





Abstract
Human S ilent I nformation R egulator T ype 1 (SIRT1) is an NAD + -dependent deacetylase protein which is an intermediary of cellular metabolism in gene silencing and aging. SIRT1 has been extensively investigated and shown to delay senescence; however, less is known about the regulation of SIRT1 during aging. In this study, we show that the peroxisome proliferator-activated receptor-γ (PPARγ), which is a ligand-regulated modular nuclear receptor that governs adipocyte differentiation and inhibits cellular proliferation, inhibits SIRT1 expression at the transcriptional level. Moreover, both PPARγ and SIRT1 can bind the SIRT1 promoter. PPARγ directly interacts with SIRT1 and inhibits SIRT1 activity, forming a negative feedback and self-regulation loop. In addition, our data show that acetylation of PPARγ increased with increasing cell passage number. We propose that PPARγ is subject to regulation by acetylation and deacetylation via p300 and SIRT1 in cellular senescence. These results demonstrate a mutual regulation between PPARγ and SIRT1 and identify a new posttranslational modification that affects cellular senescence.
INTRODUCTION
Diverse genetic and environmental factors affect longevity in a wide variety of organisms ( 1 ). Cellular senescence, also known as replicative senescence, is a process through which cells lose their proliferative potential after a limited number of population doublings (PDs). This is accompanied by a specific set of changes including growth cessation, morphologic changes and appearance of senescence-associated beta-galactosidase (SA- β- gal) activity.
SIRT1 (sirtuin 1) (Sir2 histone deacetylase) is an important determinant of longevity that plays a role in life-span regulation in diverse species ( 2–5 ). Caloric restriction (CR) can greatly increase life span in mammals ( 6 ), and the mammalian Sir2 ortholog, SIRT1, may mediate this effect of CR by affecting peroxisome proliferator-activated receptor γ (PPARγ), PGC1-α, p53, forkhead transcription factors (FOXO) and p300, just to name a few ( 7–11 ). In addition, SIRT1 activity has also been associated with tumorigenesis ( 12 ). As such, it is conceivable that SIRT1 may be a ‘double-edged sword’ that requires tight regulation. Despite the fact that there has been extensive study of SIRT1 function, the regulation of SIRT1 is less well understood.
PPARγ is a transcription factor that belongs to the ligand-activated nuclear receptor superfamily. PPARγ plays an important role in the induction of cellular differentiation and the inhibition of cell growth by promoting cell-cycle arrest ( 13 , 14 ). In addition, PPARγ promotes cellular senescence by inducing p16 INK4α (CDKN2A), which is an important cell-cycle inhibitor ( 15 ). A previous study in our laboratory showed that SIRT1 promoted cell proliferation and antagonized cellular senescence in human diploid fibroblasts ( 16 ).
Considering our previous work and that of others, we asked the question whether ligand-activated PPARγ regulates SIRT1 expression at the transcriptional level or the protein level, and how it functions in the cell. Our results demonstrated that SIRT1 complexes with PPARγ via its core deacetylase catalytic activity domain, and both SIRT1 and PPARγ bind directly to the promoter of SIRT1 itself and thus control homeostatic SIRT1 expression. We also found that the transcriptional activity of PPARγ is regulated through deacetylation by SIRT1. This association of SIRT1 and PPARγ together with the transcriptional modulation of SIRT1 appears to be important in the senescence process controlled by PPARγ. This study therefore shows evidence that the longevity-regulating protein SIRT1 is under the direct control of PPARγ and that the role of PPARγ in regulating cellular senescence involves a complex feedback loop including SIRT1. Our results also suggest that PPARγ acetylation may play an important role in regulation of SIRT1 during cellular senescence.
MATERIALS AND METHODS
Plasmids, antibodies and regents
The SIRT1 promoter was isolated by polymerase chain reaction (PCR) and spanned −1048 to +900 bp, which contained the entire SIRT1 promoter CpG island. The promoter was inserted into the Bgl II/Hind III sites of the pGL3 basic luciferase expression vector (Promega). A series of fragments from this sequence were PCR-amplified or treated with restriction endonucleases. Expression plasmids encoding human SIRT1 (pBABE-SIRT1), human SIRT1 H363Y (deacetylase domain mutation) and human PPARγ (pBABE-PPARγ) were constructed according to the manufacturer’s guideline. The upstream (encompassing the coding region for amino acids 1–253) and downstream (encompassing the coding region for amino acids 496–747) regions of pcDNA3.1-ΔMyc-SIRT1 were constructed by PCR amplification of the pcDNA3.1-SIRT1 plasmids, using the primers 5′-ATGGCGGACGAGGCGGCCCT-3′; 5′-AAGTTTGGCATATTCTTGCACTCTTGCAGT-3′; 5′-GAATATGCCAAACTTGAATATGCCAAACTT-3′ and 5′-GATACTAAACAAACTACCTATCAAG-3′, respectively. These two PCR products were mixed, annealed and amplified in a third PCR reaction using primers 5′-GGATCCATGGCGGACGAGGCGGCCCT-3′ and 5′-GCGGCCGCGATACTAAACAAACTACCT A -3′. The final PCR product encoding ΔMyc-SIRT1 was cut with BamH I/Not I, and ligated into pcDNA3.1. Site-directed mutagenesis was performed using the QuikChange protocol (Stratagene). Antibodies which were used included anti-SIRT1 (Abcam ab32441), anti-Sir2 (Upstate 07-131), anti-p300 (Upstate 05-257), anti-PPARγ (Upstate 07-466), anti-actin (Abcam ab11003), anti-FLAG antibody (Sigma M2), anti-Myc (Santa Cruz TA-01) and anti-acetyl-lysine (Cell Signalling). The SuperSignal west picochemiluminescent substrate was obtained from Pierce Biotechnology (Rockford, IL). Sirtinol (Sigma).
Cell lines and animals
Human embryonic lung diploid fibroblast 2BS cells (obtained from the National Institute of Biological Products, Beijing, China) were originally isolated from female fetal lung fibroblast tissue and have been characterized previously ( 17 ). 2BS cells are considered to be young at PD30 or below and to be fully senescent at PD55 or above. Human WI-38 embryonic lung diploid fibroblast cells (ATCC number: CCL75) were obtained from the Chinese Academy of Sciences (Shanghai, China). WI-38 cells are considered to be young at PD25 or below and to be fully senescent at PD50 or above. 2BS cells, WI-38 cells, HeLa cells and Phoenix cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM), with 10% fetal calf serum (Sigma) at 37°C in a 5% CO 2 atmosphere. Young (3–4 months) and aged (18–22 months) BALB/c mice were maintained on a 12-h light/dark cycle with food and water available ad libitum . All animal studies were performed in accordance with the guidelines set forth by the Peking University Animal Ethics Committee.
Retroviral infection was performed as described ( 18 ). In experiments presented in Figure 7 , Phoenix cells were transfected with either pBABE, pBABE-SIRT1, pBABE-SIRT1 H363Y , pSUPERretro (Oligoengine) or pSUPERretro-PPARγ RNAi (5′-GCCCTTCACTACTGTTGAC-3′) ( 19 ) using lipofectamine 2000 (Invitrogen). After 48 h of transfection, the retrovirus containing medium was collected, filtered, treated with polybrene (1 μg/ml) and used to infect young 2BS target cells. Infected cells were selected for resistance to puromycin (2.5 μg/ml) and G418 (200 μg /ml), and then pooled.
Luciferase reporter assay
HeLa cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and were transfected with the indicated plasmids by using Lipo transfection reagent (Invitrogen). Cell lysates were prepared with the Dual Luciferase reporter assay kit (Promega Corp., Madison, WI, USA). A rennilla plasmid was included to control for efficiency of transfection. After transfection, the cells were incubated in DMEM supplemented with 10% FBS for 42 h. Luciferase activity was measured and normalized using renilla activity in the same sample. Luciferase and renilla assays were performed in triplicate, and experiments were repeated at least three times.
Immunoprecipitation, glutathione S-transferase pull-down and western blot
Cells were lysed in modified RIPA nuffer [1 mM ethylenediaminetetraacetic acid EDTA, 150 mM NaCl, 1 mM NaF, 1 mM Na 3 VO 4 , 1 m MPMSF, 0.25% deoxycholic acid (sodium salt), 1% NP-40, 1 µg/ml leupeptin, 1 µg/ml aprotatin, 150 mM Tris–HCl at pH 7.4] centrifuged for 10 min at 14 000 g and the insoluble debris was discarded. Protein concentration of each sample was determined by BCA Protein Assay Reagent (Pierce). Cell lysate (500 µg protein) was immunoprecipitated using the indicated antibodies overnight, and 50 μl of protein A-agarose beads were added to the lysate and rotated at 4°C for 2 h. The beads were washed extensively with cold phosphate-buffered saline (PBS), boiled in sodium dodecyl sulfate (SDS) sample buffer, fractionated with SDS-polyacrylamide gel electrophoresis (PAGE) and probed with the corresponding antibodies. To evaluate for SIRT1-PPARγ binding, HeLa cells were transfected with 7.5 μg of SIRT1-PPARγ plasmids using calcium chloride and cells were lysed for immunoprecipitation (IP) after 48 h.
Glutathione S-transferase (GST) and the GST-fusion proteins GST-PPARγ were produced in Escherichia coli DH5α and bound to glutathione-Sepharose beads according to the manufacturer’s instructions (Amersham Pharmacia Biotech).
Transfection of short-interfering RNA
The short-interfering RNA (siRNA) sequences targeted human PPARγ (GeneID: 5468) or SIRT1 (GeneID: 23411), and the transcripts sequences were 5′-GCCCTTCACTACTGTTGACtt-3′ ( 19 ) or 5′-CGTCTTATCCTCTAGTTCTtt-3′ ( 20 ). A scrambled sequence referred to a si-NC was provided by company and used as negative control (NC). Cells were transfected with siRNA using lipofectamine-RNAiMAX according to the manufacturer’s instructions (Invitrogen).
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed using the Chromatin Immunoprecipitation Assay kit (Upstate, New York, NY, USA) according to manufacturer’s instruction. In brief, 1 × 10 6 cells were cross-linked by adding formaldehyde directly to cell culture media and incubating for 10 min at 37°C. Cells were washed twice with cold PBS, and cells were scraped and resuspended in 200 µl of SDS lysis buffer. Chromatin was then sonicated to an average length of 0.5 kb with three 30-s pulses at maximum power. Chromatin extracts were diluted 10-fold in dilution buffer and preincubated for 30 m at 4°C with 50 µl of Salmon Sperm DNA/protein A-agarose. Twenty microliters of diluted supernatant was kept for isolation of input DNA and quantitation of the DNA in different samples. After pelleting the agarose by brief centrifugation, 2 µg of anti-SIRT1 antibody or anti-PPARγ (test group) or 2 µg of anti-β-actin (unrelated antibody control) was added to the supernatant fraction and incubated overnight at 4°C with rotation. In addition, we performed an IP omitting the antibody by incubating the supernatant fraction with Salmon Sperm DNA/protein A-agarose for 1 h at 4°C. Fifty microliters of Salmon Sperm DNA/protein A-agarose was then added, and the mixture was incubated for 1 h at 4°C to collect the antibody/antigen–DNA complex. The chromatin bound to the protein A-agarose beads was eluted in 500 µl of freshly prepared elution buffer (1% SDS, 0.1 M NaHCO 3 ). After reversing the cross-linking, the samples were deproteinized and phenol–chloroform-extracted, and DNA was ethanol-precipitated using glycogen as a carrier. Pellets were resuspended in 50 µl of TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0) for PCR analysis.
In re-ChIP experiments, endogenous PPARγ, SIRT1or p300-chromatin complexes from HeLa cells were first immunoprecipitated using PPARγ, SIRT1or p300 antibodies. The complexes were eluted by incubation for 30 min at 37°C in 10 mM DTT, diluted 10 times in re-ChIP buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl at pH 8.0) and subjected to a second ChIP. Coprecipitated chromatin was analyzed by PCR for the presence of SIRT1 promoter DNA between −592 and −163 bp upstream of the SIRT1 ATG start codon using 5′-CAACGTATTTCAGGGAGCT-3′ (−592) and 5′-CCACAACACTACGGGTCA-3′ (–163) primers.
Real-time PCR methods and data analysis
Total RNA was isolated from HeLa,WI-38 and 2BS cells using an RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions and subjected to reverse transcription followed by both quantitative real-time and semi-quantitative PCRs using the High Capacity cDNA Archive Kit (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s protocols. Real-time PCR was performed using Assays-on-Demand Gene Expression Products (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's instructions, with minor modifications and run on an ABI 7300 Real-time PCR System (Applied Biosystems). Each PCR was assembled using 96-well MicroAmp Optical plates (AB Applied Biosystems) with a total volume of 15 µl containing 1.5 µl cDNA templates, 1 µM of each primer and 17.5 µl of 2×SYBR Green Master Mix and brought to final volume with RNase-free water. Thermal reaction cycles of 50°C for 2 min, 95°C for 10 min and 40 repetitions of 95°C for 15 s and 60°C for 1 min were used. Real-time PCR data were analyzed using the ΔΔC T method, normalizing the Ct values of the indicated gene to the Ct values of GAPDH relative to a control sample.
Site-directed mutagenesis
Substitution mutations were generated using the QuikChange mutagenesis kit (Stratagene) according to the manufacturer’s instructions. Synthetic oligonucleotides that contained the desired bases were used. The sequence of mutations for nucleotides at positions −337 and −336 (GG-CC) of the 5′-flanking region of the SIRT1 promoter was 5′-CCGGCGGTAGTGATTTA CC GTCAGTTTGAAAGAGAAGTTG-3′ (GG-CCmutation underlined); DNA that incorporated the desired mutations was transferred into XL1-Blue supercompetent cells. Plasmid DNA was prepared and mutations were confirmed by sequencing.
SA- β -gal analysis
For SA- β -gal staining, the stably transfected cells and normal 2BS cells were washed twice with PBS, fixed for 3–5 min at room temperature in 3% formaldehyde and washed again with PBS. Cells were then incubated overnight at 37°C without CO 2 in a freshly prepared staining buffer (1 mg/ml X-gal, 40 mM citric acid/sodium phosphate, pH 6.0, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl 2 ) ( 21 ).
PPARγ acetylation
To assess the level of PPARγ acetylation, HeLa cells were transfected with a Flag-PPARγ expression vector and where indicated, an expression vector encoding p300. Acetylation status was determined by immunoprecipitation of 1 mg of protein lysate in RIPA Lysis buffer using an antibody directed against the Flag-epitope tag. Levels of acetylated PPARγ were subsequently assessed with an anti-acetyl lysine antibody (Cell Signaling). After 24 h transfected cells were treated with 100 μM Sirtinol [Sigma; dissolved in dimethyl sulfoxide (DMSO)]. Cells were incubated with Sirtinol or transfected with SIRT1-targeting siRNA oligonucleotides for the last 24 h prior to harvest, after which they were used for subsequent experiments. For the in vitro deacetylation reaction, immunoprecipitated PPARγ was incubated with IP-purified SIRT1.This reaction was performed in 20 mM Tris, pH 8.0, for 2 h at 30°C in the presence or absence of the indicated concentration of nicotinamide adenine dinucleotide (NAD) or nicotinamide. After incubation with IP-purified SIRT1, the level of acetylated PPARγ was determined by western blot analysis and the protein was identified using an anti-acetyl lysine antibody.
Statistical analysis
Analysis of variance (ANOVA) was performed where appropriate, followed by the Student’s t -test. In all cases, P- values <0.05 were considered significant.
RESULTS
Expression and transcription levels of SIRT1 declined in senescent WI-38 cells and in lung, fat and heart tissues from senescent Balb/c mice
The replicative life span in yeast cells is sensitive to the expression level of SIR2 ( 2 ). Using WI-38 cells (Normal human fibroblast cell line) as a known cellular model of replicative senescence, we evaluated the level of SIRT1 protein in young (18PDs) and senescent (45PDs) WI-38 cells by western blot analysis. The SIRT1 expression level in cell extracts decreased as the number of cell PDs increased, as shown in Figure 1 A. Similarly, we compared the level of SIRT1 in tissues from young adult Balb/c mice (4 months of age) to those of older Balb/c mice (18–24 months). As shown as in Figure 1 C, SIRT1 levels decreased significantly with age in the lung, fat and heart from these mice.
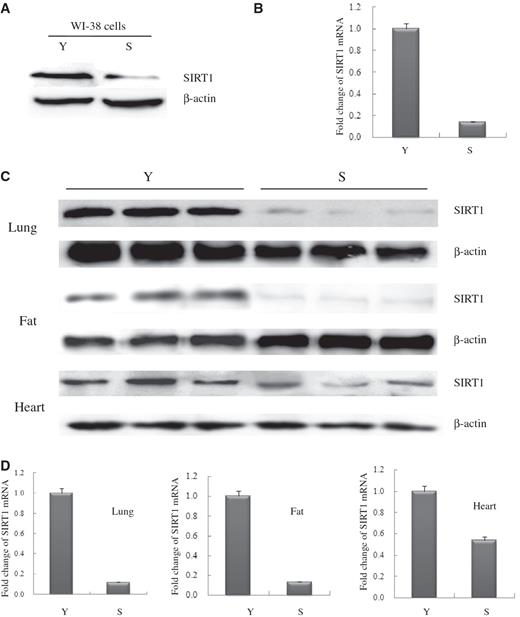
Alterations of SIRT1 mRNA and protein expression levels were determined in senescent WI-38 cells and animal tissues. ( A ) Western blot of extracts from young-15PDs (Y) and senescent-45PDs (S) WI-38 human lung fibroblasts, with actin as a loading control. Western blotting was performed using specific antibodies against SIRT1 as indicated. ( B ) Quantitative real-time PCR analysis of SIRT1 mRNA levels isolated from Y or S WI-38 cells. GAPDH transcript was used as a control. The data presented are average values obtained from triplicate data points from a representative experiment ( n = 3), which was repeated three times with similar results. ( C ) Western blot analysis of SIRT1 expression in extracts from lung, fat and heart tissues in young (Y) and senescent (S) Balb/c mice, with actin as a loading control ( n = 3 per group). ( D ) Real-time PCR analysis of SIRT1 mRNA derived from lung, fat and heart tissues in young and senescent Balb/c mice, GAPDH transcript was used as a loading control ( n = 3 per group).
To determine whether changes in the SIRT1 protein level are due to changes in SIRT1 transcription, we measured the level of SIRT1 messenger RNA (mRNA) in the same cells and tissues (including lung, fat and heart samples) via quantitative real-time reverse transcriptase-PCR (RT-PCR). We found a significant correlation between the amount of SIRT1 transcript and the age of the animal in these tissues. These results, which are shown in Figure 1 D, are consistent with our findings in cultured cells ( Figure 1 B). Our results suggested that the regulation of SIRT1 might be at the transcription levels.
PPARγ directly regulates SIRT1 transcription
As the SIR2 expression level critically influences life expectancy of both yeast and worm species ( 2 , 22 ), homeostatic regulation of this protein would seem essential. We thus wanted to determine whether PPARγ can mediate such homeostasis through direct transcriptional regulation of the SIRT1 gene. Western blot analysis revealed that PPARγ over expression or treatment with troglitazone, which is a PPARγ agonist, significantly reduced expression of SIRT1 in HeLa cells, and the greatest decrease was observed when cells both overexpressed PPARγ and were treated with troglitazone ( Figure 2 A, upper panel). To further verify this finding, we silenced the PPARγ gene and examined SIRT1 protein levels. Western blot assay revealed markedly enhanced SIRT1 expression in the PPARγsiRNA-transfected cells compared with NC-transfected cells ( Figure 2 A, lower panel). Moreover, PPARγ-agonist treatment did not reduce SIRT1 levels in PPARγsiRNA-transfected cells ( Figure 2 A, lower panel).
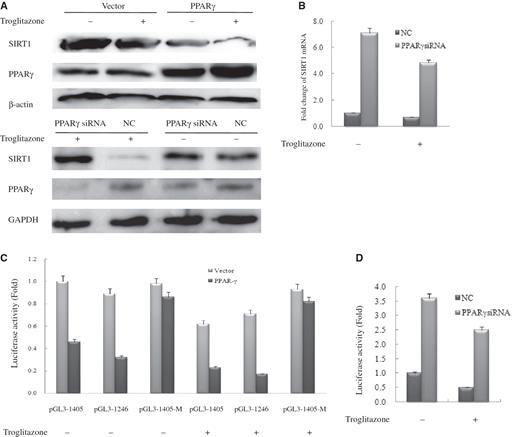
PPARγ directly regulates SIRT1 transcription. ( A ) Western blot analysis of SIRT1 and PPARγ expression in vector-transfected and PPARγ-transfected or NC-transfected and PPARγsiRNA-transfected HeLa cells with 20 μM troglitazone or DMSO (vehicle) for 48 h. Western blotting was performed using specific antibodies against SIRT1 and PPARγ as indicated. β-Actin or GAPDH was used as a loading control; NC, negative control. ( B ) Real-time PCR analysis of SIRT1 expression in NC-transfected and PPARγsiRNA-transfected HeLa cells with20 μM troglitazone or DMSO (vehicle) for 48 h. Each experiment was performed at least three times. Each bar depicts data from three independent PCR reactions (mean ± SD). ( C ) HeLa cells were transfected with expression plasmids pcDNA3.1 (vector) or pcDNA–PPARγ (PPARγ) together with wild-type SIRT1–Luc or mutant SIRT1–Luc. ( D ) HeLa cells were transfected with NC and PPARγsiRNA together with wild-type SIRT1–Luc. Cells were subsequently treated with 20 μM troglitazone. Luciferase activity was then measured 48 h after treatment. Values are the mean ±.SD of triplicate data points from a representative experiment ( n = 3), which was repeated three times with similar results.
Could the observed reduction in the level of SIRT1 have been caused by changes at the transcription level? To test this, we analyzed the levels of SIRT1 mRNA in PPARγsiRNA -transfected cells. The results are shown in Figure 2 B for (NC)-transfected and PPARγsiRNA -transfected cells. In parallel with protein levels, the level of SIRT1 mRNA was reduced by PPARγ in a ligand-dependent manner as measured by real-time quantitative PCR. Taken together, these results showed that the changes in the level of SIRT1 protein occur at the transcription level.
To further characterize these changes, we cloned a 1.9-kb SIRT1 promoter fragment which contained the PPARγ-binding site’s putative peroxisome proliferator response element (PPRE) as described in detail below, to construct a luciferase reporter (Luc). Successive deletions of the SIRT1 promoter revealed that the activity of the 1405- and 1246-bp fragments of the SIRT1 promoter increased four times relative to that of the plasmid alone in HeLa cells ( Supplementary Data, Figure 2 ). It was of interest that both these two regions contain PPRE. Subsequently, we analyzed the effects of PPARγ on the expression of the SIRT1 reporter gene, in which the luciferase gene is driven by an SIRT1 promoter. HeLa cells were transfected with various combinations of plasmids that expressed pcDNA3.1, pcDNAPPARγ or oligoPPARγsiRNA, or an NC together with both wild-type SIRT1-Luc and mutant SIRT1-Luc. Cells were then treated with troglitazone (20 μM). Reporter activity was reduced by treatment with PPARγ agonist, and this activity was 5.9-fold lower in cell lysates that contained PPARγ compared with those that did not contain PPARγ in the presence of troglitazone ( Figure 2 C). Mutation of SIRT1-Luc resulted in incomplete reduction of SIRT1 promoter activity in the presence or absence of troglitazone ( Figure 2 C). Identical results were obtained in cells transfected with PPARγsiRNA ( Figure 2 D). Thus, ligand-activated PPARγ directly regulates SIRT1 transcription.
SIRT1 interacts with PPARγ through its deacetylase catalytic domain
Protein–protein and DNA–protein interactions were next analyzed to further evaluate the relationship and interaction of SIRT1 and PPARγ. Cellular interaction between SIRT1 and PPARγ was confirmed by use of the IP assay. HeLa cells were transfected with epitope-tagged constructs encoding Myc-SIRT1 or Flag-PPARγ. IP with anti-Myc antibody, and subsequent western blotting (WB) with anti-PPARγ or anti-Flag antibody ( Supplementary Data, Figure 3 A) showed that SIRT1 can interact with PPARγ in vivo . This was also confirmed by reciprocal IP ( Supplementary Data, Figure 3 B). We also transiently expressed an epitope-tagged mutant form of SIRT1 (SIRT1 H363Y ) together with PPARγ in HeLa cells. This mutant allele specifies an amino acid substitution at residue 363 of the SIRT1 protein, replacing the histidine normally found in this position by tyrosine. This substitution would result in alteration of the highly conserved catalytic site of the SIRT1 protein, the loss of its deacetylase activity and the acquisition of a dominant-negative function. In contrast to the interaction of wild-type protein seen with PPARγ, analysis with SIRT1 H363Y resulted in reduced levels of co-immunoprecipited PPARγ [ Supplementary Data, Figure 3 A (lower panel) and B]. As shown in Supplementary Data Figure 3 A and B, IP of PPARγ from lysates demonstrated association with wild-type SIRT1, but this association was reduced by SIRT1 H363Y . To confirm that this reflected a direct physical interaction, we employed an in vitro interaction assay with recombinant GST–PPARγ. Based on the result of this GST pull-down assay ( Figure 3 A), SIRT1 interacts directly with PPARγ in vitro , which raises the possibility that PPARγ may be a substrate for SIRT1 deacetylase. Therefore, to identify the region of SIRT1 that is responsible for the SIRT1–PPARγ interaction, we generated a deletion mutant of SIRT1 designated for this study as ΔMyc-SIRT1. This mutant consists of ligation of the N-terminal and C-terminal sequences without the catalytic core domain. Our results showed that the SIRT1 catalytic domain is required for PPARγ to recruit SIRT1 ( Figure 3 B). Collectively, these data indicate that the SIRT1 catalytic domain is necessary and sufficient for the interaction of PPARγ and SIRT1.
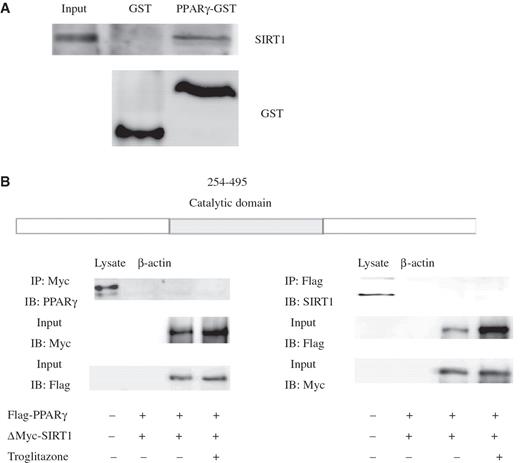
The catalytic domain of SIRT1 is required for the physical interaction of PPARγ and SIRT1. ( A ) Glutathione S-transferase (GST) pull-down assay shows that with PPARγ interacts directly with SIRT1 in vitro . Three milligrams of either purified GST or GST-PPARγ protein immobilized on glutathione 4B beads was incubated with in vitro translated SIRT1 for 2 h. After extensive washing, the beads were subjected to SDS–PAGE and probed with monoclonal SIRT1 antibody (upper panel). The lower panel shows the result of probing with monoclonal GST antibody. ( B ) HeLa cells were co-transfected with expression plasmids encoding Flag-tagged full-length PPARγ and deletion mutants of SIRT1. Immunoprecipitation and immunoblotting were performed 48 h post-transfection as indicated (+, present; –, absent).
PPARγ interacts with and recruits SIRT1 to the SIRT1 promoter template allowing SIRT1 to self-regulate
Using the ChIP assay, we found that endogenously expressed PPARγ specifically binds to the PPRE cluster on the SIRT1 promoter in HeLa cells ( Figure 4 A). As we had shown that SIRT1 interacts with PPARγ through its catalytic core domain, we speculated that these two proteins might form a functional complex in which SIRT1 may participate in the control of its own transcription. Indeed, the ChIP assay using SIRT1 antibody indicated that endogenous SIRT1 was present on its own promoter in HeLa cells ( Figure 4 A). This raised the possibility that exogenous SIRT1 influences endogenous SIRT1 transcription by forming a self-regulating feedback loop ( Figure 4 B and C). To confirm this hypothesis, we performed sequential ChIP assays, consisting first of a round of ChIP with PPARγ antibody, followed by elution of the pull-down complex, and a second round of immunoprecipitation with SIRT1 antibody. The bound DNA was then amplified for 5′ region SIRT1-binding sites. Sequential ChIP assays using SIRT1 antibody first, followed by PPARγ antibody, were also carried out. In both of these ChIP-upon-ChIP assays, endogenous PPARγ and SIRT1 co-localized on the 5′ SIRT1 promoter site, suggesting that PPARγ recruits SIRT1 to SIRT1 promoter ( Figure 4 D). As NC, PCR amplification using GAPDH primers failed to yield a significant signal ( Figure 4 A and D). Moreover, dose-dependent knockdown of PPARγ using SIRT1 antibody significantly reduced the SIRT1 ChIP signal, confirming that the binding of SIRT1 to its own promoter is mediated by PPARγ ( Figure 4 E). Neither the unrelated antibody control (β-actin) nor the NC (no antibody sample) had amplification products. Taken together, these data suggested that SIRT1 and PPARγ can form a stable transcription inhibiting complex that binds to the SIRT1 promoter and directly repress SIRT1 transcription.
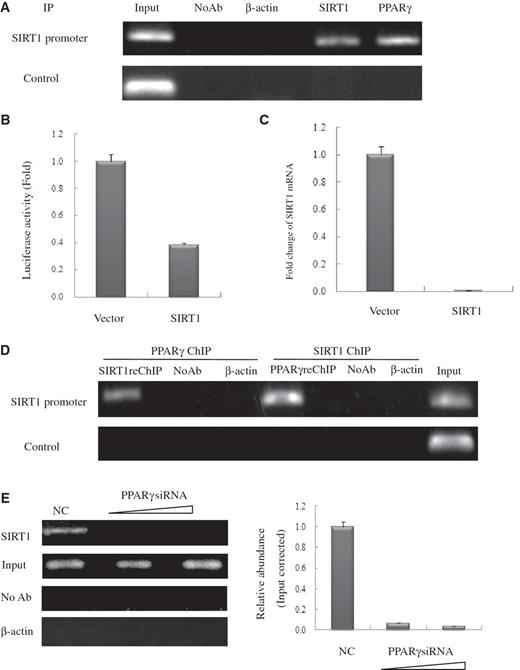
PPARγ interacts with the endogenous SIRT1 promoter and recruits SIRT1 in HeLa cells. ( A ) Soluble chromatin was prepared from HeLa cells for ChIP analysis of the SIRT1 promoter using antibodies indicated. Precipitated DNA samples were amplified by PCR using a pair of primers that amplify the –592 bp to –163 bp region with GAPDH as an unrelated primers control. ( B ) Luciferase reporter assay of the SIRT1 promoter in SIRT1 transfected HeLa cells. HeLa cells were transfected with expression plasmids vector and SIRT1 as shown, together with SIRT1–Luc. Luciferase activities were measured 48 h after treatment. Values are the mean ± SD of triplicate data points from a representative experiment ( n = 3), which was repeated three times with similar results. ( C ) Real-time quantitative PCR analysis of endogenous SIRT1 mRNA levels in SIRT1-transfected HeLa cells. HeLa cells were transfected with 10 µg of vector or pCDNA3.1-SIRT1. GAPDH transcript was used as a internal control. The error bar represents 1 SD. ( D ) ChIP-upon-ChIP assay showing the colocalization of PPARγ with SIRT1 at the SIRT1 promoter. Soluble chromatin was first immunoprecipitated with rabbit SIRT1 antibody, and the eluted product was re-immunoprecipitated with rabbit PPARγ antibody. ChIP-upon-ChIP assay was also performed with immunoprecipitation in reverse order, that is, PPARγ ChIP followed by SIRT1ChIP. ( E ) PPARγ recruites endogenous SIRT1 to its promoter in PPARγ knockdown cells in a dose-dependent manner. Real-time quantitative PCR verified the occurrence of recruitment. HeLa cells transfected with 10- or 20-nM PPARγ siRNA (triangle) were processed for ChIP uing SIRT1 antibody. The PCR primers amplified the SIRT1 promoter region as indicated in (A). For negative controls, a sample that did not contain antibody (No Ab) was immunoprecipitated, and antibody against β-actin was used as an unrelated antibody control.
SIRT1 binds p300 and deacetylates PPARγ in vivo and in vitro
Previous findings have indicated that PPARγ levels remain constant in young and senescent cells, but do not explain why the transcriptional activity of PPARγ increases in senescent cells. However, posttranslational modifications are known to affect PPARγ transcriptional activity ( 15 ). Moreover, SIRT1 acting as a protein deacetylase has been shown to deacetylate numerous transcriptional regulators, including p53, AR and Erα. Therefore, as PPARγ binds to the SIRT1 deacetylatase catalytic domain, we were prompted to ask whether human PPARγ is also subject to lysine deacetylation. This was tested, using IP with PPARγ antibody from HeLa cell lysate and an antibody that recognizes acetyl-lysine for WB, and low levels of acetylated PPARγ were found ( Figure5 A, upper panel). Treatment with sirtinol (100 μM), which is a known inhibitor of SIRT1 for 24 h ( 23 ), led to a substantial increase in PPARγ acetylation. Similar results were obtained in SIRT1siRNA-transfected cells, which resulted in reduced levels of endogenous SIRT1 ( Figure 5 A, lower panel). It is also recognized that protein acetylation is generally mediated by acetyltransferases such as the protein p300, and SIRT1 and p300 can interact ( 24 ) (also Supplementary Data, Figure 4 ) to form a complex and bind the SIRT1 promoter ( Figure 5 B). This raised the possibility that PPARγ could be acetylated by the acetyltransferase p300. As shown in Figure 5 C, the expression of p300 significantly increased PPARγ acetylation in contrast to p300DN (an acetyltransferase-impaired mutant-p300 dominant-negative form). Wild-type p300 strongly induced PPARγ acetylation; however, an enzymatically deficient mutant p300 (DN) failed to do so ( Figure 5 C). In fact, the level of PPARγ acetylation showed a clear correlation with the amount of transfected p300. Subsequently, we tested whether SIRT1 could deacetylate PPARγ. As shown in Figure 5 A and E, sirtinol treatment strongly induced PPARγ lysine acetylation but this effect was abolished by treatment with trichostatin A (TSA), which is inhibitor of class I and II histone deacetylases (HDACs) to which class III HDACs (SIRT1) are not sensitive, indicating PPARγ can be specifically deacetylated by SIRT1.
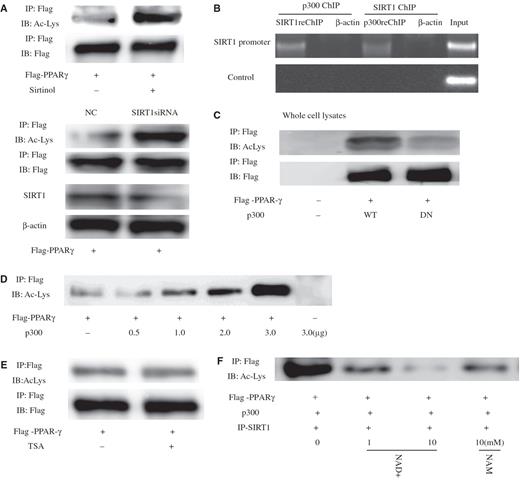
SIRT1 deacetylates PPARγ in vivo and in vitro . ( A ) Levels of PPARγ acetylation are increased by the SIRT1 inhibitor sirtinol or siRNA Knockdown of SIRT1. Where indicated, cells were treated for 24 h with 100 μM sirtinol or transfected SIRT1-targeting siRNA oligonucleotides for 24 h. PPARγ was immunoprecipitated (IP) from protein lysates using the Flag-epitope tag, and levels of acetylated PPARγ were determined by western blot (WB). ( B ) SIRT1 and p300 form complexes targeting SIRT1 to promoters. Untransfected HeLa cells were analyzed with sequential ChIP assay using SIRT1 and p300 antibodies. The PCR primers amplified the SIRT1 promoter region from –592 to –163 bp with GAPDH as an unrelated primers control, and antibody against β-actin was used as an unrelated antibody control. ( C ) Expression of p300 stimulates PPARγ acetylation. Levels of acetylated PPARγ were assessed in the presence of co-transfected p300 or an acetyltransferase-impaired mutant (p300DN). ( D ) Levels of PPARγ acetylation as a function of increasing amounts of transfected p300. ( E ) Endogenous PPARγ was acetylated in response to trichostatin A (TSA) for 8 h. Endogenous PPARγ was immunoprecipitated and subjected to western blot using antibodies directed against acetylated-lysine residues. The blot was reprobed for the total amount of PPARγ. ( F ) SIRT1 deacetylates PPARγ in vitro in an NAD-dependent manner. HeLa cells were transfected with expression plasmids encoding Flag-PPARγ and p300, and PPARγ was immunoprecipitated from protein lysates using the Flag-epitope tag. SIRT1 from cells overexpressing SIRT1 was purified by immunoprecipitation and added to immunoprecipitated PPARγ for 1 h in the presence or absence of indicated concentrations of either nicotinamide adenine dinucleotide (NAD) or NAM (nicotinamide). Levels of acetylated PPARγ were determined by western blot analysis using an antibody that recognizes acetyl-lysine (Ac-Lys) residues.
To determine whether PPARγ can also be deacetylated by SIRT1 in vitro , PPARγ and p300 vector were immunoprecipitated, and IP-purified SIRT1 was added to these immunoprecipitates. As shown in Figure 5 F, in the presence of NAD purified SIRT1 was able to deacetylated PPARγ. However, addition of NAD did not reduce PPARγ acetylation in the absence of SIRT1 ( Supplementary Data, Figure 5 ). Subsequently, as shown in Figure 6 , levels of acetylated PPARγ in young 2BS or WI-38 cells decreased significantly in comparison with those in senescent 2BS or WI-38 cells. Taken together, these data indicate that PPARγ was a deacetylase target of SIRT1 and acetylation levels of PPARγ elevated during aging.
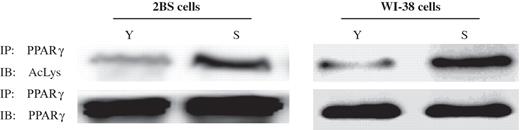
Acetylation levels of PPARγ in young and senescence 2BS and WI-38 cells. PPARγ was immunoprecipitated from protein lysates using PPARγ antibody, and acetylation levels of PPARγ were determined by western blot analysis using an antibody that recognizes acetyl-lysine (Ac-lys) residues.
SIRT1 deacetylase activity influenced appearance of senescence-associated features in 2BS cells
Cellular senescence is characterized by elevated levels of endogenous β-gal activity at pH 6.0 which may be identified by assay with X-gal which gives a blue color reaction. Young (22 PDs) and senescent (61 PDs) 2BS cells were analyzed as controls. In order to further verify that PPARγ can modulate senescence via SIRT1 deacetylase, we evaluated the role of SIRT1 deacetylase activity in mediating the senescence effects of PPARγ-shRNA in 2BS cells. Young 2BS cells infected with both SIRT1 and PPARγ-shRNA or SIRT1 H363Y and PPARγ-shRNA were monitored ( Figure 7 A). The results showed that 2BS with enhanced expression of both wild-type SIRT1 and PPARγ-shRNA displayed lower frequency of SA-β-gal staining. However, there were no significant difference in SA-β-gal activity in the mock vectors-infected cells as compared with SIRT1 H363Y and PPARγ-shRNA-infected cells ( Figure 7 B). As expected, both wild-type SIRT1 and PPARγ-shRNA-infected cells displayed young phenotypes ( Figure 7 B). Taken together, these data indicate that SIRT1 deacetylase activity caused resistance to cellular senescence.
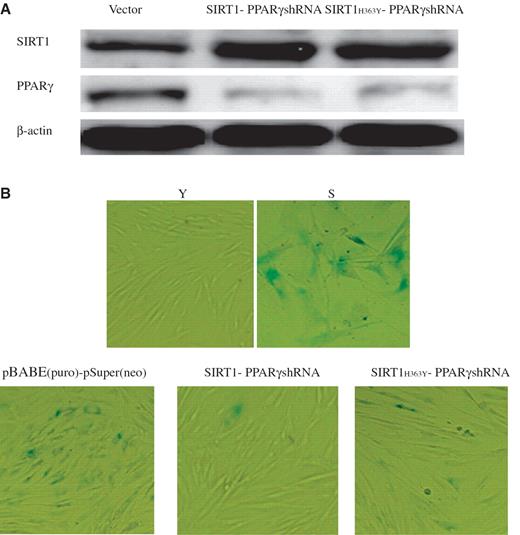
SIRT1 deacetylase activity influenced appearance of senescence-associated features in 2BS cells. 2BS cells infected with the plasmids pBABE (puro) and pSuper(neo) or pBABE-SIRT1 and pSuper-PPARγshRNA or pBABE-SIRT1 H363Y and pSuper-PPARγshRNA were analyzed for β-gal sataining which is a classical biomarker for senescence. ( A ) Western blot analysis of the SIRT1 and PPARγ protein levels in 2BS cells exposed to the indicated perturbations. Western blotting was performed using specific antibodies against SIRT1 and PPARγ as indicated. The β-actin lane is the loading control. ( B ) β-Gal sataining (blue) in 2BS cells exposed to the indicated preturbations. Stably infected 2BS cells expressing empty vector or both SIRT1 and PPARγshRNA or both SIRT1 H363Y and PPARγshRNA, were passaged until one (cells expressing empty vector) underwent senescence. Young 2BS (22PDs) cells were used as a negative control. Senescent 2BS (61PDs) cells were used as a positive control.
DISCUSSION
Senescence is the state or process of aging at the cellular level, and this process is thought to be related both to age-related diseases and to tumorigenesis ( 12 ). SIRT1 is a member of the highly conserved gene family of sirtuins, which were originally discovered in yeast as factors required for gene silencing. Sirtuins have NAD + -dependent deacetylase and adenosine diphosphate (ADP)-ribosyl transferase activity, which modulate the yeast replicative life span by suppressing genome instability through chromatin modification.
In this study, we explored the relationship between changes in the level of the mammalian homolog SIRT1 and senescence both in culture and in the aging process in mice. Both protein and mRNA levels of SIRT1, which is the mammalian ortholog of SIR2, were significantly higher in young WI-38 cells than those in senescent cells. Similarly, the amount of SIRT1 transcript is reduced in older tissues of lung, heart and fat. Although it has been reported that the level of SIRT1 mRNA does not decrease significantly with passage in IMR90 cells ( 25 ), it is likely this is because a different cell line was used, leading to differences in results. Sasaki et al . ( 25 ) reported no significant correlation between the amount of SIRT1 transcript and the age of the animal for brain, testis and thymus tested ( 25 ). In addition, we found SIRT1 transcript levels do not significantly differ in brain and kidney ( Supplementary Data, Figure 8 ). These inconsistent data regarding SIRT1 transcript patterns might be to the various roles of SIRT1 in different tissues during the progression of successive passages. Our findings suggest the possibility that SIRT1 may be modulated at the transcription level in the process of aging.
It is recognized that PPARγ activation promotes cellular senescence ( 15 ). This raised a question as to whether activated PPARγ serves to regulate SIRT1 and, if so, whether PPARγ and SIRT1 interact physically or functionally, and how this association influences the function of SIRT1 and PPARγ during aging. Our results demonstrated that SIRT1 is transcriptionally regulated by PPARγ, and also demonstrated that exogenous SIRT1 can inhibit endogenous SIRT1 at the transcription level.
Some studies have suggested that SIRT1 functions by repressing PPARγ activity ( 7 ). Our results show that not only does SIRT1 function by repressing PPARγ, but conversely PPARγ also represses SIRT1, thus forming a negative feedback loop. At the same time, this negative feedback effect and the fact that PPARγ and SIRT1 form a complex also raised the possibility that SIRT1 exerts negative self-controlling feedback.
More specifically, our results showed that SIRT1 complexes with PPARγ via its catalytic domain, and also showed that both SIRT1 and PPARγ can bind to the promoter of the SIRT1 gene and thus control homeostatic SIRT1 expression in the process of aging. SIRT1 has deactylase activity, and this strongly suggested the possibility that regulation of PPARγ activity is associated with deacetylation. Point mutation at the catalytic domain (H363Y) of SIRT1 greatly diminished the SIRT1–PPARγ interaction and demonstrated that an SIRT1 deletion mutant cannot bind to PPARγ. Moreover, the central deacetylase domain of SIRT1 is required for PPARγ binding. We were therefore led to evaluate whether PPARγ may be subject to acetylation modification and deacetylation by SIRT1, and found this is indeed the case.
The mechanism by which acetylation alters PPARγ activity is not clear. However, it has been demonstrated in general that acetylation can modify transcription factor activity by increasing the DNA-binding activity, stability or interaction with other transcription components. Studies of SIRT1 have shown that its functions are extremely extensive, including interaction with important nuclear receptors such as ERα and AR, and research has demonstrated acetylation of ERα and AR increases their transcriptional activation and protein stability ( 26–30 ). To determine why the transcriptional activity of PPARγ increases as cell ages ( 14 ), we analyzed the acetylation state of PPARγ in both young and senescent 2BS and WI-38 cells. As expected, the levels of PPARγ acetylation increased as cells aged ( Figure 5 ). SIRT1 protein directly interacts with PPARγ to physically form a complex, and this interaction might possibly control the acetylation and deacetylation balance status of PPARγ protein, thus influencing PPARγ transcriptional activity. However, we cannot at this point determine whether SIRT1 deacetylation of PPARγ is the only pathway by which PPARγ activity is regulated in the process of aging.
It has been reported that SIRT1 and p300 can interact ( 24 , 31 ), and results in this study also support this finding ( Supplementary Data, Figure 4 ). Moreover this type of interaction is similar to those which have been founded between SIRT1 and protein targets other than p300. For example, SIRT1 has been observed to bind separately to both p53 and Foxo3a ( 9 , 32–34 ), and evidence shows that p53 and Foxo3a can also bind to each other ( 35 ). SIRT1 may therefore be an important component of a number of distinct transcriptional complexes potentially acting as a scaffold to tether various members of the complex together. In addition, it is possible that SIRT1 functions as a bridge, coordinating metabolic status with transcription of key target genes with NAD dependent regulation of the activity of SIRT1.
SA- β -gal activity can distinguish senescent cells from quiescent or terminally differentiated cells and act as a biomarker of senescent cells ( 20 ). Our previous observations showed that silencing PPARγ decreased SA- β -gal activity ( 14 ) and SIRT1 overexpression led to a reduction of SA- β -gal activity ( 15 ). We want to know whether it changes senescent-like phenotype (influenced SA- β -gal activity) when we infect both SIRT1 and PPARγshRNA or both SIRT1 H363Y (dominant-negative form of SIRT1 deacetylase) and PPARγshRNA plasmids into young 2BS cells. Our results suggested that SIRT1 deacetylase activity influenced appearance of senescence-associated features in 2BS cells ( Figure 7 B).
In summary, our present data show that the longevity-regulating protein SIRT1 is under the direct control of PPARγ and that the role of SIRT1 in regulating cellular senescence potentially involves a negative feedback loop in which both PPARγ and SIRT1 participate as depicted in the model in Figure 8 . Various polyunsaturated fatty acids, which are the major dietary constituents, are specific ligands for PPARγ ( 36 ) and SIRT1 catalytic activity can be modulated by NAD and small molecular organic compounds. Based upon this model, PPARγ and SIRT1 may together engage in a complex set of circular interactions to ensure that SIRT1 exerts normal homeostatic control of cellular responses in aging and environmental conditions. Given the capacity shown by SIRT1 to repress PPARγ activity, it is clear that SIRT1 can provide a novel transcriptional and posttranscriptional regulation loop for PPARγ. Under normal physiological conditions, PPARγ represses SIRT1 transcription, thereby allowing p300 acetylation of PPARγ to enhance the function of this protein and promote aging. In addition, SIRT1 may also regulate PPARγ by a combination of PPARγ deacetylation and promoter targeting.
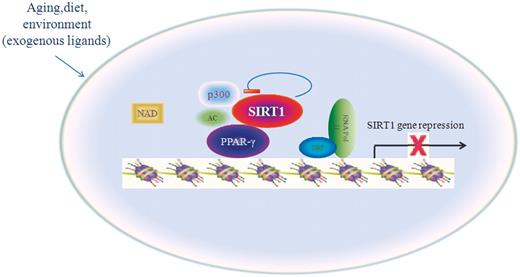
Model for the role of PPARγ in the cellular senescence. Reciprocal-type regulation of PPARγ and SIRT1 is proposed for modulation of cellular senescence in response to environmental factors. PPARγ represses the transcription of SIRT1, SIRT1 deacetylates PPARγ posttranscriptionally and PPARγ trans-inhibits SIRT1.
In conclusion, our study demonstrated that activated PPARγ represses SIRT1 gene expression during aging, in part by deacetylation. Previous research has shown that fatty acids can influence the transcriptional activity of PPARγ. In our research, PPARγ promoted senescence by reducing SIRT1 expression in a ligand-dependent manner and SIRT1 activity can be modulated by NAD and small molecular organic compounds. The model suggested by our study may offer an opportunity to observe gene–environment interactions associated with cellular senescence in health and disease.
FUNDING
Funding for open access charge: The National Basic Research Programs of China (No. 2007CB507400); National Natural Science Foundation of China (No. 30671064).
Conflict of interest statement . None declared.
ACKNOWLEDGEMENTS
We would like to express our appreciation to Dr Fuyuki Ishikawa (University of Cambridge) and Dr. Eric Lam (Imperial College School of Medicine at Hammersmith Hospital) for providing us SIRT1 cDNA expression constructs (pcDNA3.1-Myc-SIRT1 and pcDNA3.1- SIRT1 H363Y ). We thank Dr. Wengong Wang for helpful proposals.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments