





Abstract
Several sequences forming G-quadruplex are highly conserved in regulatory regions of genomes of different organisms and affect various biological processes like gene expression. Diverse G-quadruplex properties can be modulated via their interaction with small polyaromatic molecules such as pyrene. To investigate how pyrene interacts with G-rich DNAs, we incorporated deoxyuridine nucleotide(s) with a covalently attached pyrene moiety (Upy) into a model system that forms parallel G-quadruplex structures. We individually substituted terminal positions and positions in the pentaloop of the c-kit2 sequence originating from the KIT proto-oncogene with Upy and performed a detailed NMR structural study accompanied with molecular dynamic simulations. Our results showed that incorporation into the pentaloop leads to structural polymorphism and in some cases also thermal destabilization. In contrast, terminal positions were found to cause a substantial thermodynamic stabilization while preserving topology of the parent c-kit2 G-quadruplex. Thermodynamic stabilization results from π–π stacking between the polyaromatic core of the pyrene moiety and guanine nucleotides of outer G-quartets. Thanks to the prevalent overall conformation, our structures mimic the G-quadruplex found in human KIT proto-oncogene and could potentially have antiproliferative effects on cancer cells.
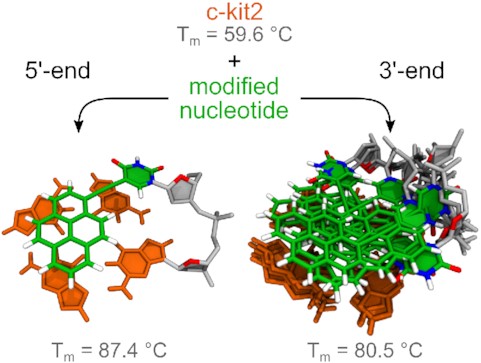
Different stacking modes of pyrene at 5′ and 3′-ends of the c-ki2 G-quadruplex.
INTRODUCTION
G-rich sequences are not randomly distributed throughout the human genome. They are concentrated in regions implicated in essential cellular processes such as initiation of DNA replication, transcription and telomere maintenance (1,2). Due to their biological significance, extensive effort has been directed towards eliciting a therapeutic response by manipulating non-B-DNA structures found in telomeric and promoter regions (3,4). The KIT receptor is a transmembrane protein that belongs to a family of growth factor receptors with intrinsic tyrosine kinase activity and participates, upon activation by endogenous ligands, in a variety of physiological processes such as cell growth, proliferation, migration and survival (5). Specific mutations and/or overexpression of the KIT proto-oncogene have been implicated in several human cancers including gastrointestinal stromal tumours, pancreatic cancer, melanoma and haematological neoplastic diseases (6,7). Due to its role in the pathogenesis of cancer, KIT is an attractive target for anti-cancer treatment (8–10). The human KIT proto-oncogene promoter contains three G-rich regions, c-kit1, kit* and c-kit2, which are capable of folding into non-B-DNA structures, specifically G-quadruplexes (11,12). These four-stranded structures consist of stacked G-quartets, planar arrangements of four guanines held together by Hoogsteen hydrogen bonds organized around a central cation (13). Structures of G-quadruplexes are highly polymorphic and their fold is influenced by various factors such as oligonucleotide sequence, loop arrangements, crowding conditions and presence of other biomolecules. The ability of c-kit1, kit* and c-kit2 sequences to fold into G-quadruplexes has been demonstrated recently, and their high-resolution structures are available in the Protein Data Bank (14–19). The three G-rich sequences in KIT are clustered and spaced only few nucleotides apart. Interestingly, the function of each G-quadruplex unit is strongly affected by formation or disruption of the remaining two (20). Furthermore, kit* and c-kit2 interact and form a tandem G-quadruplex unit (21). The promoter segment comprising kit* and c-kit2 contains putative binding sites for Sp1 and AP2 transcription factors (12,22). Apart from binding to a double-stranded consensus motif, Sp1 can also bind to a G-quadruplex (22). Considering that Sp1 binding is critical for activity of the human KIT promoter (23), highly stable G-rich oligonucleotides mimicking G-quadruplexes from KIT could be used as decoys to sequester these proteins and modulate KIT expression.
During the last decade extensive effort has been directed towards developing a variety of DNA probes based on functionalized oligonucleotides (24). Approaches for manipulating promoter G-quadruplex structures in situ often utilize small molecules that effect their thermodynamic properties and/or interfere with potential protein–DNA interactions (13). G-quadruplexes are especially suitable for targeting with planar organic compounds with an extended aromatic core structure that can stack through π–π interactions on outer G-quartets (25,26). Unfortunately, development of new non-covalent binders is hindered by serious selectivity issues (27). Alternatively, polyaromatic groups can be covalently attached to short DNA oligonucleotides at desired position(s) and delivered to cells where they elicit a therapeutic response.
Pyrene is a polycyclic aromatic hydrocarbon, which versatility can be extended with chemical modifications. Thanks to its unique fluorescence properties, pyrene-modified oligonucleotides can act as microenvironment-sensitive fluorescent probes (28,29), excimer- or exciplex-generating molecules (30–36), nucleic acid complex intercalators (37–40) and π–π stacking hydrophobic moieties (41,42). Photophysics of pyrene-labelled compounds has been studied extensively, whereas only a limited number of studies focused on the structure of pyrene-conjugated nucleic-acid-based constructs. Recently, we have shown that incorporation of a pyrene-conjugated uridine building block into the thrombin binding aptamer (TBA) can lead to thermal stabilization and increased resistance against nuclease degradation under human serum conditions (43). Although TBA is often used as a model system, its antiparallel chair-like topology is unique. We have also shown that short pyrene-conjugated oligonucleotides can recover the G-quadruplex fold of G-rich oligonucleotides after oxidative damage (44). On the other hand, Xodo et al. utilized a pyrene-conjugated G-rich sequence derived from the KRAS oncogene as a stable G-quadruplex decoy for sequestering transcription factors (45). A reduction of pancreatic tumour xenografts in mice supported the decoy approach as a promising cancer therapy.
In the current study, we focus on the G12T/G21T mutant of promoter c-kit2 sequence, d(CGGGCGGGCGCTAGGGAGGGT), (hereby referred to as c-kit2) as a representative of a monomeric parallel G-quadruplex. In parallel G-quadruplex topologies with propeller loops, outer G-quartets are more exposed to the solvent compared to G-quadruplexes with diagonal or edge-type loops. We hypothesized that large exposed surface areas of guanine nucleobases would be ideal for stacking with polyaromatic moieties such as pyrene. Introduction of a pyrene-conjugated nucleotide, 5-(1-pyrenylethynyl)-2′-deoxyuridine (Upy), in c-kit2 was expected to increase thermal stability. However, as the pyrene moiety in Upy is covalently tethered to the uracil nucleobase, careful positioning of this bulky building block will likely to be required not to perturb the G-quadruplex structure. Positions in the structure with more degrees of freedom such as overhangs and long non-structured loops should tolerate substitutions with Upy without causing alterations in G-quadruplex topology. We individually substituted Upy at terminal positions and all positions of the pentaloop in the c-kit2 sequence and evaluated the effect of pyrene moieties on the G-quadruplex forming ability, structure and thermodynamic stability. Upy at all positions adjacent to the core of G-quadruplex was expected to stack on outer G-quartets and result in thermodynamic stabilization. However, substitutions in the pentaloop caused structural polymorphism. On the other hand, introduction of pyrene moieties to individual or both termini of the c-kit2 sequence resulted in highly stable G-quadruplex structures. Unexpectedly, pyrene moiety at the 3′ terminus featured increased dynamics that hindered detailed NMR analysis. Therefore, we had to bridge the gap by state-of-the-art simulation techniques to describe its complex structural properties with atomistic resolution.
MATERIALS AND METHODS
Sample preparation
The 5-pyrene-dU-CE phosphoramidite and other standard phosphoramidites were purchased from Glen Research. Isotopically labelled phosphoramidites were purchased from Cambridge Isotope Laboratories. All oligonucleotides were synthesized on a K&A Laborgeraete H-8 DNA/RNA Synthesizer employing standard phosphoramidite chemistry. Natural abundance oligonucleotides were prepared in DMT-on mode, deprotected with AMA solution (1:1 mixture of aqueous 30% ammonium hydroxide and 40% aqueous methylamine) at 65°C for 30 min and purified with Glen-Pak cartridges. During the purification process failed sequences were eliminated and the DMT was subsequently removed. The solvent was evaporated and samples were dissolved in 100 mM LiCl following desalting using FPLC. Site-specific 10% 15N- and 13C-labelled samples were prepared in DMT-off mode, deprotected with AMA solution at 65°C for 30 min, dissolved in 100 mM LiCl and purified with FPLC. All oligonucleotides were dissolved in a solution containing 10% D2O and 20 mM KCl, 5 mM K-phosphate buffer (pH 7.0) with DNA concentration ranging from 0.5 to 1.0 mM, if not stated otherwise. NMR titration of 100 μM c-kit2, c-kit2_py1 and c-kit2_py21 samples with the sodium salt of 1-pyrenesulfonic acid and hydroxypyrene was performed at 0.0, 0.5, 1.0, 2.0, 3.0 and 4.0 molar equivalents. NMR samples with different ratios of oligonucleotide and hydroxypyrene were prepared by dissolving dry hydroxypyrene in DMSO-d6 and titrating 15 mM hydroxypyrene solution in DMSO-d6 into an aqueous solution of the oligonucleotide. Concentrations of DMSO-d6 used in this study did not cause any spectral differences, which could account for structural changes of DNA influenced by DMSO-d6.
Oligonucleotide concentration was determined by measuring ultraviolet absorbance at 260 nm using the Varian CARY-100 BIO UV-VIS UV/VIS Spectrophotometer with 1.0 cm light path cells. An extinction coefficient of 202 200 M−1 cm−1 was calculated with the nearest neighbour method for the non-substituted c-kit2 oligonucleotide and used for all samples.
UV melting profiles
Samples for UV melting experiments were prepared by diluting NMR samples with a blank solution containing 20 mM KCl and 5 mM K-phosphate buffer (pH 7.0). UV melting experiments were performed on a Varian CARY 3500 UV-VIS spectrophotometer with the Cary Win UV Thermal program using 1.0 cm light path cells. Samples were heated/cooled at 0.5°C min-1 in the range of 10–95°C, and absorbance at 295 nm was measured at 0.5°C steps. UV melting experiments were replicated 5 × for each oligonucleotide. T1/2 of each measurement was determined from the first derivation of A295 versus temperature plot. Final T1/2 was calculated as the average of 5 measurements. The van’t Hoff enthalpies (ΔHm) were determined by fitting the slope of melting curves at T1/2 and using the equation |${\rm{\Delta \ }}{H_m} = \ - 4RT_m^2( {\delta \alpha /\delta T} )$| (46).
Fluorescence spectroscopy
Fluorescence properties of 150 μM c-kit2, c-kit2_py1, c-kit2_py21 and c-kit2_py1/21. G-quadruplexes in 20 mM KCl and 5 mM K-phosphate buffer (pH 7.0) solution were measured on QuantaMaster (PTI) spectrofluorometer at 25°C using a quartz cuvette with 1.0 cm path length and excitation wavelength of 330 nm.
NMR experiments
NMR data were collected on Bruker AVANCE III HD and AVANCE NEO 600 MHz NMR spectrometers equipped with cryogenic probes in the temperature range from 0 to 80°C. For suppression of water signals, the excitation sculpting pulse sequence element was used. NMR spectra were externally referenced to Sodium trimethylsilylpropanesulfonate (DSS). Translation diffusion coefficients were obtained with the use of a stimulated echo experiment using bipolar gradients. DOSY was performed at 15°C due to reduced resonance overlap and a favourable population ratio. 1D 15N-edited HSQC and 2D 13C-edited HSQC experiments were performed with 15N-, 13C-labelled oligonucleotides. 2D NOESY spectra were acquired at mixing times 70, 100, 150, 200 and 250 ms, 2D TOCSY at mixing time 65 ms and 2D ROESY at mixing time 120 ms. 1H-13C HSQC spectra measured on natural abundance samples were used to distinguish between syn and anti conformations of glycosidic bonds. NMR spectra were processed and analysed using TopSpin (Bruker). Resonance assignment and integration were done with Sparky (NMRFAM) software (47).
Interproton distances were calculated from 250 ms NOESY spectra, which were chosen from NOE build-up curves with C9 H5-H6 distance used as a reference (2.479 Å). Based on calculated distances, cross-peaks were divided into four categories (strong, medium, weak, very weak), which corresponded to plateaus in applied flat bottom potentials (1.8–3.6 Å, 2.6–5.0 Å, 3.5–6.5 Å, 4.5–7.5 Å) during simulated annealing. Hydrogen bonds within G-quartets were restrained (>3.0 Å). Further, potassium cations were placed in between adjacent G-quartets and restrained to guanine O6 atoms (>4.0 Å) as a physically sound alternative to planarity restraints. Harmonic potential was applied above and below these ranges with a force constant of 20 kcal mol−1 Å−2.
Flat bottom potentials for dihedral angles χ were -90° to 90° for syn and 90° to 270° for anti nucleotides. Restraints were also applied to backbone dihedral angles α (−120° to 120°), β (150° to 210°), γ (30° to 90°), δ (130° to 190°), ϵ (170° to 300°) and ζ (−120° to 120°). Employed force constant was 200.0 kcal mol−1 rad−2. Sugar puckering was restrained via ν0, ν1, ν2, ν3 and ν4 dihedral angles, which were calculated from pseudorotation phase set between 144° and 180° using standard AMBER procedure. Dihedral angle definition is shown in Supplementary Figure S1. All sugar puckers were determined to be of S-type (C2’-endo) based on 2D TOCSY NMR spectra.
Molecular dynamics
A simulated annealing protocol was used for c-kit2_py1 using GPUs and pmemd.cuda 18.0 and OL15 force field (48–51) with Generalized Born/Surface Area implicit solvent model (igb = 1). Additionally, Upy was fitted with one extra pseudo-torsion term to correctly describe rotation of pyrene moiety and uracil nucleobase with respect to the linker (Supplementary Data, Force Field Parameterization, Supplementary Figure S2). A starting structure was prepared by substituting Upy into position 1 of c-kit2 NMR structure (PDB ID 2KYP). Before each run, the structure was annealed at 1200 K for 200 ps using all restraints except NOE distance restraints that involved Upy1 to remove the bias from a single starting structure (Supplementary Figure S3). Simulation following annealing was 200 ps long and consisted of several phases: 5 K → 1200 K (1 ps), 1200 K (39 ps), 1200 K → 600 K (50 ps), 600 K → 300 K (50 ps), 300 K → 0 K (60 ps) and final geometry optimization (5 000 steps). In total, 1000 repeats were performed. Ten structures with the lowest sum of potential and restraint energy were further refined by molecular dynamics (MD) in the explicit solvent.
Each selected structure was immersed into a truncated octahedral box filled by the TIP3P water with potassium and chloride ions. Resulting concentration of KCl was approximately 0.14 mM and radius of the largest inscribed sphere was about 24 Å. Temperature and pressure were kept at 300 K and 100 kPa, respectively. NOE and hydrogen bond distance restraints as well as χ torsion angles and torsion angles within the ribose ring were active during the refinement. After 1 μs of simulation, the systems were cooled to 0 K during 10 ns at a constant volume and then geometry optimized (5000 steps) leading to the final NMR ensemble. Alongside, a same set of simulations was performed without any restraints to assess their effect on the obtained structure.
Molecular dynamics simulations of c-kit2_py21 closely followed settings used during refinement of the NMR structures in the explicit solvent (vide supra). The initial configurations of c-kit2 and c-kit2_py21 systems were taken from the previous tests (Supplementary Data, Testing Molecular Dynamics, Supplementary Figures S4–S6). Collected trajectories were 1 μs long. Because exchange between states observed in NMR is far beyond the timescale of regular MD, we decided to investigate possible stacking modes of Upy in c-kit2_py21 using biased MD providing also free energies of sampled configurations. The bias was introduced along geometrical parameters (collective variables, CVs) developed by the analysis of the testing runs and are shown in Supplementary Figure S7 including used restraint. Biased MD was done in a modified pmemd program coupled with PMFLib (https://pmflib.ncbr.muni.cz) using CPUs due to complexity of employed CVs and PMFLib design.
Corresponding free energy surface was calculated by the adaptive biasing force (ABF) method (52) enhanced by the multiple-walker approach (MWA) (53). The calculated mean forces were integrated using Gaussian process regression (GPR) (54) to get the final free energy landscape. ABF/MWA simulation accumulated 3 μs of sampling, which ensured converged free energy. Standard deviations (errors) of the free energy were calculated by Gaussian process from standard deviations of mean forces. Statistical inefficiency due to correlation in time series was evaluated by optimizing a GPR hyperparameter, which was achieved by maximizing logarithm of marginal likelihood. Free energies are reported with confidence intervals provided at three standard deviations (99.7% confidence level). More details on the procedure are provided in (55).
Analysis of simulations
Trajectories were processed and analysed using the cpptraj module of AMBER and the CATs package. Further, CATs were connected with PMFLib and served for finer filtering of the ABF/MWA trajectories by values of collective variables and calculated free energies. Thermodynamic states corresponding to the local minima found on the free energy surface were represented by ensembles of structures sampled in the ABF/MWA trajectories that were ≤0.3 kcal mol−1 (0.5RT at 300 K) above the free energy minima. If a free energy minimum contained differing conformations, structures were clustered. Visual representation of each thermodynamic state was prepared by randomly selecting and partially optimizing a structure from these ensembles. Average structures could not have been used due to flexibility of loop nucleotides. All structures were rendered using VMD (56).
RESULTS
Position of Upy substitution influences c-kit2 fold
Terminal positions and all positions of the pentaloop in c-kit2 were individually substituted with Upy and the capability of constructs to form G-quadruplex was evaluated through examination of 1D 1H NMR spectra (Supplementary Figures S8 and S9). Oligonucleotides c-kit2_py9, c-kit2_py10, c-kit2_py11, c-kit2_py12 and c-kit2_py13 (hereby referred to as loop Upy analogues) exhibited broad NMR resonances suggesting formation of multiple structures in solution and/or aggregation (Supplementary Figure S8). On the other hand, favourable spectral properties were attained with oligonucleotides c-kit2_py1, c-kit2_py21 and c-kit2_py1/21 (hereby referred to as terminal Upy analogues), in which Upy was substituted at the 5′ and/or 3′ terminus of c-kit2 sequence (Figure 1 and Supplementary Figure S9). Guanine imino resonances of c-kit2_py1 and c-kit2_py21 were unambiguously assigned with the help of 15N-edited HSQC spectra acquired on partially (10%) residue-specifically 15N-enriched oligonucleotides (Supplementary Figures S10 and S11). Imino region of the c-kit2_py1 spectrum with its 12 resonances indicates formation of a G-quadruplex structure with three G-quartet planes. Differences in linewidths amongst imino resonances of c-kit2_py1 (Δν1/2 ≈ 7 Hz for G2 and 15 Hz for G6) suggest different levels of exposure to exchange with bulk solvent and/or increased local dynamics (vide infra). Moreover, a set of broader, low intensity signals (<10%) suggests the presence of an additional minor structure. The major and minor structures of c-kit2_py1 exhibit different translational diffusion coefficients of 9.3 × 10−11 and 7.4 × 10-11 m2 s−1 at 15°C, respectively, which suggest their monomeric and dimeric nature (Supplementary Figure S12). 1H NMR spectrum of c-kit2_py21 exhibits nine major guanine imino resonances in the range from δ 11.0 to 12.0 ppm and four lower intensity imino resonances in the range from δ 10.3 to 11.0 ppm. Interestingly, one major and two lower intensity imino resonances were assigned to G20 suggesting presence of three distinct conformations that are in slow exchange on the NMR time scale. Two upfield imino resonances of lower intensity in the spectrum of c-kit2_py21 were assigned to G4 and G8, while no imino resonance for G16 was observed at 25°C. Favourable spectral properties were also attained when c-kit2 was double substituted at positions 1 and 21 (Figure 1 and Supplementary Figure S9). Imino region of c-kit2_py1/21 spectrum contains two sets of resonances in the range from δ 10.2 to 11.9 ppm, which suggest presence of major (>85%) and minor structures (<15%). Imino resonances corresponding to the major structure were assigned to 11 G-quartet forming guanines, while no imino resonance for G16 was observed at 25°C. Interestingly, three imino resonances were assigned to G20 suggesting presence of three distinct conformations in the major structure of c-kit2_py1/21.
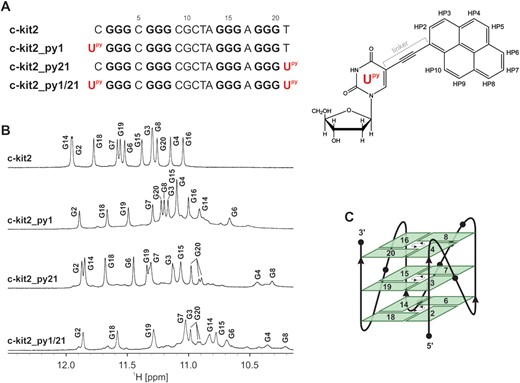
(A) Sequences of c-kit2 and terminal Upy analogues (Upy highlighted in red) (left). Guanines involved in G-quartet formation are in bold. Chemical formula of 5-(1-pyrenylethynyl)-2′-deoxyuridine (Upy) with atom numbering of pyrene moiety (right). (B) Imino regions of 1D 1H NMR spectra of c-kit2 and terminal Upy analogues with assignment. Spectra were acquired in 90% H2O and 10% D2O, 20 mM KCl and 5 mM K-phosphate buffer, pH 7 on a 600 MHz NMR spectrometer. Oligonucleotide concentrations ranged from 0.7 to 1.0 mM. (C) Schematic representation of the c-kit2 promoter G-quadruplex topology. Hydrogen-bond directionality is indicated with arrows in the centre of each G-quartet.
Robustness of the c-kit2 fold was evaluated by extending the parent sequence and terminal Upy analogues by up to three nucleotides in each direction. Additional nucleotides at the 5′- or 3′-end of c-kit2 resulted in only minor chemical shift changes of imino resonances indicating that the G-quadruplex fold is not perturbed (Supplementary Figures S13 and S14). While extensions at 5′-end of all three terminal Upy analogues resulted in occurrence of multiple structures in solution and/or aggregation (Supplementary Figures S15–S17), extensions at 3′-end provided well-resolved 1D 1H NMR spectra with a single major structure in solution (Supplementary Figures S18–S20). Interestingly, addition of nucleotide(s) at the 3′-end of Upy21 in c-kit2_py21 and c-kit2_py1/21 resulted in sharpening of upfield G4 and G8 imino resonances (Supplementary Figures S19 and S20). This suggests reduced dynamics of Upy21 most likely caused by stacking with 3′-flanking nucleotide(s).
UV absorbance and fluorescence spectra of c-kit2 and its terminal Upy analogues were acquired in the wavelength region characteristic for the pyrene moiety (Supplementary Figure S21). Expectedly, c-kit2 does not exhibit any fluorescent properties and any absorbance above 320 nm. On the other hand, all three terminal Upy analogues show absorption between 360 and 420 nm being two times higher for c-kit2_py1/21 than for c-kit2_py1 and c-kit2_py21. Respective fluorescence spectra exhibit a fluorescence maximum near 450 nm. Compared to c-kit2_py1/21, fluorescence maximum of c-kit2_py1 and c-kit2_py21 is blue-shifted by 13 and 5 nm, respectively. While c-kit2_py21 exhibits only slightly higher fluorescence intensity than c-kit2_py1/21, c-kit2_py1 exhibits the highest intensity of all terminal Upy analogues (Supplementary Figure S21).
Upy substitutions can result in substantial thermal stabilization
UV-melting experiments showed that all terminal Upy analogues exhibit higher thermal stability compared to parent c-kit2. While the melting profile of c-kit2 is classified as monophasic, melting profiles of c-kit2_py1 and c-kit2_py21 are more complex and display two transitions (Figure 2). Low temperature transitions observed at approximately 55.0 and 60.0°C for c-kit2_py1 and c-kit2_py21, respectively, exhibit small absolute changes in absorbance and may correspond to melting of minor species. High temperature transitions occur at 87.4 and 80.5°C for c-kit2_py1 and c-kit2_py21, which represents a 27.8 and 20.9°C increase compared to c-kit2, respectively (Supplementary Table S1). Expectedly, c-kit2_py1/21 exhibits an even larger gain in thermal stability. However, its melting temperature could not be extracted from UV data due to limitations related to the boiling point of the solvent (Supplementary Figure S22). We observed a hysteresis in the melting profiles of the parent c-kit2 and all terminal Upy analogues indicating that the structural transition is slow relative to the temperature gradient. A simple thermodynamic analysis revealed that the pyrene moiety at the 5′-end (c-kit2_py1) is enthalpically favourable compared to the 3′-end (c-kit2_py21) by 9.2 kcal mol−1 (Supplementary Table S1).
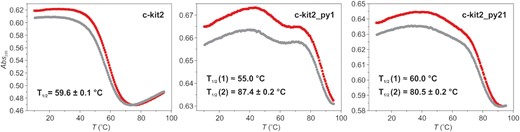
UV melting profiles of c-kit2, c-kit2_py1 and c-kit2_py21. Data were acquired for ∼10 μM DNA with 20 mM KCl and 5 mM K-phosphate buffer, pH 7. UV-melting experiments were initiated by equilibrating samples at 10°C for 10 min before being heated to 95°C (red curve) then after 10 min at 95°C the temperature was reduced to 10°C (grey curve). Temperature was ramped at 0.5°C/min and absorbance was followed at 295 nm. T1/2 values were extracted form red curves.
Although none of the substitutions in the pentaloop of c-kit2 results in well-resolved NMR spectra (Supplementary Figure S8), three loop Upy analogues (c-kit2_py9, c-kit2_py10 and c-kit2_py13) exhibit higher T1/2 values than c-kit2. The highest stabilization by 16.5 and 5.5°C is observed for Upy substitutions at positions 9 and 13, respectively (Supplementary Figure S22). Interestingly, these correspond to the 5′- and 3′-ends of the pentaloop in the parent G-quadruplex structure. On the other hand, Upy substitutions at positions 11 and 12 result in 1.5 and 15.0°C decrease in T1/2 (Supplementary Figure S22).
Parallel fold is retained upon Upy substitutions
Imino, aromatic and anomeric resonances of c-kit2_py1 and c-kit2_py21 were assigned by analysing NOE connectivities (Figure 3 and Supplementary Figure S23). Assignment of guanine aromatic protons was confirmed through acquisition of 13C-edited HSQC spectra using partially (10%) residue-specifically isotopically enriched oligonucleotides (Supplementary Figure S24). The majority of imino, aromatic and anomeric resonances of c-kit2_py1/21 could be assigned through sequential connectivities and by comparing their chemical shifts with those for c-kit2_py1 and c-kit2_py21. Sequential walks (H1’n-H8/H6n+1) in NOESY spectra of all terminal Upy analogues were interrupted at G4-C5, C5-G6, G16-A17 and A17-G18 steps due to large interproton distances resulting from single nucleotide double-chain-reversal loops. Furthermore, a lack of C9H6-G8H1’, G10H8-C9H1’ and C11H6-G10H1’ cross-peaks in NOESY spectra of all terminal Upy analogues was attributed to dynamic behaviour of the pentaloop. NOESY spectra of c-kit2_py1 contained intense exchange cross-peaks between major and minor components, which complicated the signal assignment. However, they could be unequivocally distinguished from true NOE cross-peaks with the help of ROESY spectra (Supplementary Figure S25).
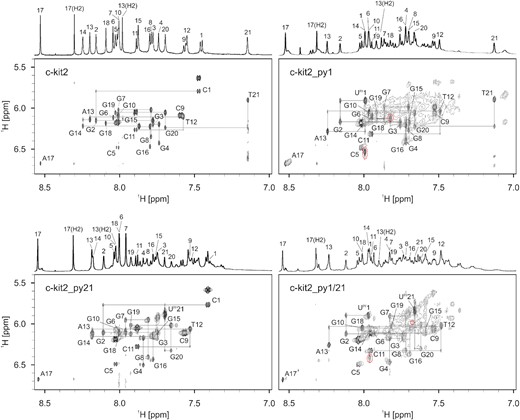
Aromatic-anomeric regions of 2D NOESY spectra (τm = 250 ms) with traced sequential walks for c-kit2 and terminal Upy analogues. The intra-nucleotide H8/H6(n)-H1'(n) cross-peaks are labelled. Red dashed circles in the spectra of c-kit2_py1 and c-kit2_py1/21 mark exchange cross-peaks between different species. Spectra were acquired in 90% H2O and 10% D2O, 20 mM KCl and 5 mM K-phosphate buffer, pH 7 on a 600 MHz NMR spectrometer. Oligonucleotide concentrations ranged from 0.7 to 1.0 mM.
Examination of inter-nucleotide NOE connectivities between imino-imino and imino-aromatic protons revealed that topology of the parent c-kit2 G-quadruplex remained unperturbed despite introduction of Upy substitutions. NMR data show that all terminal Upy analogues adopt a parallel G-quadruplex topology consisting of G2-G6-G14-G18, G3-G7-G15-G19 and G4-G8-G16-G20 quartets (Figure 1C). All guanines forming the G-quadruplex core adopt anti glycosidic conformations and G-quartets exhibit counter-clockwise hydrogen bond donor-acceptor directionalities. The parallel G-strands of terminal Upy analogues form four medium width grooves and are linked by three double-chain-reversal loops.
c-kit2_py1 features a single stacking mode of Upy
1H NMR resonances of the pyrene moiety in the monomeric G-quadruplex adopted by c-kit2_py1 were found in the range from δ 7.5 to 8.4 ppm. Although pyrene resonances are heavily overlapped with aromatic resonances of nucleobases, their unique spin-spin coupling network (resulting in eight doublets and one doublet of doublet) allowed us to unequivocally identify and assign them with TOCSY and DQF-COSY experiments. An intense H6-H1' cross-peak in a NOESY spectrum acquired with a short mixing time of 70 ms suggested the syn conformation for Upy1, whereas remaining nucleotides in c-kit2_py1 adopt anti conformations. The syn conformation of Upy1 is supported by a higher-frequency shift of its C6 resonance (δ13C ≈ 145 ppm) with respect to δ13C ≈ 135 ppm observed for guanine nucleotides (Supplementary Figure S26) (57).
Quantum mechanical calculations revealed that rotation of the pyrene moiety with respect to the uracil nucleobase of Upy1 is not free and two coplanar conformations (M1 and M2) are preferred (Supplementary Figure S2). Conformation M1 is characterized by a short distance between Upy1H6 and HP2. Rotation of the pyrene moiety with respect to the uracil nucleobase by 180° results in conformation M2 with Upy1H6 facing towards HP10 (see Figure 1). Observation of the Upy1H6-HP10 NOE cross-peak in NMR spectra showed that Upy1H6 is positioned closer to HP10 than to HP2, which suggest that Upy in c-kit2_py1 adopts conformation M2. This experimental observation does not agree with quantum mechanical calculations, which showed slight energetic preference for conformation M1 most likely due to the stabilizing contact HP10-O4 and the absence of other supramolecular interactions in our isolated model. Besides Upy1H6-HP10 cross-peak, we observed five inter-nucleotide NOE contacts involving pyrene protons, which defined orientation of the pyrene moiety and its stacking with the G2-G6-G14-G18 quartet (Supplementary Figures S23 and S27).
A solution-state structure of c-kit2_py1 was determined on the basis of NMR observables using a restrained simulated annealing protocol. A series of 1000 simulations resulted in a well converged set of structures. Ten structures of c-kit2_py1 with the lowest sum of potential and restraint energy (NMR ensemble) were selected and refined via explicit solvent simulation before further analysis (Figure 4A). c-kit2_py1 forms a well-defined G-quadruplex consisting of three G-quartets, parallel strand orientations and propeller-type loops. The C9-G10-C11-G12-A13 pentaloop in c-kit2_py1 (average RMSD of pentaloop 1.62 ± 0.86 Å) retains its propensity for internal stacking. Due to the Upy substitution at position 1, the C1:A13 non-canonical base pair, observed in the parent c-kit2, cannot be formed. Consequently, the G2-G6-G14-G18 quartet is available for stacking interaction with the pyrene moiety and uracil nucleobase of Upy1. The pyrene moiety was found to be located predominantly above G6 and G14, while the uracil nucleobase is located mostly above G18 (Figure 4B). The value of Upy1 glycosidic torsion angle χ averages to 34° for ten structures of the NMR ensemble, which agrees with the experimentally determined syn conformation. Interestingly, both pyrene moiety and uracil nucleobase of Upy1 exhibit lateral motion above the G2-G6-G14-G18 quartet (Figure 4C). All sugars in c-kit2_py1 adopt the S-type conformation (Supplementary Table S2).
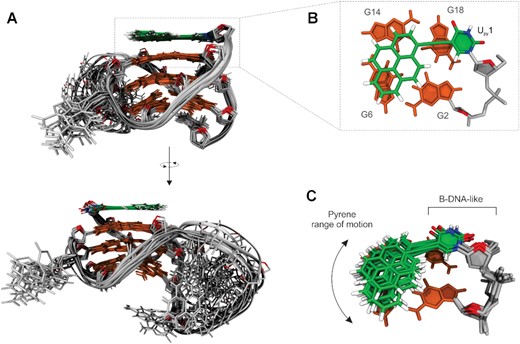
Solution-state structure of c-kit2_py1. (A) Superposition of 10 lowest energy structures from the NMR ensemble refined in the explicit solvent. (B) Details of Upy1 stacking on the top of G2-G6-G14-G18 quartet. (C) Superposition of Upy1 conformations demonstrating range of motion sampled by the pyrene moiety, while deoxyuridine linker adopts a B-DNA-like conformation. Upy1 is shown in green and G-quartets in orange. Loops and backbone are in grey with sugar O4’ atoms highlighted in red.
Structures of the c-kit2_py1 NMR ensemble were also subjected to 1 μs long unrestrained explicit solvent MD simulations. Stability of the systems without restraints suggested good convergence of the NMR ensemble (Supplementary Figure S28). Fast Upy1 syn to anti transitions were the only observed difference compared to the distance restrained trajectories. The less populated (23%) anti conformation results from rotating the sugar 5′ terminus of Upy1 towards G2 (Supplementary Figure S28C) and might represent a short-lived higher energy state. Overall, the Upy1 syn–anti ratio in the unrestrained simulation (χ averaged to 58° for syn and -164° for anti) is in line with the NOE-based assessment (Supplementary Figure S29). All sugars averaged to the S-type conformation but with a wider range of values (Supplementary Table S3). Elimination of restrains also resulted in pronounced dynamic behaviour of the pentaloop during MD simulations (average RMSD of pentaloop 3.65 ± 0.55 Å). We observed transient stacking of the 3′-end residue of the pentaloop (A13) with the pyrene moiety and of the 5′-end residue (C9) with the G4-G8-G16-G20 quartet (Supplementary Figure S28A). A nucleus-independent chemical shift (NICS) approach showed that G6 and G14 imino resonances shift to lower frequencies upon stacking of the pyrene moiety (Supplementary Figure S30 and Supplementary Table S4). This agrees with NMR data where imino resonances of G6 and G14 in c-kit2_py1 were found shielded with respect to those in c-kit2 (Figure 1B). However, not so clear agreement was seen for the ensemble refined with restraints. This suggests that the time-averaged restraints might not capture well transient positions of pyrene. Although, exact values of NMR shifts could not be reproduced due to limitation of the computational method (Supplementary Table S4).
Upy stacking modes in c-kit2_py21 are in dynamic equilibrium
Presence of three distinct stacking modes of Upy21 with the G4-G8-G16-G20 quartet was identified in 1H NMR spectra of c-kit2_py21 measured above 40°C. These exhibited three sets of imino resonances for G20, G16 and G8, and two (possibly overlapped) resonances for G4 (Figure 5A and Supplementary Figure S31A). Additionally, we observed doubling of G3 and G19 imino resonances from the central G-quartet (Supplementary Figure S31A). Two out of three stacking modes exhibited a downfield C6 chemical shift (δ13C ≈ 145 ppm) for the Upy21 residue, which suggest its syn glycosidic conformation (57). On the other hand, the last Upy21C6-H6 cross-peak exhibited a chemical shift of δ13C ≈ 135 ppm suggesting its anti glycosidic conformation (Figure 5B).
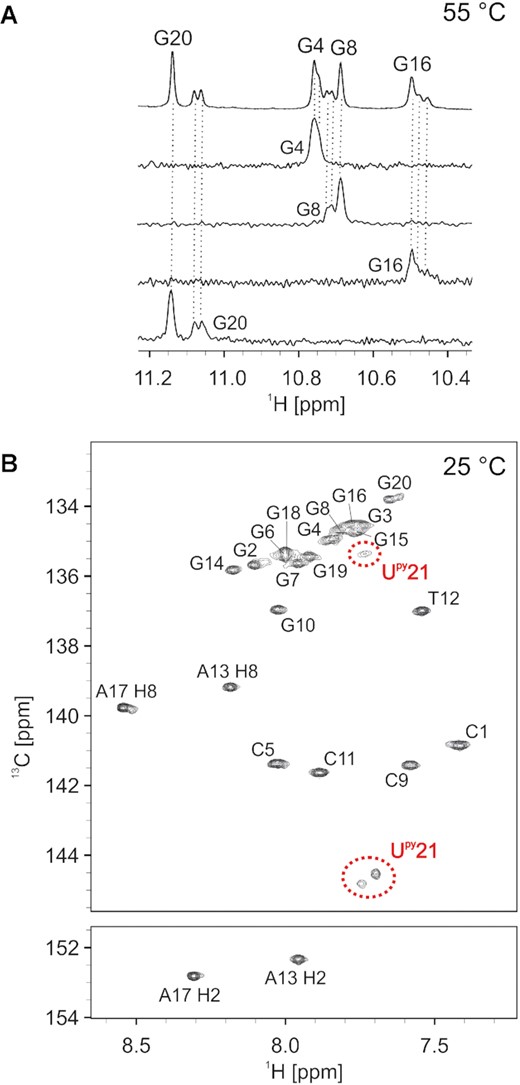
(A) Imino region of G4-G8-G16-G20 quartet of a 1D 1H NMR spectrum of c-kit2_py21 with 1D 15N-edited HSQC spectra below. Spectra were acquired at 55°C. Oligonucleotide concentration varied from 0.6 to 0.9 mM. (B) 2D 1H-13C HSQC spectrum of c-kit2_py21 acquired at 25°C with assignment. Three signals observed for Upy21 are marked by red dashed circles. The spectrum was acquired with 90% H2O and 10% D2O, 20 mM KCl, and 5 mM K-phosphate buffer, pH 7, on a 600 MHz NMR spectrometer.
Analysis of NOESY spectra (Figure 3) revealed that c-kit2_py21 adopts the same parallel G-quadruplex topology consisting of three G-quartets as the parent c-kit2. However, all imino resonances of the G4-G8-G16-G20 quartet were found to be shifted upfield in c-kit2_py21 compared to c-kit2, which suggest that all distinct conformations of Upy21 feature a shielding effect of the stacked pyrene moiety. This was supported by a single NOE contact between the pyrene moiety and G8H2’. Unfortunately, dynamic equilibrium between distinct stacking modes of Upy21 resulted in poor chemical shift dispersion and lack of NOE contacts between Upy21 and the G-quadruplex core. Consequently, position of the pyrene moiety with respect to the core of the c-kit2_py21 G-quadruplex could not be determined on the basis of NMR observables. In order to obtain a more detailed picture on dynamic processes in c-kit2_py21, we built a model from the parent c-kit2 structure (PDB ID: 2KYP) with Upy21 positioned above the G4-G8-G16-G20 quartet and performed a set of MD simulations (20 μs in total, Supplementary Figures S4–S6). G-quadruplex structure of c-kit2_py21 was stable and the pyrene moiety remained stacked during all simulations. While orientation of the pyrene moiety was found to be dependent on the starting conformation and did not change during MD (suggesting a high energy barrier), the uracil-sugar moiety was able to rotate with respect to the linker. We found a geometrical description, i.e. collective variables, discriminating both rotation of pyrene moiety and rotation of uracil-sugar moiety with respect to the linker (Supplementary Figure S7).
Next, we explored multiple conformations of Upy21 in c-kit2_py21 via biased MD. We calculated the free energy surface (Supplementary Figure S32) from which we extracted a minimum free energy path (Figure 6). Interestingly, five free energy minima (thermodynamic states) were predicted on the path (1, 2, 2*, 1* and 3 in Figure 6), in contrast to three distinct conformations observed experimentally by NMR (Figure 5). However, both computational and experimental results suggested that one state is preferred. Calculated free energies transferred to 94% Boltzmann population of state 1 at 300 K.
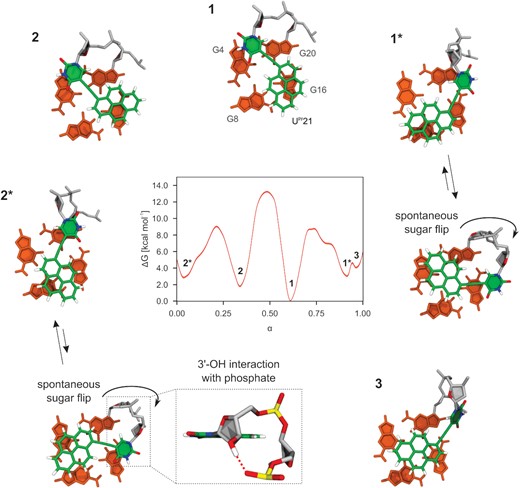
Minimum free energy path connecting five detected thermodynamic states 1, 1*, 2, 2* and 3 with representative structures showing different stacking modes of pyrene moiety in c-kit2_py21. Minimum free energy path was extracted from the free energy surface shown in Supplementary Figure S32. Dimensionless parameter α describes position along the path.
States 1 and 2 have the pyrene moiety positioned mostly above G16 (Figure 6), which agrees with shielding of its imino resonance with respect to c-kit2. Upy21 backbone conformation in 1 and 2 remains relaxed B-DNA-like as in the case of c-kit2_py1. State 1 differs from 2 by a 180° rotation of pyrene moiety around the linker. Interestingly, the most stable state 1 has an identical Upy21 conformation as the quantum mechanical global minimum in vacuo, which was not the case for c-kit2_py1. States 1* and 2* are distinguished from 1 and 2, respectively, by rotating uracil-sugar moiety with respect to the linker by 180°. This rotation brings the pyrene moiety above G8 (Figure 6) and explains the NMR shielding of the G8 imino resonance of c-kit2_py21. State 3 differs from the rest by featuring a non-planar pyrene-uracil arrangement that is stabilized by interaction of the uracil nucleobase with the preceding phosphate (Figure 6). However, population of 3 is most likely insignificant due to its higher energy.
Surprisingly, a detailed structural analysis revealed that states 1* and 2* are represented by two different conformations, which are undistinguishable by employed collective variables. Without biasing this degree of motion, Upy21 sugar spontaneously flipped on the backbone and started to interact with the preceding phosphate via its 3′-OH group. 1* and 2* ensembles were split between non-flipped and sugar-flipped conformations with approximate ratios of 40:60 and 60:40, respectively. This suggests a low energy barrier for the interconversion and similar free energies. The sugar-flip brings the pyrene moiety above G4, which agrees with shielding of the G4 imino resonance in NMR spectra of c-kit2_py21. If only B-DNA-like structures 1 and 2 were populated, G4 would not experience shielding. Furthermore, substantial backbone reorganization in 1* and 2* could explain doubling of G19 and G3 imino resonances (Supplementary Figure S31A).
In total, we identified seven different stacking modes of Upy21 as representative structures of the calculated free energy minima (Figure 6). However, sets of NMR resonances could not be assigned to individual stacking modes of Upy21 due to complex averaging of all motions.
Observation of three sets of imino resonances for G20, G16, G8 and G4 in temperature-dependent 1D 1H NMR spectra of c-kit2_py1/21 suggested preservation of distinct dynamics of Upy on opposite sides of the G-quadruplex core in the double-substituted terminal Upy analogue (Supplementary Figure S33). This was corroborated by overlaying 13C-edited HSQC spectra of c-kit2_py1, c-kit2_py21 and c-kit2_ py1/21 (Supplementary Figure S34). While the chemical-shift dispersion of cross-peaks corresponding to pyrene moieties showed to be almost identical in both carbon and proton dimensions in all three HSQC spectra, the majority of cross-peaks in c-kit2_ py1/21 was slightly shifted towards lower frequencies in the 1H dimension compared to c-kit2_py1 and c-kit2_py21 (Supplementary Figure S34).
Introduction of Upy21 reduces structural heterogeneity
The c-kit2 G-quadruplex dimerizes with time, while c-kit2_py1 and c-kit2_py21 remain unaffected even after several months in solution (Supplementary Figure S35). Compared to the sequence found in the human KIT gene, the c-kit2 oligonucleotide and its Upy analogues contain two G-to-T substitutions, which are known to reduce heterogeneity and suppress formation of dimeric species. Interestingly, we found that introduction of Upy at position 21 of the genomic KIT sequence eliminates heterogeneity and formation of dimeric species without the need for any G-to-T substitution (Supplementary Figure S36). On the other hand, samples with Upy at position 1 are highly polymorphic in the absence of G-to-T substitutions (Supplementary Figure S36).
Free pyrene exhibits a binding preference for G-quadruplex structure
Titration of a c-kit2 G-quadruplex solution with a sodium salt of 1-pyrenesulfonic acid resulted in shifting and broadening of several c-kit2 imino resonances in 1D NMR spectra (Supplementary Figure S37). Changes were detected for the G4-G8-G16-G20 quartet with a low-frequency shift of all guanine imino resonances and a maximum of 0.07 ppm for G4 (Supplementary Figure S40). On the other hand, minor high-frequency shifts were observed for methyl and aromatic resonances of T21 and aromatic resonances of G20 and C9. No new signals suggesting formation of new G-quadruplex species arose during the titration. Similar behaviour was observed during titration of c-kit2_py1. We detected broadening and shielding of imino resonances from the G4-G8-G16-G20 quartet, and deshielding of T21CH3, T21H6 and G20H8 resonances during titration of c-kit2_py1 (Supplementary Figure S38). Absolute values of chemical shift differences were smaller than in the case of c-kit2 with the maximum of 0.05 ppm for G4 and G20 (Supplementary Figure S40). Surprisingly, there were no observable changes in NMR resonances of the c-kit2_py21 G-quadruplex during its titration with the sodium salt of 1-pyrenesulfonic acid (Supplementary Figures S39 and S40).
Since 1-pyrenesulfonic acid is negatively charged we have also tested the interaction with the neutral hydroxypyrene. Titration of c-kit2 and c-kit2_py1 solutions with hydroxypyrene resulted in significant shifting and broadening of imino resonances assigned to the G4-G8-G16-G20 quartet (Supplementary Figures S41 and S42). On the other hand, no shifting and broadening of imino resonances was observed during titration with c-kit2_py21 (Supplementary Figure S43). However, we detected sharpening of G4 and G8 imino resonances as well as appearance of G16 imino resonance with increasing concentration of hydroxypyrene in the c-kit2_py21 solution. Similar sharpening was observed in 1H NMR spectra measured above 30°C (Supplementary Figure S31A).
Our data suggest that 1-pyrenesulfonic acid and hydroxypyrene interact with the c-kit2 G-quadruplex and its analogues only when the G4-G8-G16-G20 quartet is available for interaction. While stacking of the 1-pyrenesulfonic acid and hydroxypyrene to the G2-G6-G14-G18 quartet might be energetically favourable, this G-quartet in c-kit2 and c-kit2_py21 is blocked by the C1:A13 non-canonical base pair or already stacked with the covalently attached pyrene moiety in c-kit2_py1.
DISCUSSION
Polyaromatic moieties can be employed for modulating G-quadruplex properties via their stacking with G-quartets. We recently showed that a single residue substitution with a pyrene conjugated nucleotide (Upy) in TBA can cause a moderate increase in the thermal stability of the G-quadruplex structure (43). However, the chair-like topology of TBA consisting of two G-quartets is unique and those results cannot be generalized and easily extrapolated to modulate properties of promoter G-quadruplexes. On the other hand, the genomic c-kit2 sequence originating from the KIT proto-oncogene exhibits features typical for G-quadruplex forming sequences in human promoter regions. Genomic c-kit2 contains G-tracts of different lengths and additional Gs in loop forming regions, which result in multiple G-quadruplex structures in solution (58). Different mutations were employed in order to reduce the level of structural polymorphism in c-kit2. Herein, we focused on G12T/G21T mutant of genomic c-kit2 sequence forming a single monomeric three-quartet G-quadruplex, as a representative of parallel G-quadruplex structures.
We individually incorporated Upy at all positions in the pentaloop and terminal positions of the c-kit2 sequence. While some of the loop substitutions resulted in destabilization of the G-quadruplex structure, most of them caused an increase in thermal stability. Due to extended structure of Upy, stacking of pyrene moiety is less likely if Upy is substituted in the middle of the loop. Expectedly, a notable stabilization effect was achieved, when Upy was placed at 5′- or 3′-ends of the pentaloop. We believe that this is due to partial and structurally heterogenous π–π stacking interactions of the pyrene moiety with adjacent G-quartets. A substantial increase in thermal stability of up to 27.8°C was achieved when terminal C1 and T21 were individually substituted with Upy. Initial observations suggested that pyrene moieties in c-kit2_py1 and c-kit2_py21 G-quadruplexes are exposed to similar environments, which resulted in only minor differences in fluorescent properties and thermal stability. However, a detailed analysis revealed major differences in structural dynamics of Upy between the two terminal analogues. While c-kit2_py1 appeared structurally rigid with one well-defined stacking mode of the pyrene moiety, c-kit2_py21 demonstrated Upy in multiple conformational states. In total, we identified seven ensembles with distinct stacking modes of Upy21. Although one stacking mode is predominant, remaining six likely serve as a pool of structures, which play a role in the stabilization of the c-kit2_py21 G-quadruplex. Differences in thermal stability of c-kit2_py1 and c-kit2_py21 might stem from different efficacy of π–π stacking interactions between the pyrene moiety and adjacent guanine nucleobases. Considering its size, the pyrene moiety can effectively stack on two guanines of an adjacent G-quartet to maximize π–π stacking interactions. While the pyrene moiety in c-kit2_py1 is located above two guanine nucleobases, this is not the case in c-kit2_py21 where it was found to stack only on one guanine of an adjacent G-quartet. Therefore, less efficient stacking in c-kit2_py21 results in slightly lower thermal stability and larger structural heterogeneity compared to c-kit2_py1.
Efficient stacking of the pyrene moiety requires a planar stacking partner. Therefore, we analysed planarity of G-quartets in c-kit2, c-kit2_py1 and c-ki2_py1 by comparison of root-mean-square deviation (RMSD) from a plane, which was best-fitted individually to each G-quartet. The 5′ G-quartet was uniformly found to be the most planar, whereas the 3′ G-quartet is the least planar in all three systems (Supplementary Figure S44, c-kit2_py1: average RMSD of 5′ G-quartet 0.28 ± 0.06 Å, middle G-quartet 0.35 ± 0.06 Å, 3′ G-quartet 0.50 ± 0.07 Å). Positioning of a fully planar, fused ring system of pyrene next to a more planar 5′ G-quartet in c-kit2_py1 was found to be advantageous and correlated with tighter stacking of Upy1. On the other hand, the less planar 3′ G-quartet in c-kit2_py21 probably promoted conformational tumbling and various stacking modes of Upy21. We believe that the contrast between structural dynamics of Upy1 and Upy21 might stem from an intrinsic asymmetry of c-kit2 G-quartets. This way Upy acts as a probe for local G-quadruplex dynamics, which is true especially for c-kit2, where outer G-quartets are exposed and Upy interactions with propeller loops are minimized. This is a vice-versa effect to the binding of ligands that comprises unfused aromatic rings to G-quadruplexes, where ligand planarity is key for efficient stacking (59). Similar conclusions can be drawn from comparing Upy substitutions in TBA and c-kit2. While TBA G-quartets are protected by two TT and one TGT loop, bulky Upy was able to sandwich between G-quartets and loop residues. However, the maximum thermal stabilization achieved for TBA was 6°C, whereas c-kit2 exhibits up to 29°C increase. In the case of TBA, stabilization gained by Upy stacking appears to have been largely lost by steric clashes with the loop nucleotides. Modulation of G-quadruplex properties via covalent attachment of polycyclic aromatic hydrocarbon molecules, therefore, needs to consider mostly local geometry as the stability of different conformations depends on the balance between stacking, deformation of the linker residue and steric interactions.
Incorporation of two pyrene moieties into the TBA sequence results in disruption of the hybrid G-quadruplex structure or oligomerization facilitated by pyrene–pyrene interactions (43). On the other hand, the parallel c-kit2 G-quadruplex can accommodate two pyrene moieties at opposite sides of the G-quadruplex, which does not perturb the overall topology. Sandwiching the G-quadruplex core between two pyrene moieties provided additive gain in thermal stabilization compared to single substitutions, while preserving distinct dynamic behaviour of polyaromatic groups on opposite sides of the structure. c-kit2_py1/21 was expected to show propensity for dimerization/oligomerization via pyrene–pyrene stacking. Surprisingly, we did not observe any higher-order structure formation in c-kit2_py1/py21 despite the high concentration needed for NMR studies (mM range). Additionally, we showed that single incorporation of Upy at position 21 of genomic c-kit2 region (with no G-to-T substitution) prevents formation of the dimeric species. Therefore, covalent attachment of a polyaromatic group at the appropriate position in the sequence can restrict formation of multimeric G-quadruplex species and reduce structural heterogeneity as an alternative to guanine substitutions or modulation of sample conditions.
KIT mutations and overexpression not only cause dysfunction leading to tumours (60–63), but aberrant expression of KIT is associated with poor prognosis in patients with lung cancer (64). Unfortunately, regulation of KIT expression is complex and relatively little is known about regulatory factors that are involved. Clinical drugs directly targeting c-KIT kinase generally become less effective due to the appearance of c-KIT secondary mutations (8,65). Therefore, alternative ways of regulation of KIT expression are needed. Higher-order arrangement of the entire G-rich domain in the core-promoter region of the KIT gene is expected to regulate protein recruitment, in particular transcription factors. The specificity protein 1 (Sp1) is a prototype member of a family of related transcription factors that modulate gene expression (66). Sp1 with the binding site located in the promoter portion comprising c-kit2 and kit*, generally functions as a transcriptional activator, which contributes to the proliferative and metastatic tumour phenotypes (66). Considering that Sp1 recognizes the G-quadruplex DNA structure as an alternative binding motif (22), we believe that modified oligonucleotides mimicking KIT G-quadruplexes have potential to saturate Sp1-binding sites and prevent the protein from binding to the promoter in genomic DNA. After transfer into the cells, our engineered oligonucleotides could down-regulate KIT transcription by sequestration of Sp1 and consequently decrease cell growth. Besides that, interaction of so-called G4 decoys with transcription factors might expand on our knowledge about the processes involved in regulation of KIT proto-oncogene transcription. Using covalently attached ligands could reduce frequently occurring off-targeting of multiple G-quadruplexes commonly observed for non-covalent therapeutic binders. Spatial distribution of fluorescent G-quadruplexes and their complexes with transcription factors could be visualized in cells with fluorescent microscopy. Furthermore, detailed structural data could also prove useful for studying fluorescence with high-level ab initio computational methods.
In conclusion, using Upy substitutions we have achieved a considerable thermal stabilization of the c-kit2 G-quadruplex while preserving its original topology. Three substituted c-kit2 G-quadruplexes varying in the accessibility of outer G-quartets expand the portfolio of possible interactions with biological targets. Additionally, high thermal stability of our structures allows their use as platforms for attachment of additional functional groups (24). We believe that development of approaches to improve stability of genomic G-quadruplexes and of methods for their characterization could bring improved therapeutics for treatment of cancer or other G-quadruplex related disorders.
DATA AVAILABILITY
Atomic coordinates and list of chemical shifts for the c-kit2_py1 NMR structures have been deposited with the Protein Data bank under accession number 7NWD.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We are very grateful to Dr. Petr Kulhanek for numerous consultations of the computational part of the work and to Dr. Jan Novotny for fruitful discussions about some NMR experiments. We would like to thank the CERIC-ERIC Consortium for access to their experimental facilities.
FUNDING
This work was supported by the European Programme H2020 MSCA ITN [grant number 765266-LightDyNAmics project]. The authors also acknowledge financial support from the Slovenian Research Agency [grants P1-0242 and J1-1704] and the project CEITEC 2020 [Grant No. LQ1601] with financial support from the Ministry of Education, Youth and Sports of the Czech Republic (MEYS CR) under the National Sustainability Programme II. CIISB, Instruct-CZ Center of Instruct-ERIC EU consortium, funded by MEYS CR infrastructure project LM2018127, is gratefully acknowledged for the financial support of the measurements at the Core Facility Josef Dadok National NMR Center. The computational resources were supported by MEYS CR from the Large Infrastructures for Research, Experimental Development and Innovations project “e-Infrastructure CZ” [LM2018140]. Funding for open access charge: H2020 Marie Skłodowska-Curie Actions [765266].
Conflict of interest statement. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments