





Background
Fetchner syndrome (FTNS, OMIM 153640) is an autosomal dominant disorder characterized by the association of macrothrombocytopaenia, leukocyte inclusions, sensorineural deafness, cataracts and nephropathy often leading to end-stage renal disease (ESRD) [ 1 ]. Epstein syndrome (EPTS, OMIM 153650) presents the same features of FTNS, with the exception of leukocyte inclusions and cataracts [ 2 ]. Recently, it was shown that both FTNS and EPTS derive from mutations of MYH9 , the gene for the heavy chain of non-muscle myosin IIA [ 3,4 ]. The same gene was found to be also responsible for two non-syndromic forms of inherited macrothrombocytopaenia with leukocyte inclusions, May-Hegglin anomaly (MHA, OMIM 155100) and Sebastian syndrome (SBS, OMIM 605249) [ 3 ]. Further studies demonstrated that patients initially diagnosed as having MHA/SBS can subsequently develop nephropathy, deafness and/or cataracts, while affected relatives of FTNS or EPTS patients present for all their lifetime a clinical picture indistinguishable from that of MHA/SBS subjects [ 5 ]. Moreover, immunofluorescence studies showed that leukocyte inclusions containing the MYH9 protein are present not only in MHA/SBS and FTNS, but also in all patients with EPTS [ 5 ]. Therefore, it was clear that FTNS, EPTS, MHA and SBS are not distinct entities, but rather they represent different clinical presentations of a single disease, characterized by the constant finding of congenital thrombocytopaenia and leukocyte inclusions, and possible development of nephropathy, deafness and/or cataracts in childhood or adult life. For this entity, the term MYH9 -related disease ( MYH9 -RD) has been proposed [ 5 ].
Nephropathy occurs in ∼30% of MYH9 -RD patients [ 6 ]. It presents at a juvenile age with proteinuria, sometimes causing nephrotic syndrome, with or without microhaematuria. In most patients, the nephropathy progresses early to loss of renal function, until reaching ESRD requiring dialysis or kidney transplantation before the fourth decade of life [ 1 , 5 , 6 ]. The MYH9 protein was found to be expressed in podocytes, mesangial cells and tubular epithelia [ 4 ], but pathogenesis of nephropathy caused by MYH9 mutations is unknown, and no therapy has been proposed so far.
Here we report the first four patients with genetically confirmed MYH9 nephropathy treated by pharmacological blockade of the renin–angiotensin system (RAS), and suggest that this approach is effective in this disorder and could modify the clinical course of the disease.
Case reports
Patients 1, 2 and 3
A 39-year-old male (patient 1) was admitted to our renal unit in 2001 because of non-nephrotic range proteinuria (1230 mg/24 h) and mild renal failure (serum creatinine 1.8 mg/dl, GFR 62.3 ml/min). He had a medical history of idiopathic thrombocytopaenia since infancy (platelet count 17 × 10 9 /l). The diagnostic suspicion of FTNS was confirmed through demonstration of giant platelets and typical ‘Döhle-like’ leukocyte inclusions by the examination of peripheral blood smear, and sensorineural deafness by audiometric assay [ 5 ]. Cataracts were excluded by ophtalmological evaluation. Diagnosis was definitively confirmed by mutational screening of MYH9 , which identified the 4270G>C mutation resulting in the 1424D>H substitution in the MYH9 protein [ 5 ]. Combination therapy with ramipril, 10 mg/day, plus telmisartan, 80 mg/day, was started. The last clinical evaluation was performed after 68 months of continuous treatment. Main clinical findings at the time immediately before starting therapy and at the last evaluation are summarized in Table 1 . Figure 1 shows the results of 24-h proteinuria measurements before and during treatment. The relatives of patient 1 were also investigated. His 42-year-old brother (patient 2) presented with proteinuria (1570 mg/24 h), mild renal failure (serum creatinine 1.2 mg/dl, GFR 83.1 ml/min), thrombocytopaenia, leukocyte inclusions, sensorineural deafness and a history of bilateral congenital cataracts [ 5 ]. Therapy with ramipril plus telmisartan at the same dosages as for the propositus was then started. The propositus’ 15-year-old son (patient 3) presented with thrombocytopaenia, leukocyte inclusions and sensorineural hearing loss [ 5 ]. At the age of 18 years, he developed stable proteinuria (768 mg/24 h), so treatment with ramipril, 10 mg/day, plus losartan, 50 mg/day, was started (Table 1 and Figure 1 ). In both affected relatives, the diagnosis of FTNS was confirmed by identification of the 4270G > C MYH9 mutation.
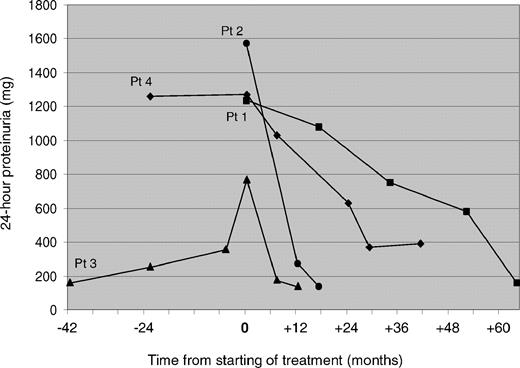
Results of measurements of 24-h proteinuria before and during treatment by RAS inhibition in four patients with MYH9 nephropathy. Patients 1, 2 and 3 were treated by combination therapy with ramipril plus an angiotensin receptor blocker, while patient 4 was treated by ramipril alone. Pt = patient.
Main clinical features of four patients with MYH9 nephropathy treated by RAS blockade at the time immediately before starting therapy (baseline) and at the last clinical evaluation
| Baseline | Last clinical evaluation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | Length of treatment (months) | Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | |
| Patient 1 | 1231 | 1.8 | 62.3 | 140/95 | 68 | 90 | 1.6 | 60.4 | 120/75 |
| Patient 2 | 1570 | 1.2 | 83.1 | 130/90 | 16 | 137 | 1.1 | 84.6 | 115/80 |
| Patient 3 | 768 | 0.9 | 108.7 | 120/75 | 11 | 143 | 0.7 | 110.9 | 110/75 |
| Patient 4 | 1280 | 0.9 | 110.4 | 130/90 | 40 | 390 | 0.9 | 97.0 | 120/80 |
| Baseline | Last clinical evaluation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | Length of treatment (months) | Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | |
| Patient 1 | 1231 | 1.8 | 62.3 | 140/95 | 68 | 90 | 1.6 | 60.4 | 120/75 |
| Patient 2 | 1570 | 1.2 | 83.1 | 130/90 | 16 | 137 | 1.1 | 84.6 | 115/80 |
| Patient 3 | 768 | 0.9 | 108.7 | 120/75 | 11 | 143 | 0.7 | 110.9 | 110/75 |
| Patient 4 | 1280 | 0.9 | 110.4 | 130/90 | 40 | 390 | 0.9 | 97.0 | 120/80 |
Patients 1–3 were treated by combination therapy with ramipril plus telmisartan or losartan, while patient 4 was treated by ramipril alone.
Uprot = urinary protein, SCr = serum creatinine, CrClear = creatinine clearance, BP = blood pressure.
Main clinical features of four patients with MYH9 nephropathy treated by RAS blockade at the time immediately before starting therapy (baseline) and at the last clinical evaluation
| Baseline | Last clinical evaluation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | Length of treatment (months) | Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | |
| Patient 1 | 1231 | 1.8 | 62.3 | 140/95 | 68 | 90 | 1.6 | 60.4 | 120/75 |
| Patient 2 | 1570 | 1.2 | 83.1 | 130/90 | 16 | 137 | 1.1 | 84.6 | 115/80 |
| Patient 3 | 768 | 0.9 | 108.7 | 120/75 | 11 | 143 | 0.7 | 110.9 | 110/75 |
| Patient 4 | 1280 | 0.9 | 110.4 | 130/90 | 40 | 390 | 0.9 | 97.0 | 120/80 |
| Baseline | Last clinical evaluation | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | Length of treatment (months) | Uprot (mg/24 h) | SCr (mg/dl) | CrClear (ml/min) | BP (mmHg) | |
| Patient 1 | 1231 | 1.8 | 62.3 | 140/95 | 68 | 90 | 1.6 | 60.4 | 120/75 |
| Patient 2 | 1570 | 1.2 | 83.1 | 130/90 | 16 | 137 | 1.1 | 84.6 | 115/80 |
| Patient 3 | 768 | 0.9 | 108.7 | 120/75 | 11 | 143 | 0.7 | 110.9 | 110/75 |
| Patient 4 | 1280 | 0.9 | 110.4 | 130/90 | 40 | 390 | 0.9 | 97.0 | 120/80 |
Patients 1–3 were treated by combination therapy with ramipril plus telmisartan or losartan, while patient 4 was treated by ramipril alone.
Uprot = urinary protein, SCr = serum creatinine, CrClear = creatinine clearance, BP = blood pressure.
Patient 4
A 30-year-old female was referred to our internal medicine unit in 1995 with a diagnosis of idiopathic thrombocytopaenic purpura unresponsive to both corticosteroids and splenectomy (platelet count 20 × 10 9 /l). We diagnosed MHA on the basis of platelet macrocytosis, giant platelets and ‘Döhle-like’ leukocyte inclusions at the examination of peripheral blood smear. In the year 2000, the de novo missense mutation 279C>G in the MYH9 gene, causing the 93N>K substitution in the MYH9 protein, was demonstrated [ 3 ]. The search for signs of nephropathy was performed for the first time when the patient was 36 years old. Proteinuria was 1260 mg/24 h, while serum creatinine and GFR were normal (0.9 mg/dl and 120.3 ml/min, respectively) [ 5 ]. Sensorineural hearing loss was demonstrated by audiometric examination, and cataracts were excluded by ophthalmological evaluation [ 5 ]. At the age of 38 years, given the persistence of proteinuria, therapy with ramipril, 5 mg/day, was started. The last clinical evaluation was performed after 40 months of continuous treatment with ramipril at the same dosage (Table 1 and Figure 1 ).
Discussion
Kidney impairment affects a minority of patients with MYH9 mutations, but, whenever it occurs, the clinical features are those of a nephropathy that develops at a juvenile age and has a progressive and severe evolution [ 1 , 5 , 6 ]. We recently reported a series of 108 consecutive patients with MYH9 -RD [ 6 ]. Chronic proteinuria >500 mg/day was present in 26 patients, with a mean age of 23 years at the onset. Nineteen of these subjects (73%) developed renal failure after a mean time of 32 months from the discovery of proteinuria, and 16 of these cases progressed to ESRD. Mean ages at the onset of renal failure and ESRD were 26 and 27 years, respectively [ 6 ]. The 7 proteinuric patients who did not present kidney failure were observed for 1–48 months (mean 17), and proteinuria was found to progressively increase ( n = 4) or to be stable ( n = 3) over time. Therefore, none of our 26 cases with MYH9 nephropathy experienced spontaneous improvement. A severe course of MYH9 nephropathy emerges also from the FTNS or EPTS patients previously reported in the literature [ 1,2 ].
Until now, no therapeutic options to slow down the progression to ESRD were offered to these patients. Since the blockade of RAS through the administration of angiotensin-converting enzyme inhibitors and/or angiotensin receptor blockers proved to be effective in reducing proteinuria and preserving renal function in acquired diabetic and non-diabetic nephropathies [ 7 ], we used this strategy also in two patients with genetically confirmed MYH9 nephropathy (patients 1 and 4). In both of them, we obtained a progressive reduction of proteinuria to 7% and 30% of the baseline value, respectively, and this reduction was maintained after 68 and 40 months of treatment. We subsequently treated two affected relatives of patient 1, and in both of them we observed a prompt normalization of proteinuria. In all our patients, the kidney function remained stable during treatment. In particular, patient 1 already presented an increased serum creatinine before the start of treatment, and no further progression was observed during the following 68 months of the follow-up (Table 1 ). Of course, we cannot ascertain if treatment contributed to the preservation of kidney function in these subjects. However, in view of the major predictive role of proteinuria reduction on progression of chronic nephropathies [ 8 ], we believe that such a degree and duration of the decrease of urinary protein excretion can actually have an impact on the long-term outcome of the disease. This remark is supported by the observation of a renoprotective effect of RAS blockade in some patients with Alport syndrome [ 9 ], another inherited disorder with progressive nephropathy variably associated with sensorineural deafness and cataracts. Interestingly, in this condition the best results were obtained in patients receiving an early treatment [ 9 ], and an animal model suggested that even better results could be obtained by starting treatment before proteinuria appears [ 10 ]. This approach can now be proposed also in MYH9 -RD, since a recent genotype–phenotype analysis identified the MYH9 mutations that severely affect the kidney function and invariably lead to ESRD [ 6 ]. In fact, the patients we treated presented a relatively early stage of kidney damage. However, on the basis of the good results they obtained from RAS inhibition, it seems reasonable to expect that also patients with a more advanced stage of kidney impairment could obtain some advantage from this kind of approach.
Concerning the mechanisms of the favourable effect of RAS inhibition in MYH9 -RD, no certain explanation is possible at present, since the pathogenesis of kidney damage in this disorder is still poorly known. However, previous observations suggested that the alteration of structure and actin cytoskeleton reorganization of podocytes are primarily responsible for the nephropathy caused by MYH9 mutations [ 11,12 ]. Interestingly, several studies indicated that RAS inhibition in chronic nephropathies has a direct protective effect on podocyte structure and function, which is independent of its haemodynamic effects [ 13 ]. Thus, we can speculate that the effectiveness of RAS blockade in MYH9 nephropathy could derive not only from the reduction of systemic and glomerular blood pressure, but also from a specific protective effect on the cells representing the primary target of MYH9 dysfunction.
In conclusion, renoprotective treatment by RAS blockade is effective in reducing proteinuria in patients with MYH9 nephropathy. These observations indicate the need for a larger clinical study to ascertain if this therapeutic approach can actually modify the natural history of this disease.
This work was supported by grants from the Telethon Foundation (no. GGP06177 to C.L.B.) and the Italian Ministero della Salute (to C.L.B.).
Conflict of interest statement . None declared.


{kind=link}
Comments