





Abstract
Forkhead box M1 (FOXM1) is overexpressed and activates numerous oncoproteins in tumors. However, the mechanism by which the FOXM1 protein aberrantly accumulates in human cancer remains uncertain. This study was designed to clarify the upstream signaling pathway(s) that regulate FOXM1 protein stability and transcriptional activity.
Mass spectrometry and immunoprecipitation were performed to identify the FOXM-metadherin (MTDH) interaction. In vivo and in vitro ubiquitination assays were conducted to test the effect of MTDH on FOXM1 stability. Chromatin immunoprecipitation assays were used to determine the involvement of MTDH in FOXM1 transcriptional activity. Cell invasion assays, tube formation assays, and in vivo tumor formation assays were performed to evaluate the cooperative activities of FOXM1 and MTDH during tumorigenesis.
MTDH directly interacts with FOXM1 via the N-terminal inhibitory domain of MTDH, and this interaction disrupted the binding of cadherin-1 to FOXM1, thus protecting FOXM1 from subsequent proteasomal degradation. Deleting the MTDH-binding sites of FOXM1 abolished the MTDH overexpression-mediated stabilization of FOXM1.
MTDH also bound to FOXM1 target gene promoters and enhanced FOXM1 transcriptional activity. MTDH knockdown destabilized FOXM1 and attenuated its transcriptional activity, consequently inhibiting cell cycle progression, angiogenesis, and cancer cell invasion in vitro and in vivo; these effects were abolished via forced overexpression of a stabilized mutant form of FOXM1. Thus, MTDH stabilized FOXM1 and supported the sustained activation of FOXM1 target genes.
These findings highlight a novel MTDH-regulated mechanism of FOXM1 stabilization and provide profound insight into the tumorigenic events simultaneously mediated by FOXM1 and MTDH.
The transcription factor FOXM1 is aberrantly activated in the majority of human cancers, and this characteristic is highly associated with poor clinical prognosis. FOXM1 drives the overexpression of critical oncogenes involved in malignant properties including angiogenesis, cell cycle acceleration, invasion, and drug resistance.1–3 FOXM1 also performs nontranscriptional functions: FOXM1 activates Wnt signaling in glioma stem cells and induces self-renewal.4 FOXM1 is also critical for the sustained activation of tumor growth factor-β signaling.5 In mouse models, Rosa26-Foxm1b transgenic mice harbored more and larger lung and colorectal tumors following induction with chemical reagents.6 Similarly, FOXM1 overexpression accelerated the initiation and progression of prostate adenocarcinoma in Rosa26-Foxm1/TRAMP and Rosa26-Foxm1/LADY double-transgenic mice. In contrast, urethane-mediated lung tumorigenesis was decreased in Mx-Cre/Foxm1−/− mice, and hepatocellular carcinomas induced by diethylnitrosamine/phenobarbital failed to develop in Alb–Cre Foxm1b−/− mice.7 FOXM1 protein level is also highly correlated with glioblastoma patients’ total survival.8 These observations suggest that FOXM1 also plays a crucial role in oncogenesis in humans.
FOXM1 overexpression is commonly observed in human cancers, but genetic alterations in FOXM1 are not commonly observed.9 Instead, FOXM1 could be activated by upstream oncogenes such as GLI1, heat shock factor 1 (HSF1), or hypoxia-inducible factor 1 (HIF1) via direct transcriptional activation.10–12 Alternatively, FOXM1 could be targeted by certain microRNAs at the posttranscriptional level.13,14 However, the mechanisms contributing to FOXM1 protein abundance in human cancers have not been clarified.
MTDH was first cloned as a human immunodeficiency virus-1 (HIV-1) and tumor necrosis factor alpha (TNF alpha)–inducible gene in primary human fetal astrocytes.15 It is now evident that MTDH overexpression is widespread in humans with advanced-stage cancer and correlates with poor prognosis.15–17 Gain and loss-of-function studies in vitro and in vivo revealed that MTDH is involved in multiple pathological processes of human cancer including cell proliferation, invasion, metastasis, and gene expression.17–21 MTDH expression was also reported to be negatively correlated with prognosis in glioblastoma patients.22 Mechanistically, MTDH directly interacts with the p65 subunit of NF-κB and is recruited to downstream target gene promoters.23,24 MTDH also activates the PI3K/Akt pathway via an as-yet undetermined mechanism.25 In addition, MTDH activates Wnt/β-catenin signaling via ERK42/44.26 Although MTDH overexpression alone did not induce spontaneous tumor formation in mouse models, MTDH/c-Myc transgenic mice showed highly aggressive and metastatic tumor phenotypes.27 In contrast, MTDH knockout inhibited tumor formation and metastasis, and this finding indicated that MTDH may perform critical functions in tumor progression and proliferation.28,29 Interestingly, many FOXM1 target genes are also regulated by MTDH,17,18,22 raising the question of whether MTDH is involved in FOXM1 signaling in human malignancies.
In this study, we examined whether MTDH is involved in FOXM1 protein regulation and/or stabilization. We showed that MTDH directly interacts with FOXM1 to not only stabilize FOXM1 but also enhance FOXM1 transcriptional activity. These results indicate a novel regulatory association between these 2 proteins and suggest a potential target for new therapies against glioblastoma.
Detailed Materials and Methods are available in the Supplementary information.
Materials and Methods
Cell Culture
Normal human astrocytes (NHAs) were cultured in AGM BulletKit medium. The glioblastoma and 293T cells were cultured in Dulbecco's modified Eagle’s medium supplemented with 10% fetal bovine serum (HyClone). The patient-derived cell lines were cultured as described previously.4
Lentivirus Generation and Infection
Lentiviral particles were produced using a lentivirus packaging mix (ViraPower, Life Technologies). Cells were infected with the presence of 6 μg/mL polybrene (Sigma-Aldrich).
Patient Tissue Specimens
This study was conducted using 85 randomly selected tumor samples that had been histopathologically diagnosed between 2000 and 2012. The study was approved by the ethics committee of Sun Yat-sen University, and informed consent was obtained from all subjects.
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation (ChIP) assays were performed using a ChIP assay kit (Cell Signaling). The oligonucleotides used for qPCR and ChIP are described in Supplementary Table 3.
Immunohistochemistry
Immunohistochemical analysis was performed using tissue sections of paraffin-embedded glioblastoma specimens. We quantitatively scored the tissue sections as described in the Supplementary Methods.
Bimolecular Fluorescence Complementation Assay
Wild-type or mutant FOXM1 was cloned into the bimolecular fluorescence complementation (pBIFC)-VN173/flag vector, and MTDH was cloned into the pBIFC-VC155HA vector. All images were acquired using a laser scanning confocal microscope.
Promoter Reporters and Dual-luciferase Assay
2Cells were transfected with the reporter plasmids indicated together with pRL-TK. Luciferase activity was measured using the Promega Dual-Luciferases Reporter Assay Kit.
Invasion Assay and Tube Formation Assay
Invasion chambers containing polycarbonate filters (8-μm pore size; BD Biosciences) were used with growth factor-reduced Matrigel matrix (50 μg/filter; BD Biosciences). Human umbilical vein endothelial cells were trypsinized and seeded (5 × 104 cells per well) in each well with 250 μL of conditioned medium. The chamber was incubated for 6 hours.
Flow Cytometry
The DNA contents of indicated cells were analyzed using a fluorescence-activated cell sorter (FACSVantage), and the resulting data were processed using FACS CellQuest software (Becton Dickinson).
Mass Spectrometry
The Mass spectra were analyzed using the National Center for Biotechnology Information nonredundant protein database via Mascot (Matrix Science) and SEQUEST (Thermo Fisher Scientific).
Immunoprecipitation
Cells were lysed in co-IP buffer, and the precleared supernatant was subjected to IP using primary antibodies at 4°C overnight.
Glutathione S-transferase Pull-down and in Vitro Binding Assays
6xHis-tagged and Glutathione S-transferase (GST)-tagged proteins were expressed in E. coli and purified using a Ni-His purification kit (Novagen) and GST beads, respectively. The protein complexes were washed with co-IP buffer and analyzed by Western blotting.
In Vitro Ubiquitination Assays
The in vitro ubiquitination assay was performed on an ubiquitin ligation reaction mixture and ubiquitin reaction buffer (Boston Biochem). The reactions were taken according to the manufacturer’s protocols.
Intracranial Injection
All mouse experiments were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University. Cells were intracranially injected into 4-week-old female athymic nude mice as previously described.4
Statistical Analysis
The significance of differences in the human glioblastoma data was determined using Pearson’s correlation test. The significance of differences in the in vitro data was determined using the Student t test (2-tailed). The significance of differences in the in vivo data was determined using the Mann–Whitney U test. P< .05 was considered to be significant.
Results
FOXM1 Abundance Correlates with MTDH Expression
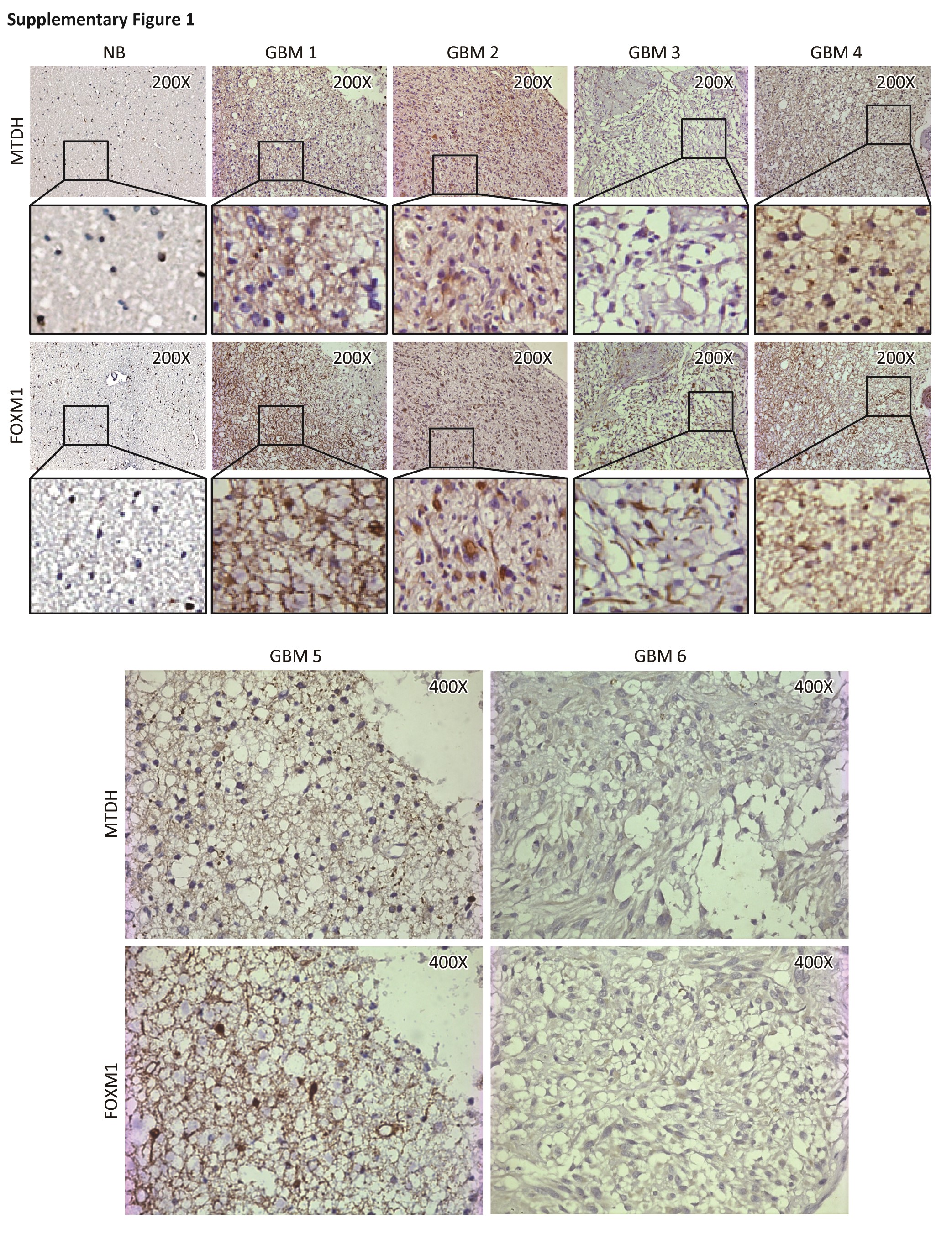
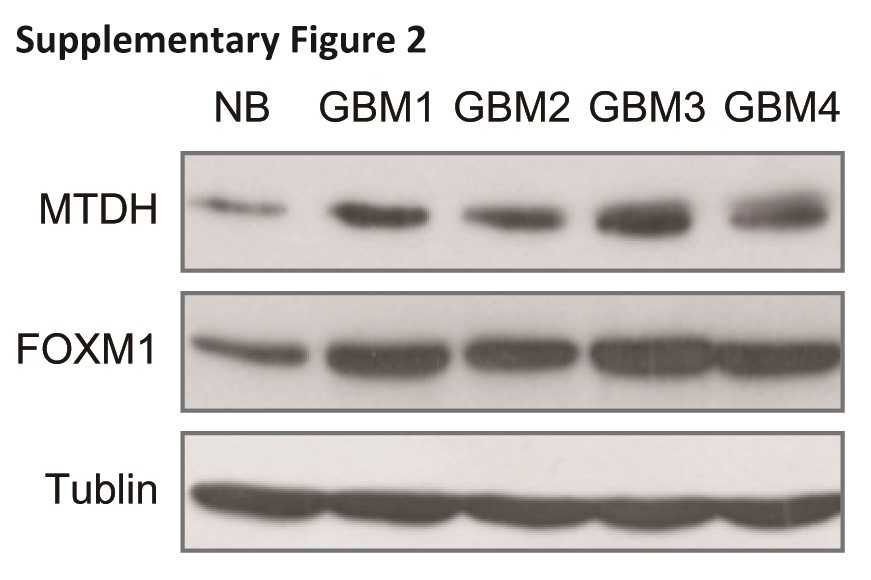
We first examined the MTDH and FOXM1 expression patterns in human glioblastoma samples. We quantified a positive correlation (R2 = 0.7607, P < .001) between MTDH and FOXM1 in terms of both distribution and abundance in 85 glioblastoma samples (Fig. 1A and Fig. S1). Western blotting of total protein extracts, taken from the same set of frozen brain tissues from 1 control and 4 glioblastoma patients as the samples used in Supplementary Fig. 1, confirmed the correlation of FOXM1 with MTDH at the protein level (Fig. S2).
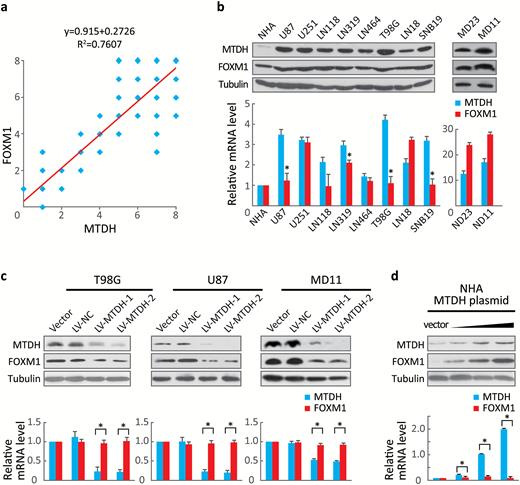
Forkhead box M1 (FOXM1)F abundance correlated with MTDH expression. (A) The 85 glioblastoma tissue sections were quantitatively scored from 0 to 8 according to the percentage of positive cells and the staining intensity. Pearson’s correlation test showed a correlation between MTDH and FOXM1 expression (R2 = 0.7607, P < .001). (B) MTDH and FOXM1 expression in NHA and several established glioblastoma cell lines were determined via Western blotting and real-time RT-PCR. Values are presented as means±SD for triplicate samples. * indicates that the cells express low FOXM1 but high MTDH mRNA levels and strongly express the FOXM1 protein. (C) The protein and mRNA levels of FOXM1 and MTDH were measured in T98G, U87, and MD11 cells stably expressing 2 independent MTDH shRNAs. Values are presented as means±SD for triplicate samples. *P < .001. (D) FOXM1 and MTDH levels were detected in NHA-overexpressing increasing doses of MTDH (0, 0.1, 1.0, or 2.0 μg). Values are presented as means±SD for triplicate samples. *P < .001.
To exclude the possibility that this positive correlation was due to tissues other than astrocytoma, we analyzed MTDH and FOXM1 expression in several established glioma- and patient-derived cell lines. A positive association between MTDH and FOXM1 protein expression was identified in these samples (Fig. 1B, upper panel). Notably, MTDH, and FOXM1 mRNA levels were not as strongly correlated as their protein levels; some cells expressing low FOXM1 mRNA levels, but high MTDH mRNA levels, also strongly expressed the FOXM1 protein (Fig. 1B, lower panel, indicated by *).
Because both the U87 and T98G cell lines showed low FOXM1 but high MTDH mRNA expression, we chose these 2 cell lines to stably express 2 independent MTDH small hairpin RNAs (shRNAs). We observed a significant (P< .001) reduction in FOXM1 protein expression but no change in FOXM1 mRNA expression in both cell lines. Additionally, stably inhibiting MTDH in MD11 patient-derived cells reduced FOXM1 mRNA and protein expression (Fig. 1C). Conversely, transfecting MTDH into NHA cells, which display low levels of both MTDH and FOXM1, dose-dependently increased FOXM1 and MTDH protein expression. However, FOXM1 mRNA expression did not change upon MTDH transfection (Fig. 1D). These results suggested that MTDH may regulate FOXM1 at the posttranslational level.
MTDH Modulates FOXM1 Protein Levels via the Proteasomal Degradation Pathway
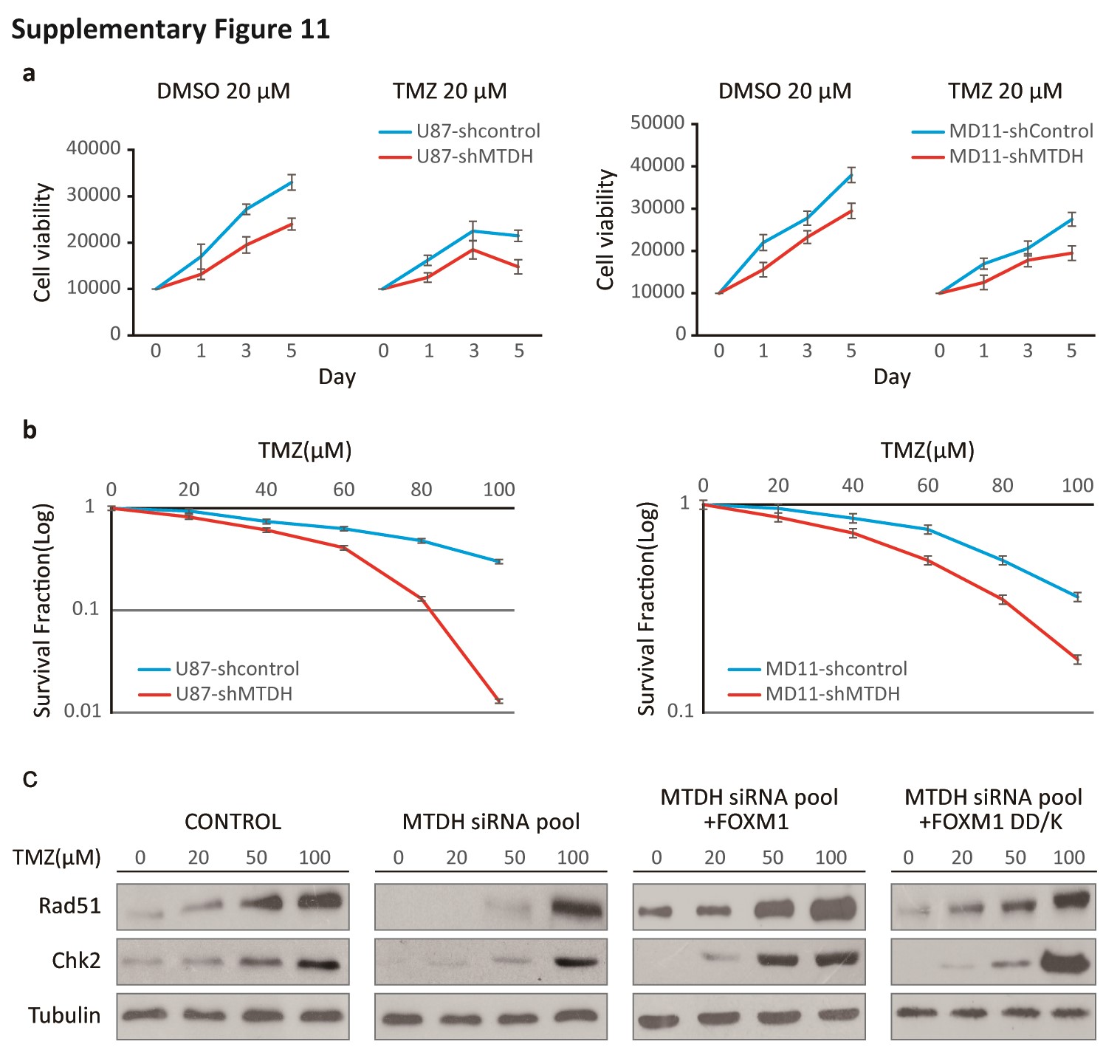
A previous report showed that FOXM1 is overexpressed during the DNA damage response.30 To explore whether MTDH silencing affects FOXM1 aggregation during DNA damage, we compared FOXM1 expression between sh-MTDH (MTDH-targeting shRNA)-transfected and control U87 cells after temozolomide (TMZ) treatment. TMZ-induced upregulation of FOXM1 was inhibited in sh-MTDH cells compared with control cells (Fig. 2A). Additionally, in the presence of cycloheximide (CHX), the half-life of FOXM1 protein in sh-MTDH U87 cells was shorter than that in control cells (Fig. 2B). To further determine whether MTDH may modulate FOXM1 protein degradation, we exposed the indicated cells to the 26S proteasomal inhibitor MG132; the calpain inhibitor ALLN was used as a control. MG132, but not ALLN, restored the FOXM1 protein levels that were suppressed by MTDH silencing (Fig. 2C).
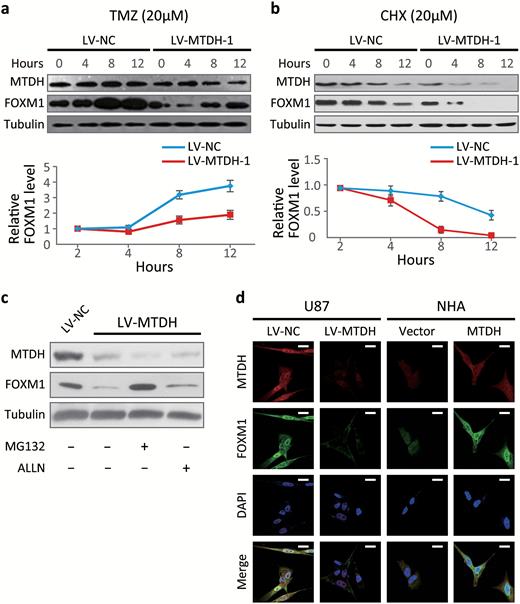
MTDH modulates the forkhead box M1 (FOXM1) protein levels by preventing the proteasomal degradation of FOXM1. (A) FOXM1 and MTDH protein were determined via Western blotting after treatment with 20 μM temozolomide (TMZ) to damage cell DNA in sh-MTDH and control U87 cells. Cells were harvested at the indicated time points following temozolomide (TMZ) treatment. (B) Cells were harvested at the indicated time points after adding cycloheximide (CHX) to inhibit new protein synthesis. (C) FOXM1 and MTDH protein levels in sh-MTDH and control U87 cells were determined after MG132 or ALLN treatment (10 μM). (D) Immunofluorescence for MTDH (Alexa 596), FOXM1 (Alexa 488), and nuclei (DAPI, blue) in sh-MTDH U87 cells, MTDH-overexpressing NHA, and control cells. Scale bar, 20 μm. Note the nuclear colocalization of MTDH and FOXM1.
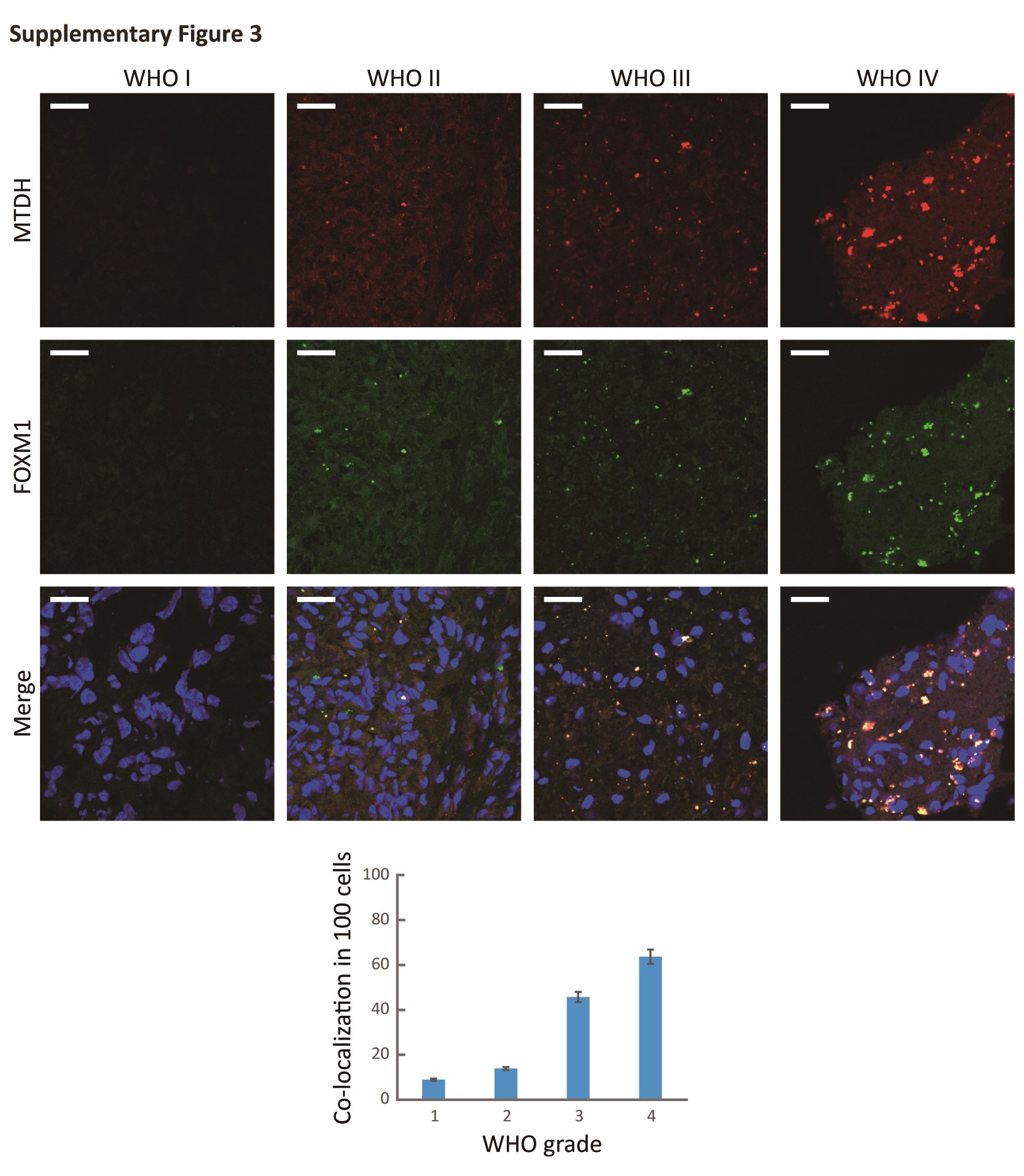
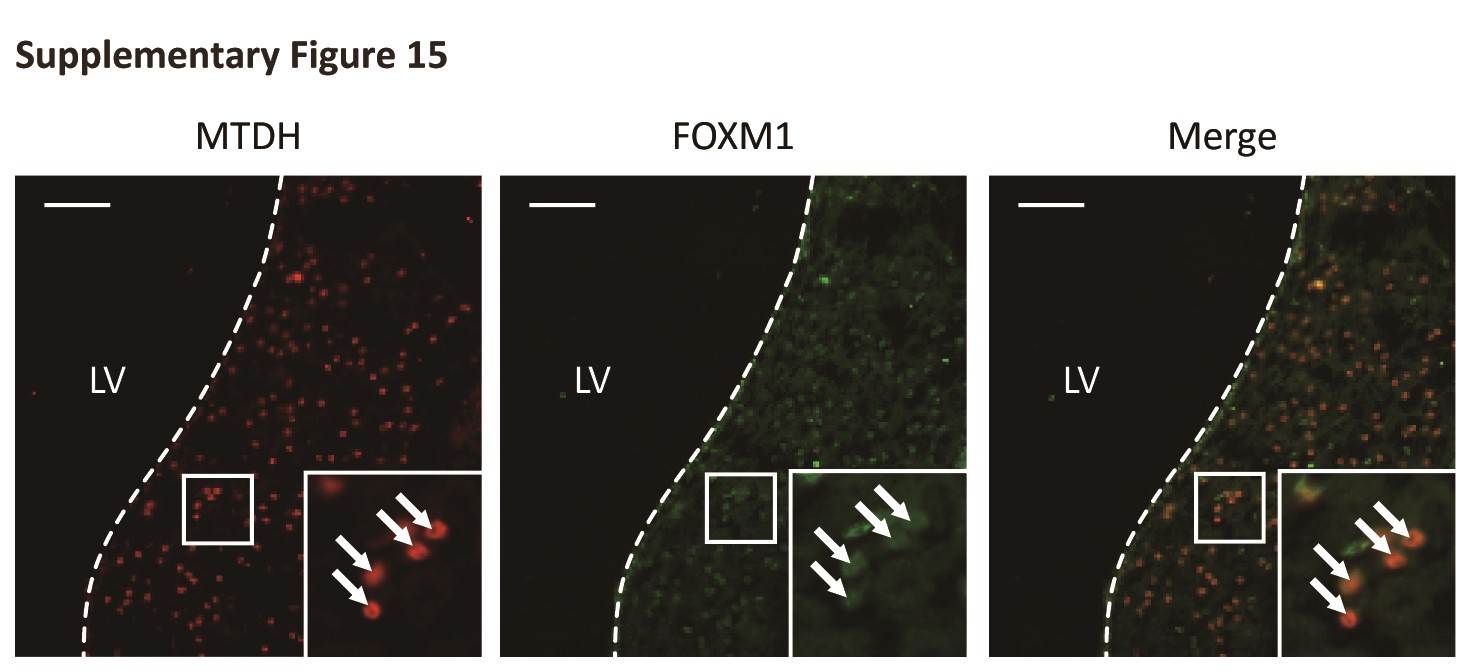
Given that MTDH and FOXM1 were strongly correlated at the protein level, we next explored their subcellular localization. We detected colocalization of MTDH and FOXM1 in both U87 and NHA cells ectopically overexpressing MTDH as well as frozen glioma samples (Fig. 2D and Fig. S3). These data suggest that MTDH modulates the FOXM1 protein levels by inhibiting FOXM1 degradation and potentially colocalizes with FOXM1 in cancer cells.
MTDH Directly Interacts with FOXM1 in Vivo and in Vitro
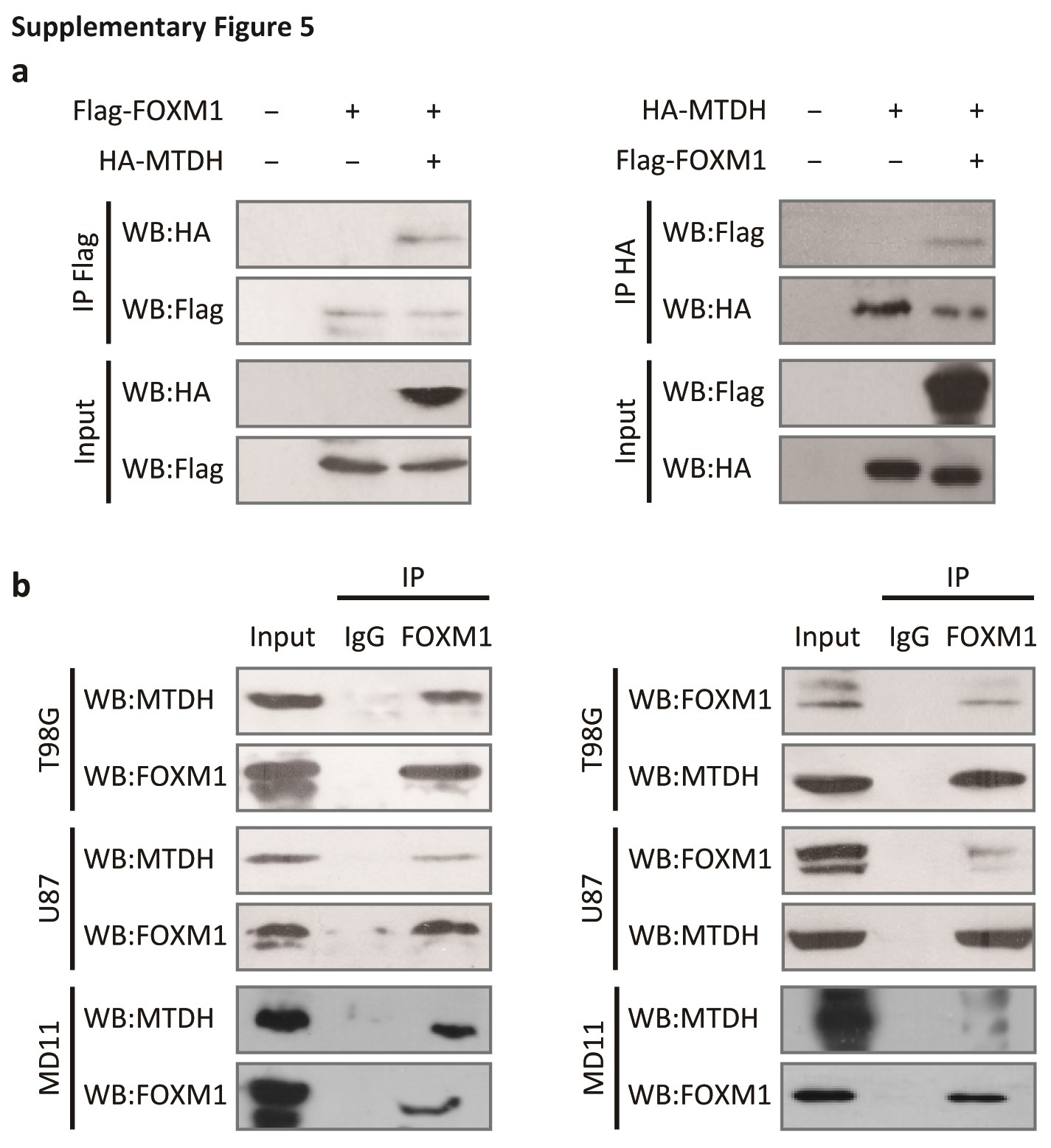
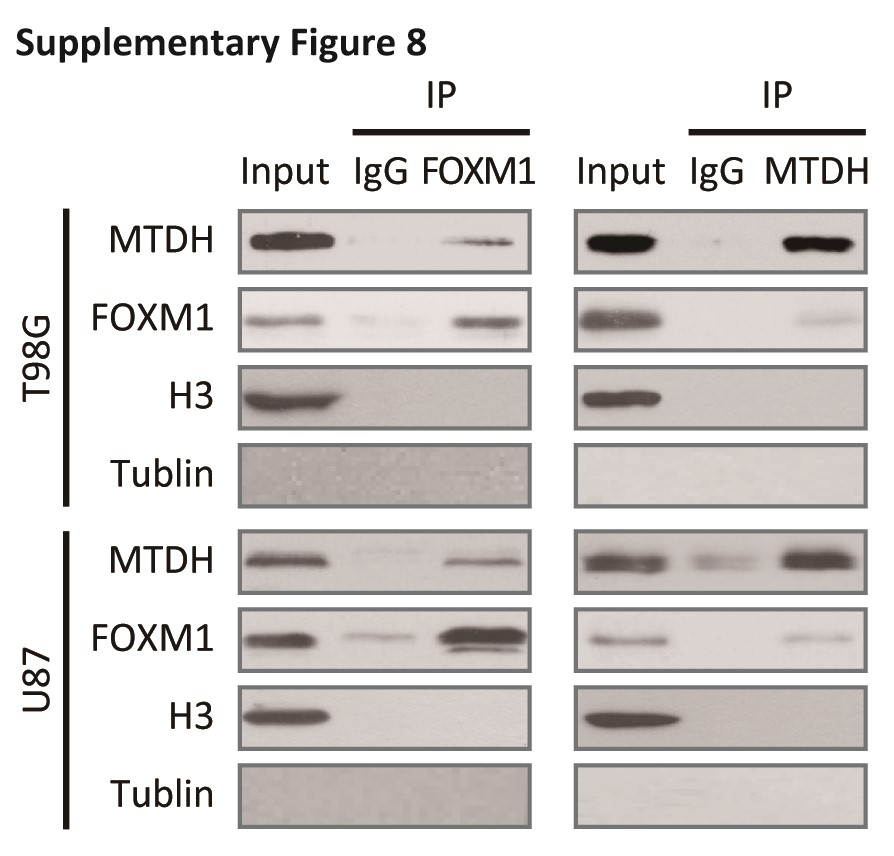
Based on the above hypothesis, we next explored the possible interaction between MTDH and FOXM1 in vivo and in vitro. A Flag-tag affinity purification procedure was performed to purify FOXM1-containing complexes, as we described previously.4 Peptide sequences corresponding to MTDH were found in the precipitated complex (Fig. 3A and Fig. S4). Furthermore, purified His-tagged FOXM1 directly bound to GST-tagged MTDH and vice versa. This result indicated that this interaction was direct (Fig. 3B). Next, we ectopically overexpressed HA-tagged MTDH and/or Flag-tagged FOXM1 in 293T cells and examined the MTDH/FOXM1 interaction in vivo (Fig. S5A). Finally, co-IP confirmed the endogenous interaction between MTDH and FOXM1 in glioma cells (Supplementary Fig. S5B).
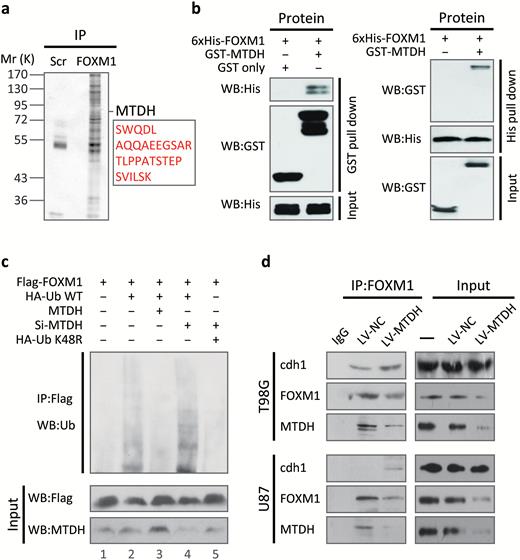
MTDH directly interacts with forkhead box M1 (FOXM1) in vitro and in vivo and inhibits FOXM1 ubiquitination by interfering with the Cdh1-FOXM1 interaction. (A) Extracts of 293T cells transfected with the Flag-tagged FOXM1 or control plasmid were subjected to immunoprecipitation (IP). The resulting protein complexes were subjected to LC-MS/MS. Detailed mass spectrometric data are shown in Supplementary Fig. 3. (B) GST and His pull-down assays were performed using purified GST-tagged MTDH and 6xHis-tagged FOXM1, respectively. (C) U87 cells were transfected with the indicated plasmids or specific siRNA. After 4 hours of treatment with MG132, cell extracts were subjected to IP. (D) Extracts of sh-MTDH and control U87 and T98G cells were subjected to IP.
MTDH Stabilizes FOXM1 by Inhibiting its Ubiquitination
We hypothesized that MTDH may affect FOXM1 degradation through their direct interaction. Therefore, we performed in vivo ubiquitination assays. As shown in Fig. 3C, co-transfection of HA-tagged ubiquitin (HA-Ub) and Flag-tagged FOXM1 slightly increased FOXM1 ubiquitination compared with the basal level (lanes 1 and 2). FOXM1 ubiquitination was significantly decreased by transfection with MTDH (lane 3). Conversely, silencing MTDH clearly increased FOXM1 ubiquitination (lane 4). This effect was reversed by transfection with K48R HA-Ub (lane 5).
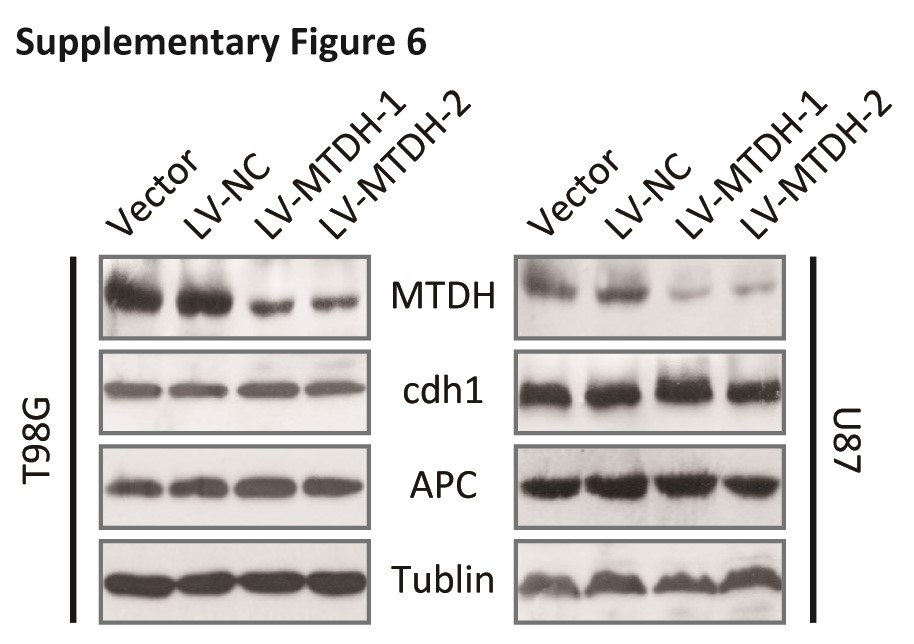
FOXM1 ubiquitination is known to require the E3 ligase APC/C and the adaptor Cdh1.31 Thus, deletion or mutation of APC/C or Cdh1, which is generally observed in cancer cells,32 may stabilize FOXM1. However, no changes in Cdh1 or APC/C were observed in sh-MTDH cells (Fig. S6). Instead, binding of Cdh1 to FOXM1 was increased in sh-MTDH cells (Fig. 3D).
Based on this evidence, we hypothesized that MTDH might possess a higher FOXM1-binding affinity than Cdh1 and that MTDH upregulation might competitively decrease the abundance of FOXM1-bound Cdh1. Therefore, we purified different truncated or mutant forms of FOXM1 and tested their binding affinity to MTDH. The His-tagged N-terminal (NT) fragment and full-length form of WT FOXM1, but not its DNA-binding domain (DBD) or C-terminal fragment, bound to GST-tagged MTDH. This result indicated that MTDH interacts with the N-terminal region of FOXM1. Furthermore, deleting the D Box (DD-Box) or the KEN box (DK-Box) of FOXM1 did not affect the FOXM1-MTDH interaction. After deleting the amino acids from 180 to 201 in FOXM1 (delta MTDH binding site, DMBS), the FOXM1-MTDH interaction was blocked (Fig. 4A).
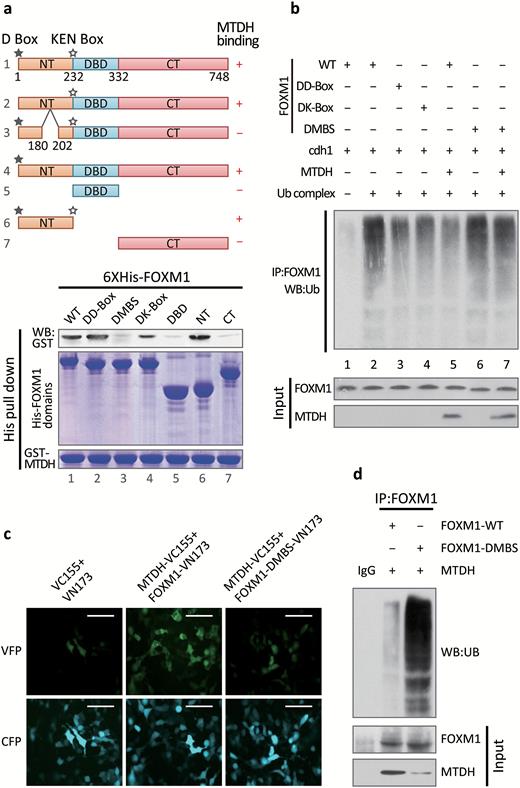
Deleting the MTDH binding site of forkhead box M1(FOXM1) destabilizes the FOXM1 protein. (A) Upper panel, diagram of the domains and mutations of FOXM1. ★, D box; ☆, KEN box; NT, N terminus; DBD, DNA-binding domain; CT, C terminus. Amino acids 180–202 constitute the delta MTDH binding site (DMBS). Lower panel, His pull-down assays were performed using purified GST-tagged MTDH and the indicated truncated or mutant forms of FOXM1. (B) The E1 ubiquitin-activating enzyme UBE1, the E2-conjugating enzyme UBE2C, the E3 ubiquitin ligase APC/C, ubiquitin, and purified GST-tagged Cdh1 were incubated with in vitro-translated MTDH and a truncated or mutant form of FOXM1. The reaction mixture was subjected to IP using an anti-Flag antibody. The reaction product was analyzed by Western blotting for ubiquitin. (C) WT or mutant FOXM1 was cloned into the pBIFC-VN173/Flag vector, and WT MTDH was cloned into the pBIFC-VC155HA vector. The indicated plasmids were co-transfected with the cyan fluorescent protein (CFP) vector into 293T cells. Scale bar, 40 μm. (D) MTDH and WT or mutant FOXM1 were stably expressed in HNA. Total cell extracts were subjected to IP.
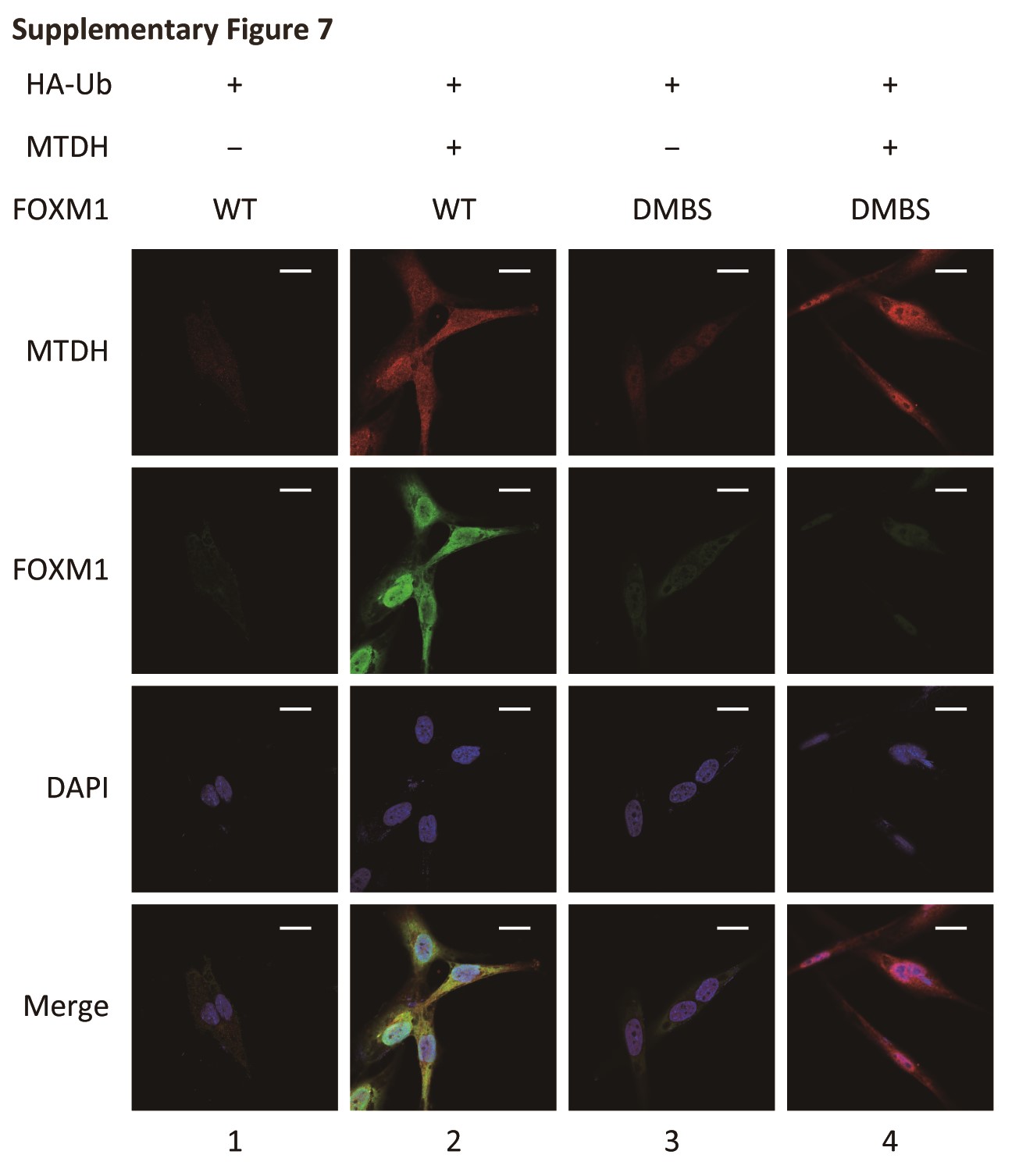
To determine whether the FOXM1-MTDH interaction is a determinant of FOXM1 ubiquitination, we next performed in vitro ubiquitination assays (Fig. 4B). Incubating ubiquitin, E1 (UBE1), E2 (UBE2C), and E3 ligases (APC/C) and Cdh1 with WT FOXM1 resulted in strong FOXM1 ubiquitination (lanes 1 and 2). However, the DD-Box and DK-Box mutants were only partially ubiquitinated (lanes 3 and 4), but FOXM1 ubiquitination was rescued by MTDH application (lanes 2 and 5). The DMBS mutant, which did not interact with MTDH, was also ubiquitinated, and its ubiquitination was not altered by MTDH application (lanes 6 and 7). These results indicated that the interaction of FOXM1 with MTDH may interfere with the recognition of Cdh1 by the KEN/D Box of FOXM1 and sequentially prevent FOXM1 ubiquitination. In NHA cells expressing excessive ubiquitin, MTDH transfection stabilized FOXM1, and MTDH and FOXM1 were colocalized (Fig. S7, lanes 1 and 2). When both MTDH and DMBS mutant FOXM1 were overexpressed, FOXM1 degradation could not be rescued (Fig. S7, lanes 3 and 4).
Next, we used the BiFC assay to validate the region of FOXM1 that binds to MTDH. Co-transfection with MTDH-VC155 and FOXM1-VN173 resulted in strong Venus fluorescent protein (VFP) signal. This observation indicated that MTDH and FOXM1 interact with each other. However, VFP florescence decreased dramatically upon co-transfection with MTDH-VC155 and FOXM1-DMBS-VN173. This finding indicated that amino acids 180–201 in FOXM1 are required for interaction with MTDH (Fig. 4C). In co-IP assay, DMBS mutant FOXM1 was much less stable during MTDH overexpression than WT FOXM1 (Fig. 4D). Thus, the MTBS binding site of FOXM1 is essential for FOXM1 binding and stabilization in vivo.
MTDH is Recruited to FOXM1 Target Gene Promoters and Enhances FOXM1 Transcriptional Activity
Because MTDH colocalized with FOXM1 in both the cytoplasm and the nucleus (Fig. 2D), we confirmed the MTDH-FOXM1 interaction in nuclear extracts (Fig. S8). To evaluate whether MTDH acts as a transcriptional enhancer,22 we performed a ChIP assay using an anti-MTDH antibody to detect FOXM1 target gene promoter regions. MTDH pulled down DNA fragments of direct target genes of FOXM1 such as VEGF, MMP-2, and Skp2 (Fig. 5A). However, MTDH did not bind to FOXM1 target gene promoters in sh-FOXM1 cells. This finding indicated that these interactions depended on FOXM1 (Fig. 5B, C).
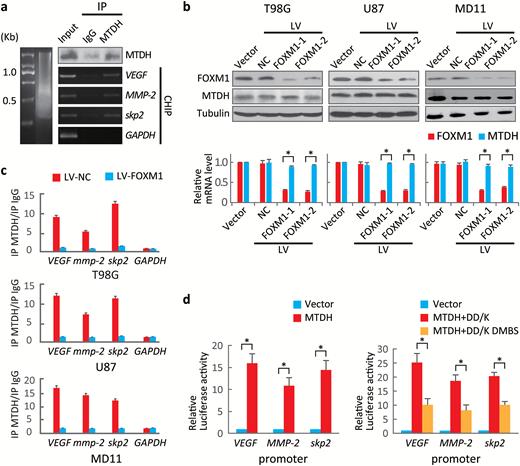
MTDH enhances forkhead box M1 (FOXM1) transcriptional activity. (A) Chromatin immunoprecipitation (ChIP) assays were performed to identify FOXM1 target gene promoters in U87 cells using anti-MTDH antibodies. The immunoprecipitated products were analyzed via standard semiquantitative PCR. (B) MTDH and FOXM1 levels in cells expressing 2 specific FOXM1 shRNAs were evaluated by Western blotting. *P < .001. (C) ChIP assays were performed on sh-FOXM1-transfected and control cells using an anti-MTDH antibody. The VEGF, MMP-2 and Skp2 promoters were amplified via semiquantitative PCR. (D) Left, dual luciferase reporter assays were used to evaluate promoter activities in 293T cells transfected with the MTDH expression or control plasmid. Right, dual luciferase reporter assays were performed to evaluate promoter activities in 293T cells carrying plasmids expressing FOXM1 lacking the D and KEN boxes (DD-box/DK-box) and FOXM1 lacking the MTDH-binding site (DMBS) as well as the D and KEN boxes. Values are presented as means±SD for triplicate samples. *P < .001.
Next, a series of luciferase assays demonstrated that MTDH enhanced the transcriptional activity of FOXM1 (Fig. 5D, left panel). The protection of MTDH against FOXM1 degradation could be due to stabilization of FOXM1 rather than enhancement of FOX1 transcriptional activity. To exclude this possibility, we used DD-box/DK-Box and DD-box/DK-box DMBS mutants. As both of these constructs are barely ubiquitinated (Fig. 4B), the differing luciferase activities of these constructs only reflected their interaction with MTDH. The DD-box/DK-box mutant displayed stronger luciferase activity than the D-box/DK-box DMBS mutant. This result indicated that the MTDH-FOXM1 interaction enhances FOXM1 transcriptional activity (Fig. 5D, right panel).
MTDH Inhibition Attenuates the Activity of FOXM1 Target Genes in Glioblastoma
To evaluate whether genes regulated by FOXM1 are also controlled by MTDH, we conducted gene expression profiling on U87 cells transfected with sh-MTDH or sh-FOXM1. The gene expression profiles after MTDH silencing was similar to that after FOXM1 silencing (Fig. 6A).
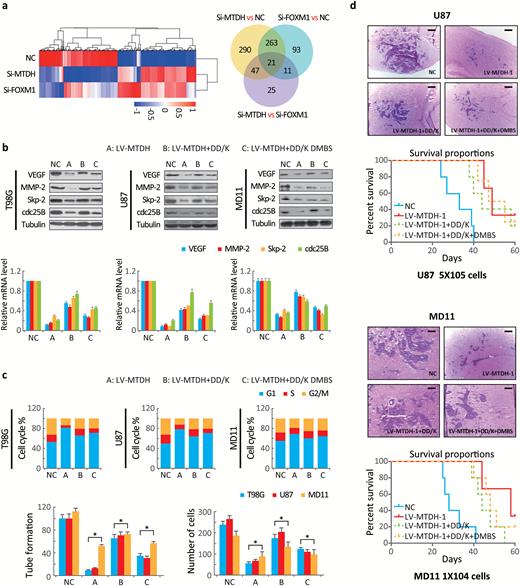
MTDH inhibition attenuates the oncogenic activity of forkhead box M1 (FOXM1) target genes, and FOXM1-stabilizing mutation prevents the MTDH inhibition-mediated attenuation of FOXM1 transcription. (A, left panel) Heatmap of the gene expression profile of U87 cells transfected with MTDH short interference (siRNA) or FOXM1 siRNA based on microarray analysis. Right panel, differential gene expression analysis comparing U87 cells expressing scrambled siRNA, siMTDH, or siFOXM1. (B) The VEGF, MMP-2, Skp2 and Cdc25B protein levels were compared among the indicated plasmid-transfected and control cells. (C) Changes in cell cycle progression, tube formation, and cell invasion among the cells indicated above were compared. Values are presented as means±SD for triplicate samples. (D) The data represent the results for 5 mice per group of 2 independent experiments. Scale bar, 10 μm. The overall survival duration of experimental mice was calculated.
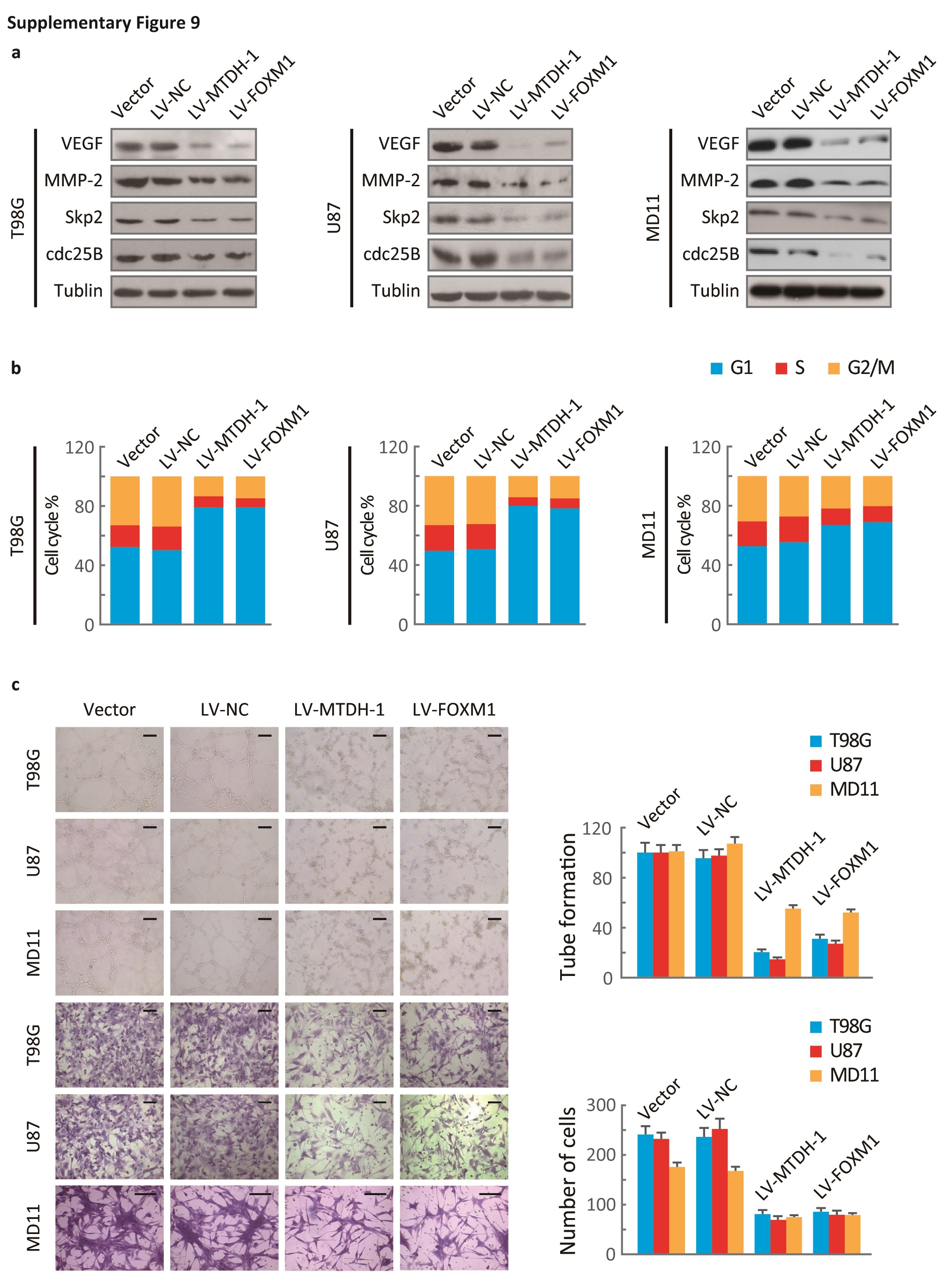
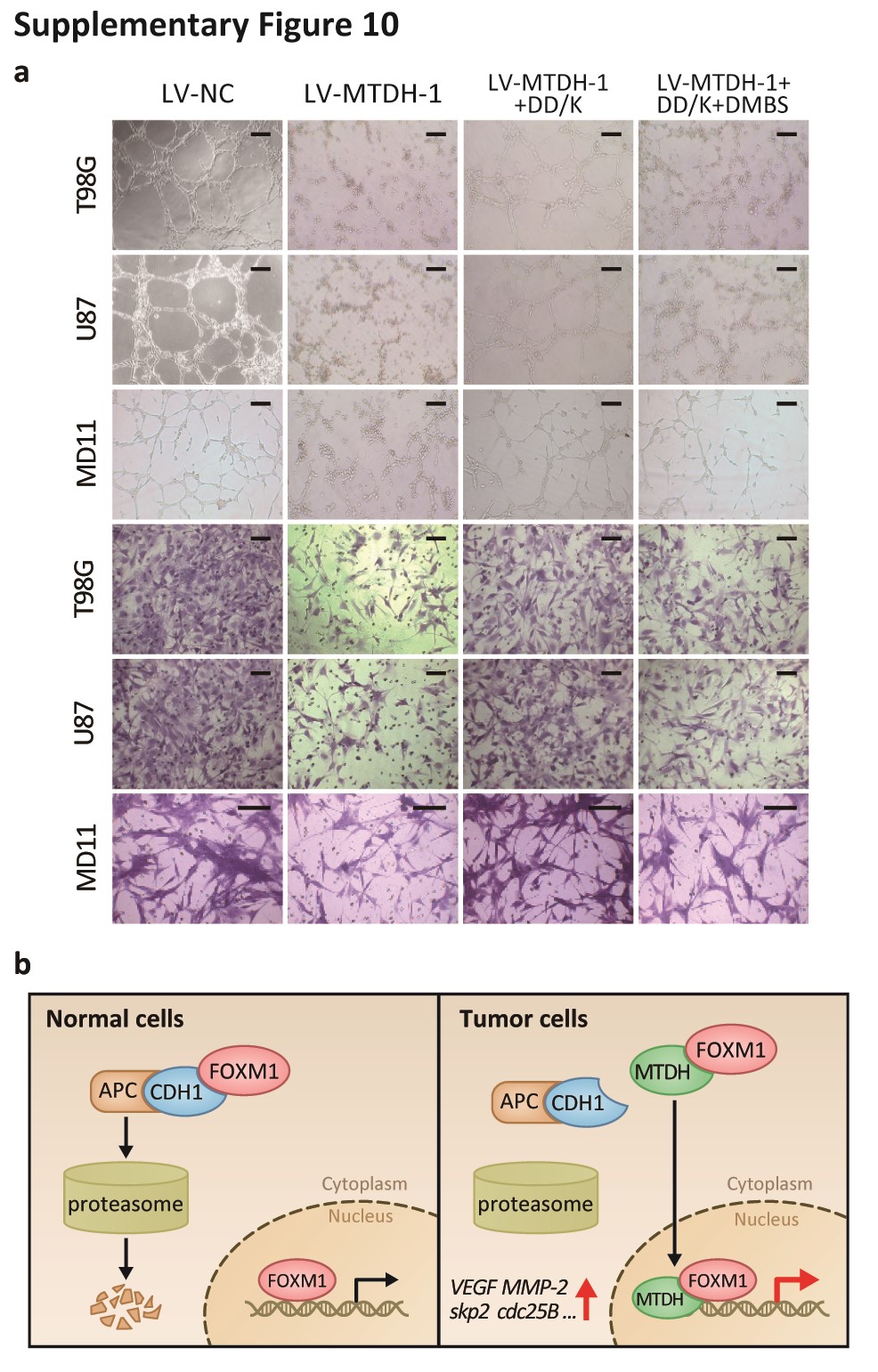
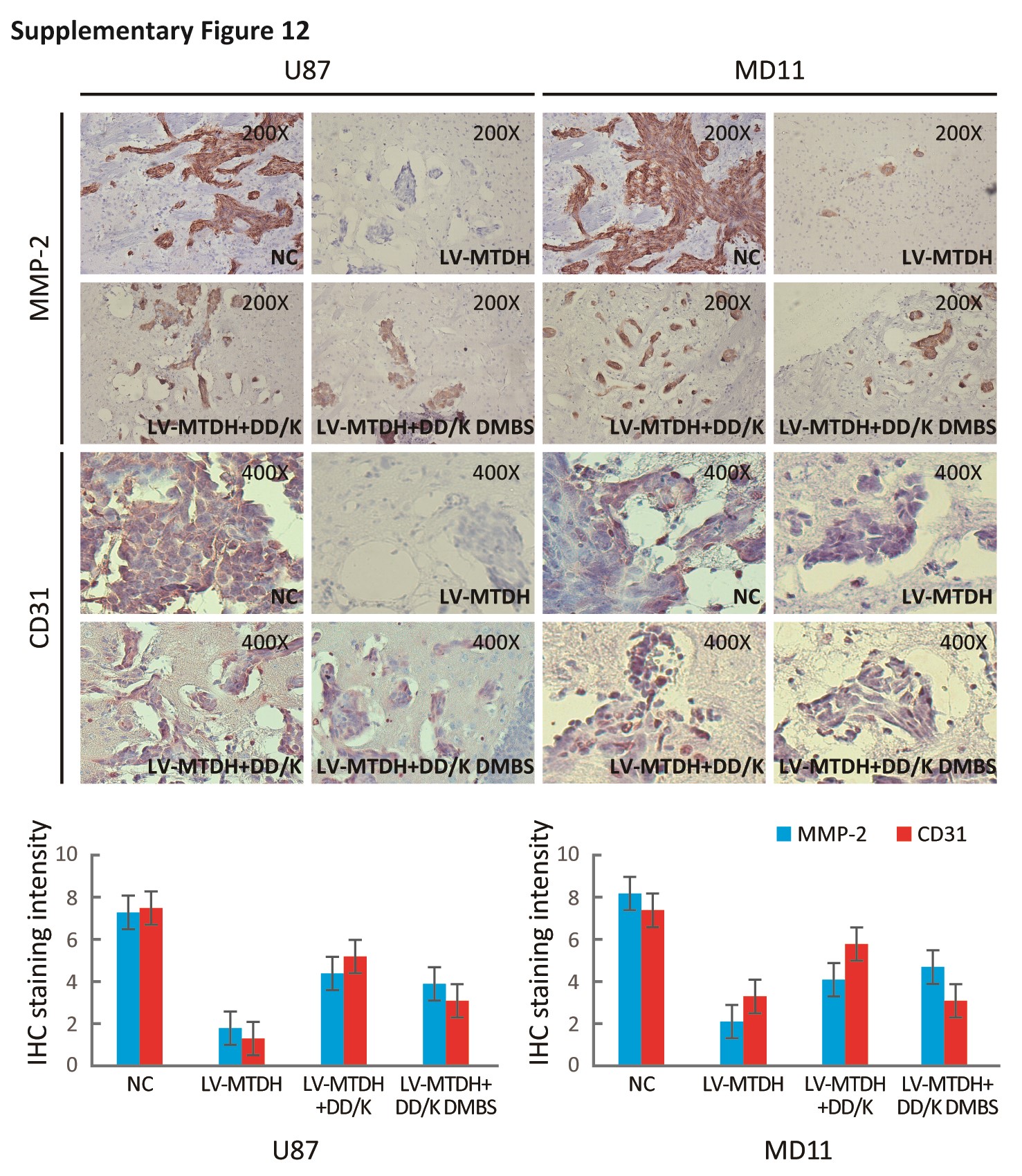
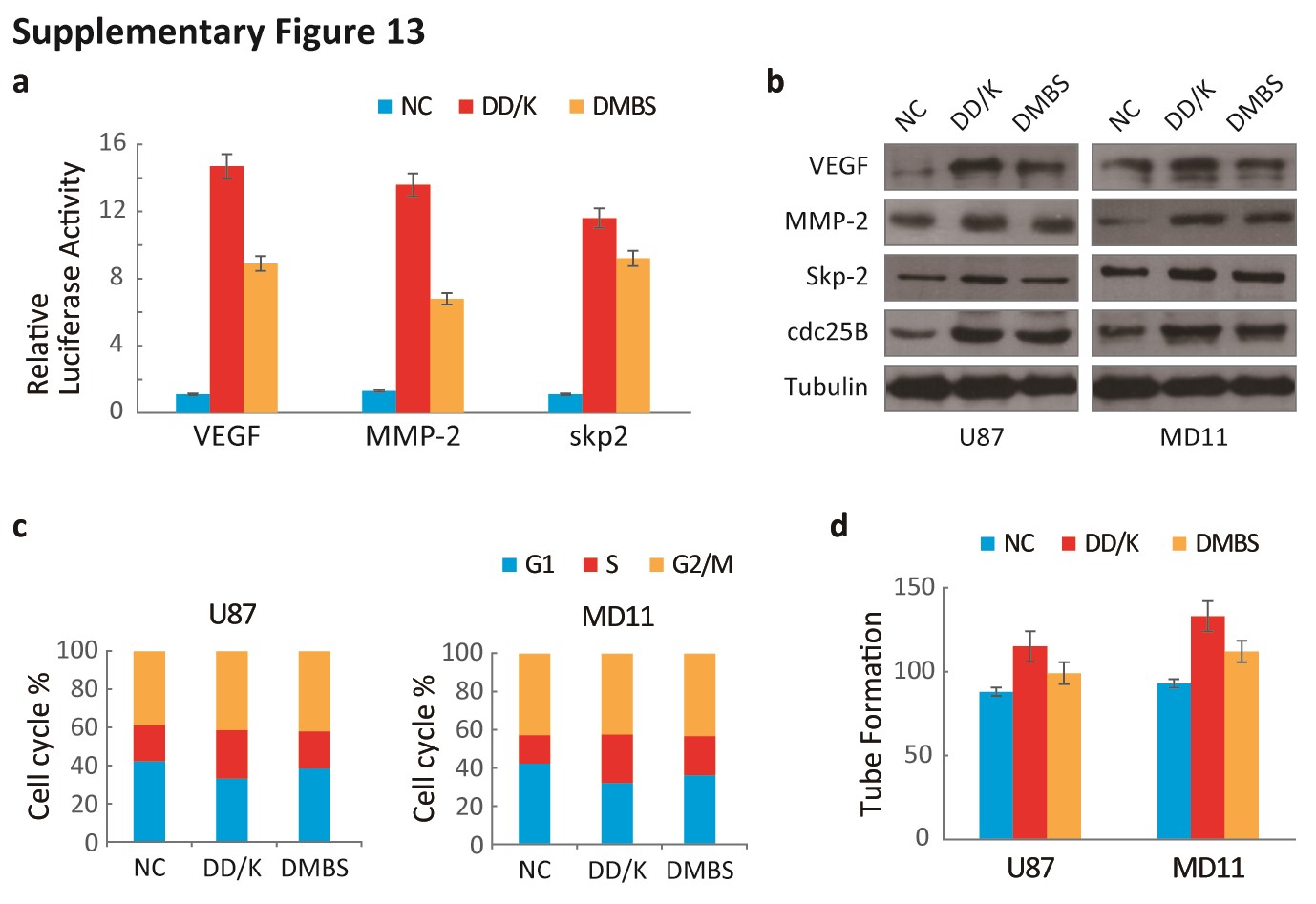
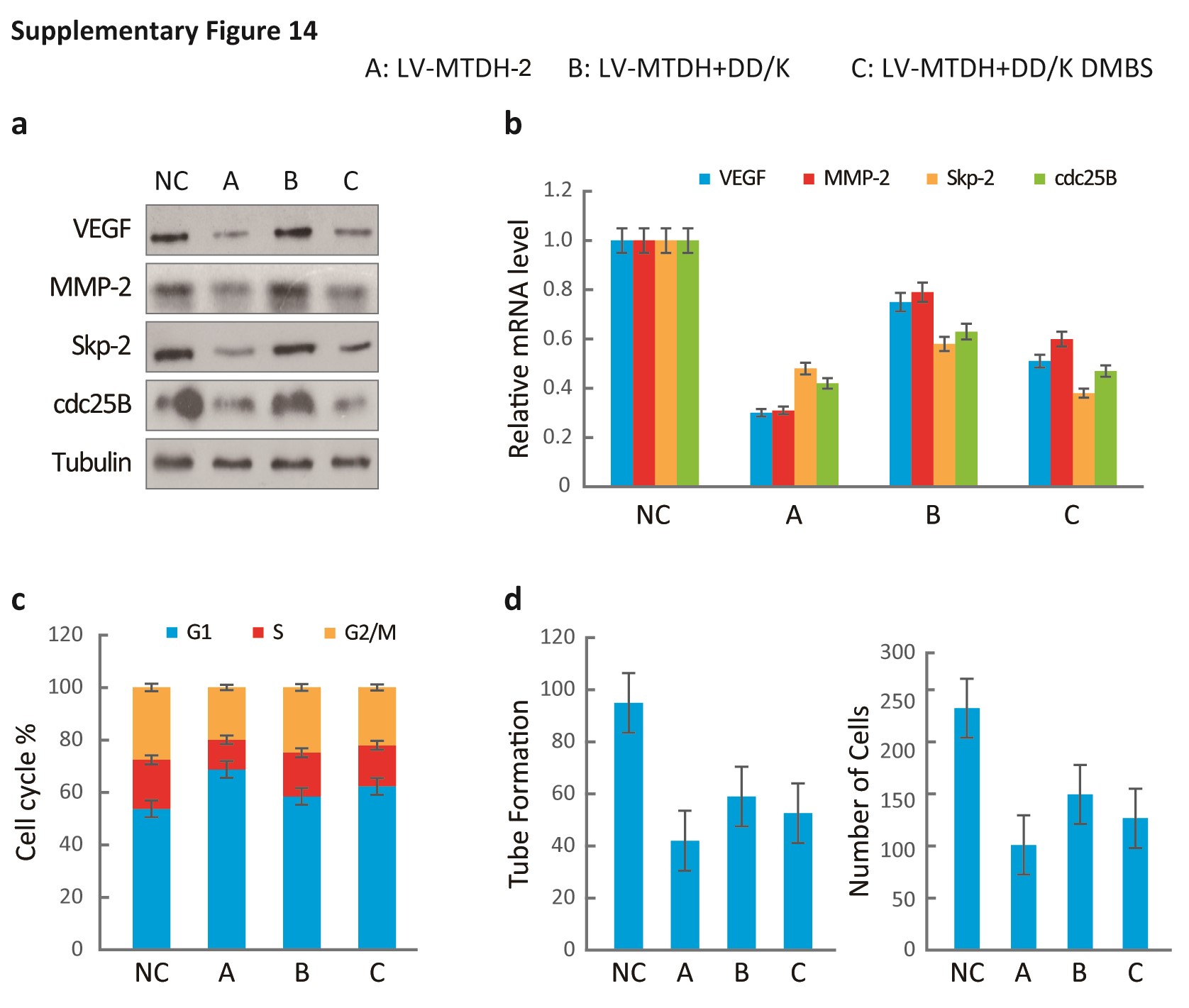
Additionally, silencing MTDH and FOXM1 independently decreased the expression of VEGF, MMP-2, Skp2, and cdc25B (Fig. S9A) and suppressed cell cycle progression, angiogenesis, and invasion (Fig. S9B, C). The DD-box/DK-box and DD-box/DK-box DMBS mutants rescued the effects of sh-MTDH. These results indicated that MTDH regulates these functions at least partially through FOXM1 (Fig. 6B). Notably, the DD-box/DK-box DMBS mutant less effectively rescued these activities than the DD-box/DK-box mutant, potentially because the DMBS mutation reduced FOXM1 transcriptional activity. These results were confirmed in biological experiments, as both mutants could restore angiogenesis, invasion, and cell cycle progression in sh-MTDH cells (Fig. S10A and Fig. 6C). Finally, stably silencing MTDH prolonged the survival of mice. Overexpressing the DD-box/DK-box mutant restored tumor malignancy and decreased the overall survival of mice. Introducing the DD-box/DK-box DMBS mutant also restored tumor growth in nude mice, although the overall survival duration of these mice was slightly longer than that of the DD-box/DK-box mutant-overexpressing mice (Fig. 6D).
Discussion
In the current study, our findings indicate that MTDH directly interacts with FOXM1 and inhibits the proteasomal degradation of FOXM1 via competition with Cdh1. Our results also suggest that MTDH increases FOXM1 transcriptional activity and enhances the expression of several genes.
FOXM1 protein levels have been thought to be regulated in part by the E3 ligase APC/C. APC/C mediates FOXM1 degradation during mitotic exit, and this process requires the recognition of the N-terminal D-box and KEN-box of FOXM1 by Cdh1. In addition to FOXM1, many mitotic genes, such as PLK1, cdc20, and aurora kinase,32–34 which produce oncogenic proteins that are frequently overexpressed in malignant tumors, are degraded by APC/C and Cdh1.35 Our findings indicate that aberrant overexpression of FOXM1 in human cancer is due to malfunction of the ubiquitination process. Specifically, MTDH displayed higher FOXM1 affinity than Cdh1, and the MTDH-FOXM1 interaction of masks the recognition of FOXM1 by Cdh1. In addition to its critical role in FOXM1 stabilization, the N-terminal domain of FoxM1 attenuates transcriptional activity via direct interaction with the C-terminal domain of FOXM1.36 The MTDH-FOXM1 interaction may also interfere with the self-interaction between the N- and C-terminals of FOXM1, thus blocking FOXM1 auto-inhibition. Because MTDH overexpression in cancer cells is observed throughout the cell cycle,37 we believe that MTDH is the key mediator of aberrant FOXM1 expression during carcinogenesis, which ultimately contributes to uncontrolled cell proliferation and tumor formation. Recent research indicated that SUMOylation of multiple sites on FOXM1, including K201, inhibited FOXM1 activity, promoted FOXM1 translocation to the cytoplasm, and enhanced APC/C- and Cdh1-mediated FOXM1 ubiquitination and degradation.38 It would be interesting to explore whether this SUMOylation is affected by MTDH-FOXM1 interaction.
MTDH has emerged as an important regulator of the development and progression of multiple types of cancer including hepatocellular carcinoma, breast cancer, prostate cancer, and glioma.26,39–41 Silencing MTDH significantly abrogated the anchorage-independent growth induced by Ha-ras/PI3K/Akt activation.25 Additionally, MTDH overexpression induced increased production of angiogenic factors, including VEGF, placental growth factor (PIGF), and fibroblast growth factor-1 (FGF1).26 MTDH also promoted invasion by activating angiopoietin-1, matrix metalloprotease-9, and HIF1.18 Previous studies showed that MTDH can bind to multiple functional proteins. MTDH interacts with the p65 subunit of NF-κB upon TNF-alpha treatment and forms a complex with NF-κB as well as with the transcriptional coactivator cyclic AMP-responsive element-binding protein (CREB)–binding protein (CBP).42 Thirkettle et al. identified that MTDH interacts with the promyelocytic leukemia zinc finger (PLZF) protein and prevents the recruitment of PLZF to the c-Myc promoter, thereby increasing c-Myc transcription.43 A more recent study showed that MTDH interacts with staphylococcal nuclease and Tudor domain containing-1 (SND1) and promotes lung metastasis.44 Given the above findings along with our present results, we infer that a major function of MTDH in tumorigenesis is its interaction with and manipulation of other pivotal oncogenic proteins.
In addition to regulating FOXM1 stability, MTDH regulates FOXM1 transcription. Previous reports showed that FOXM1 activity required multiple cofactors or post-transcriptional modifications such as CBP/p30045 and PLK1/CDK1-dependent phosphorylation of S715/724/251 on FOXM1.2,46 Our data indicated that MTDH is recruited to the FOXM1 transcriptional complex and acts as an enhancer during transcription.
Both FOXM1 and MTDH are valuable, clinically relevant prognostic markers.17,47,48 In addition, both FOXM1 and MTDH are responsible for drug resistance.48,49 Although FOXM1 is widely overexpressed in human malignancy, it is not an ideal drug target because it is required for normal cell division. Thus, interfering with the FOXM1-MTDH interaction or manipulating MTDH expression in human cancer might serve as an effective anticancer therapy.
In conclusion, this study provides novel mechanistic insight into FOXM1 protein regulation as well as the modulation of FOXM1 transcriptional activity by MTDH. Our results reveal a molecular mechanism by which FOXM1 aberrantly accumulates in proliferative human cancers. This finding suggests that the FOXM1-MTDH interaction represents a potential target for novel therapeutic strategies against glioblastoma.
Supplementary Material
Supplementary material is available at Neuro-Oncology Journal online (http://neuro-oncology.oxfordjournals.org/).
Funding
This work was supported by the National Natural Science Foundation of China (81370072, 81572477), the Natural Science Foundation of Guangdong Province for Distinguished Young Scholars (S2013050014535), the Natural Science Foundation of Guangdong Province (2014A030313183), the Pearl River New Star Science and Technology Program of Guangzhou City (2013J2200022), the Science and Technology Program of Guangzhou City (2013J4100060), and the NCI (grant R01CA157933).
Conflict of interest statement. There were no conflicts of interest to disclose.
References
Author notes
Corresponding Authors: Nu Zhang, MD, Department of Neurosurgery, The First Affiliated Hospital of Sun Yat-sen University, No. 58 Zhongshan 2 Road, Guangzhou, Guangdong Province 510080, PR China (zhangnu2@mail.sysu.edu.cn); Suyun Huang, MD, PhD, Department of Neurosurgery, Unit 1004, Room S5.8116, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, Texas 77030, USA (suhuang@mdanderson.org).
These authors contributed equally to this work.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}