6, Supplementary Material online), supporting the proposal that the expansion of I34-tRNAs was driven by the functional advantage provided by these tRNAs to translate TAPSLIVR-rich protein transcripts.
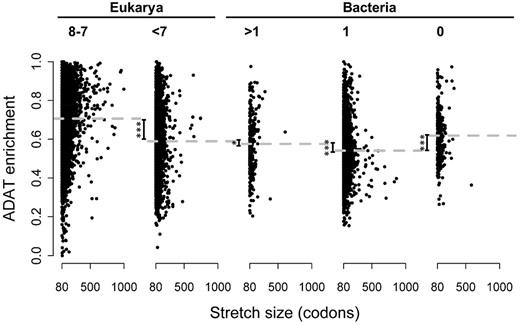
Populations of proteins enriched in ADAT amino acids as a function of A34-tRNA diversity (indicated at the top of each graph). Each dot corresponds to an amino acid stretch of length >80 with a composition of ADAT amino acids >83%. Stretches shorter than 1,000 codons were not considered.
Discussion
The process of molecular evolution that gave rise to eukaryotic ADAT from bacterial TadA was accompanied by a series of genomic transformations that modified the eukaryotic genomes at three levels: tRNA gene diversity, tRNAome composition, and codon usage of the whole transcriptome (Novoa et al. 2012). Our understanding of the chain of events involved in this process is poor and, until recently, limited to the observation that duplication of a bacterial gene coding for a monomeric deaminase with a single tRNA substrate () was the source of the two genes that, in eukaryotes, code for the heterodimeric enzyme capable of deaminating eight different tRNA isoacceptors cognate for TAPSLIVR.
It has been established that TadA evolved in bacteria from cytidine deaminases, which are universally distributed and hence likely to be ancestral to TadA. A recent in silico analysis of the bacterial distribution of A34-tRNAs and TadA genes has revealed that the elements required for I34 synthesis are not universally distributed and absent in some deep-rooted bacterial groups (Diwan and Agashe 2018). Here, we confirm this observation in Thermotogales, Spirochates, and Chloroflexi using a recurrent homology search pipeline. We also show that the vast majority of species in this set of deeply rooted bacterial orders are severely depleted of proteins rich in TAPSLIVR amino acids, with the sole exception of Chloroflexi.
Based on the widespread absence of TadA and A34-tRNA coding genes in groups of deeply rooted, thermophilic bacteria, and their different composition in terms of stretch abundance, an alternative explanation to previous proposals is that TadA and A34-tRNA were absent at the root of the bacterial kingdom. This alternative interpretation would posit that the duplication of a CDA gene leading to the emergence of TadA took place after the divergence of the most ancient bacterial clades, whereas a secondary loss took place later in those groups of bacterial species that underwent genome reduction events.
It is generally accepted that the duplication that gave rise to TadA in bacteria generated an enzyme functionally limited to , and that this tRNA was the only I34-tRNA found in prokaryotes. Here, we show unequivocally that several bacterial species contain additional A34-tRNA coding genes and that, at least in the case of O. oeni, other I34-tRNAs are present in bacteria. We have detected several genes coding for A34-tRNAs cognate for Ile, Leu, Ser, Thr, and Pro, and we have demonstrated that, in O. oeni, and , but not and , are modified to I34. Although we cannot formally rule out the possibility that the I34 modification in O. oeni is catalyzed by a different enzyme than TadA, our results indicate that the increase in the repertoire of I34-modified tRNAs probably begins with the appearance of A34-tRNAs that are initially not modified and evolve into deaminase substrates through a process of enzyme–substrate coevolution. Possibly, the hypermutable nature of the genus Oenococcus (due to the loss of the mismatch repair pathway) contributed to the increase in A34-tRNA diversity in these species (Marcobal et al. 2008).
Our analyses of O. oeni TadA indicate that this dual-substrate enzyme is fundamentally identical to other bacterial TadAs. Thus, if the deamination of in O. oeni is indeed catalyzed by TadA, the enzyme does not appear to require additional modifications to increase its tRNA substrate range. It is unclear at this stage why and are not deaminated in vivo, and whether these unmodified tRNAs are active in translation. It is likely that, in a situation analogous to the recognition of tRNAs by aminoacyl-tRNA synthetases, negative identity elements present in and but absent in prevent the deamination of the former by TadA (Giege et al. 1998).
Our results indicate that the substrate expansion that took place in the evolution from bacterial TadAs to eukaryotic ADATs could have started in bacteria. The analysis of eukaryotic genomes revealed high heterogeneity in terms of A34-tRNA diversity in SAR group of nucleated organisms (Stramenopiles, Alveolates, and Rhizaria). However, our analysis of the A34-tRNA set in the alveolate T. thermophila demonstrates that this species holds a full complement of I34-modified tRNAs cognate for TAPSLIVR. Thus, although the genomic analysis using tRNAscan-SE suggests that some SAR species exist with lower number of ADAT substrates, our experimental results show that other species within the same group have acquired a full complement of modified I34-tRNAs. We cannot rule out the possibility tRNAscan-SE is unable to identify existing A34-tRNAs in SAR genomes but, barring this possibility, our data points at the SAR group as the most variable set of eukaryotes in terms of I34 composition, either because this group contains species that represent preliminary stages in ADAT evolution or because some SAR species have lost A34-tRNAs secondarily. This might indicate that the selective pressures that keep A34-tRNA gene populations stable in plants, fungi, and metazoan are weaker among protists. Similarly, our data show that the selective pressure that drove the dramatic increase in A34-tRNA gene abundance in eukaryotic genomes did not exist in those bacteria that acquired additional I34-tRNAs.
We have shown in the past that the emergence of ADAT in eukaryotes also correlates with an increase in the abundance of TAPSLIVR-rich proteins in these organisms, and with a shift in the codon usage of the transcripts of these same proteins that favor the utilization of I34-tRNAs for their translation (Rafels-Ybern et al. 2018). TAPSLIVR-rich stretches are, in average, increased ∼5-fold in eukaryotic genomes after their numbers are corrected by genome size, and the codon usage in favor of I34-tRNAs increases gradually with the length of eukaryotic stretches, but not of bacterial ones (Rafels-Ybern et al. 2018). These observations prompted us to hypothesize that one selective advantage driving the increase of tRNA substrates of ADAT and their genomic enrichment was their contribution to the translation of TAPSLIVR-rich low-complexity transcripts. We envisage that I34-tRNAs offered a functional improvement to translational machineries in a manner comparable to the contribution of EF-P to the translation of consecutive proline codons (Doerfel et al. 2013; Ude et al. 2013; Lassak et al. 2016), or to the modifications in the tRNAome of arthropod salivary glands that allow the synthesis of the protein components of silk (Chevallier and Garel 1982; Li et al. 2015; Ribas de Pouplana et al. 2017).
The exact nature of the improvement afforded by I34 to eukaryotic translation elongation is currently unknown. Possibly, I34 impacts upon codon–anticodon recognition efficiency (Topal and Fresco 1976) and the accommodation of contiguous tRNAs within the decoding center of the ribosome (Allner and Nilsson 2011). Such contribution might be significant across the complete transcriptome and become essential during the translation of TAPSLIVR stretches. The absence of I34 could increase the risk of spontaneous ribosomal drop-off due to impaired A-site occupancy rates caused by ineffective tRNA delivery, or by a reduced concentration of available cognate tRNAs.
Among bacteria, TAPSLIVR-rich proteins are found mostly in species with a single A34-tRNA gene (), where 60% of species contain genes with TAPSLIVR stretches. This frequency is reduced to 43% in genomes with A34-tRNA diversity higher than 1, and to 26% in bacterial species that lack A34-tRNAs. It should be noted that among species lacking A34-tRNAs, TAPSLIVR stretches are almost exclusively found in the Chloroflexi phylum and are completely absent in Thermotogales. The fact that we cannot detect any correlation between codon usage preference for I34-tRNAs and the length of transcripts coding for bacterial TAPSLIVR-rich proteins suggests that I34 does not influence the ability of prokaryotes to synthesize these polypeptides.
In eukaryotes, TAPSLIVR-rich proteins are universally distributed, and up to 8-fold more abundant than in bacteria. The codon usage in eukaryotic sequences for stretch regions is biased toward triplets decoded by I34-tRNAs, and this bias increases with the length of the TAPSLIVR stretch. This codon enrichment is particularly prominent in fungi and metazoans, but whether the translational apparatuses of these species are more efficient in the synthesis TAPSLIVR-rich proteins remains to be seen.
Unlike other tRNA modifications whose levels in the tRNAome are adjustable and respond to changes in environmental conditions, I34 levels have been shown to be stable and saturated both in Saccharomyces cerevisiae and H. sapiens (Chan et al. 2010; Torres et al. 2015). Thus, it is likely that the incorporation of I34 to eukaryotic tRNAs and its genomic enrichment is not due to this modification being implicated in the regulation of stress responses (Dedon and Begley 2014; Torres et al. 2015).
Our results indicate that the evolutionary advantage conferred by the activity of ADAT was the facilitation of the synthesis of TAPSLIVR-rich proteins. Although most extant protein folds likely evolved from simple ancestral domains that combined through processes of genetic duplication (Lupas and Russell 2012), it is possible that proteins with extremely repetitive sequences could only emerge in evolution when the adequate functional improvements of the translation machinery were selected.
Materials and Methods
Definitions
ADAT amino acids were defined as those amino acids cognate to tRNAs that are modified by ADAT in most eukarya, namely TAPSLIVR. ADAT codons were defined as those codons that code for ADAT amino acids (37 codons) (supplementary table S2, Supplementary Material online, blue and dark blue). ADAT-sensitive codons were defined as the subset of ADAT codons susceptible to be recognized by modified I34 tRNAs (24 codons) (supplementary table S2, Supplementary Material online, dark blue only). ADAT stretches were defined as those regions within CDSs that are enriched in ADAT codons, as implemented in our “Running Windows” method (Rafels-Ybern et al. 2015). For each ADAT stretch, we defined ADAT enrichment as the fraction of ADAT-sensitive codons/ADAT codons. Adatness (fig. 1) of a sequence is defined as the ratio between the exponents of the e-value obtained for when this sequence is aligned by BlastP against bona fide ADAT2, ADAT3, or TadA, and the e-value obtained when the same sequence is aligned by BlastP against bona fide CDA sequences (see also “Gene and protein identification methods”). In supplementary figure S6A, Supplementary Material online, Δ1% represents the number of codons needed to increase the ADAT-enrichment by 1% based on the slope of the corresponding linear model.
Genomic Data Set Retrieval
We have analyzed the transcriptome, genome, and proteome of 150 eukaryotic species and 956 bacterial species. We analyzed a total of 3.6 Gb of bacterial genomes (3.28 million CDSs) and 18.8 Gb of eukaryotic genomes (1.79 million CDSs). Only CDSs with a start codon, a stop codon, and a number of nucleotides multiple of 3 were used in the analysis. Twelve percent of eukaryotic CDSs and 20% of bacterial CDSs were discarded because they did not fulfill one of the three requisites. Eukaryotic sequences were downloaded from the Ensembl website except for those sequences from the SAR clade. Bacterial sequences and SAR sequences were downloaded from NCBI website. Proteins were analyzed in NR database from NCBI.
Identification of Stretches by the “Running Windows” Method
To study in more detail the presence of ADAT stretches, a “Running Windows” method was applied based on similar methodologies (McDonald 1996; Hutter et al. 2006; Librado and Rozas 2009) as described (Rafels-Ybern et al. 2015). Window size was fixed to 80 codons and, as previous described (Rafels-Ybern et al. 2015), a window was considered enriched in ADAT codons when those represented >83% of the sequence. We defined an ADAT stretch as the region corresponding to a window, or a set of consecutive windows, enriched in ADAT codons. Two or more windows were considered consecutive if they overlapped in the CDS.
Statistical Analyses
Significant differences observed in figures 3 and 6 and supplementary figure S2, Supplementary Material online, were obtained using one-tail Wilcoxon test with confidence level at 0.95, using the Wilcox.Test() function from R (Novoa and Ribas de Pouplana 2012; R-Core-Team 2013). Statistical analyses shown in supplementary figure S6A, Supplementary Material online, were performed using data only from intervals with >15 samples (full data shown in supplementary fig. S6B, Supplementary Material online) and linear regressions were calculated only for groups with five or more points. Linear regressions were fitted using the lm() function as implemented in R (R-Core-Team 2013).
Gene and Protein Identification Methods
tRNA genes were predicted with tRNAscan-SE (Fichant and Burks 1991; Lowe and Eddy 1997; Nawrocki and Eddy 2013). Options -B for bacterial genomes and -G for eukaryotic genomes were used. Pseudogenes and undetermined tRNAs from standard output were discarded from the analysis. tRNA gene copy number for each organism was inferred based on the number of different A34-tRNA genes that are present for each organism.
In order to identify the most likely TadA, ADAT2, or ADAT3 protein sequence from each organism, we followed the developed the pipeline shown in supplementary figure S3, Supplementary Material online. This method allowed us to simultaneously compare the putative TadA and ADAT sequences from all the organisms in our data set. First, we implemented a protein–protein search (BlastP) using the Blast+ software (Camacho et al. 2009) using a set of bona fide TadA, ADAT2, or ADAT3 protein sequences against the NR protein sequences database (supplementary fig. S3, Supplementary Material online, step 1, HitsA) (Altschul et al. 1990). The results were filtered for our 956 bacterial organisms taking the hit with the best e-value for each species (supplementary fig. S3, Supplementary Material online, step 2). Each putative TadA, ADAT2, or ADTA3 sequence was then used as query to perform a second BlastP search against a set of bona fide CDA proteins (supplementary fig. S3, Supplementary Material online, step 3). The e-value(TadA/ADAT) and e-value(CDA) obtained (supplementary fig. S3, Supplementary Material online, step 4) were used to calculate the Adatness score of each sequence.
Phylogenetic Trees Reconstruction
From the sequences identified in the previous search, we used the T-coffee software with the option +trim_n (Notredame et al. 2000) to select a set of 47 TadA, 33 ADAT2, and 33 ADAT3 that best represented the variability of our entire data set. These sequences were aligned using T-coffee with the mode expresso (Armougom et al. 2006), and >50% gapped columns were removed from the resulting multiple sequence alignment matrix. ML trees were performed using RaxML (Stamatakis 2014). One hundred replicas were computed using the options GAMMA as a model of rate heterogeneity and LG as a model of substitutions. A consensus tree was built using the -f b option to calculate the bootstrap support values on the best ML tree from all the replicas. ML trees were plotted using the libraries ape, numDeriv, phytools, and phangorn from R (R-Core-Team 2013). The standard phylogenetic tree from figure 1 was build using the online tool PhyloT (Letunic 2015).
Experimental Procedures
RNA extraction was performed with TRIzol reagent (Ambion) following the manufacturer’s protocol. Oenococcus oeni strain PSU-1 was grown as described in Margalef-Catala et al. (2017), and cell pellets were first treated with Lysozyme (Sigma-Aldrich) following the manufacturer’s protocol and 1 ml of TRIzol reagent was added to the lysate for RNA extraction. Tetrahymena thermophila was grown as described in de Francisco et al. (2018), and cell pellets were lysed directly in 1 ml of TRIzol reagent.
The extracted RNA was ethanol precipitated, resuspended in RNase-free water, and was then treated with Turbo DNase (Thermo Fisher Scientific) following the manufacturer’s protocol. Reactions were purified with the MiTotal RNA extraction kit (Viogene) and the obtained RNA was quantified using a Nanodrop ND-1000. Inosine 34 detection by SL-ID was performed as previously described (Torres et al. 2018).The tRNA-specific bridge oligonucleotides used in this study are depicted in supplementary table S1, Supplementary Material online. Specific A34-encoded tRNA isoacceptors were PCR amplified using the oligonucleotides in supplementary table S1, Supplementary Material online, and amplicons were subjected to Sanger sequencing.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Acknowledgments
This work was supported by the Spanish Ministry of Economy and Competitiveness (grant numbers BES2013-064551 to [A.R.-Y.], and BIO2015-64572 to [L.R.d.P.]). We thank Mrs Julia Carbó for technical assistance, and the whole Gene Translation Laboratory of the IRB Barcelona for helpful discussions. We also thank Dr Juan Carlos Gutierrez for providing us with T. thermophila cell cultures.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}