





Abstract
Objectives. The paediatric idiopathic inflammatory myopathies (IIMs) are a group of rare chronic inflammatory disorders of childhood, affecting muscle, skin and other organs. There is a severe lack of evidence base for current treatment protocols in juvenile myositis. The rarity of these conditions means that multicentre collaboration is vital to facilitate studies of pathogenesis, treatment and disease outcomes. We have established a national registry and repository for childhood IIM, which aims to improve knowledge, facilitate research and clinical trials, and ultimately to improve outcomes for these patients.
Methods. A UK-wide network of centres and research group was established to contribute to the study. Standardized patient assessment, data collection forms and sample protocols were agreed. The Biobank includes collection of peripheral blood mononuclear cells, serum, genomic DNA and biopsy material. An independent steering committee was established to oversee the use of data/samples. Centre training was provided for patient assessment, data collection and entry.
Results. Ten years after inception, the study has recruited 285 children, of which 258 have JDM or juvenile PM; 86% of the cases have contributed the biological samples. Serial sampling linked directly to the clinical database makes this a highly valuable resource. The study has been a platform for 20 sub-studies and attracted considerable funding support. Assessment of children with myositis in contributing centres has changed through participation in this study.
Conclusions. This establishment of a multicentre registry and Biobank has facilitated research and contributed to progress in the management of a complex group of rare muscloskeletal conditions.
Introduction
The idiopathic inflammatory myopathies (IIMs) are a heterogeneous group of disorders that have in common a chronic autoimmune inflammatory process affecting variably muscle, skin and internal organs [1]. The childhood IIMs include JDM, juvenile PM (JPM) and other, less common, disorders [2, 3]. The incidence of JDM is ∼2–3/millions/year, with some differences between the ethnic groups [4–8]. The rarity of these conditions means that very few single centres have sufficient patient numbers to adequately power the studies of disease complications, outcomes or response to treatment.
The clinical manifestations of the IIMs differ between children and adults, and the various disease types occur with differing frequencies. In childhood, DM occurs far more frequently than PM, whereas in adults the ratio is more equal. Other forms of myositis, such as IBM, are very rare in the paediatric population. Severe complications of DM, such as vasculopathic ulceration and calcinosis, are more common in juvenile than adult-onset disease, and overlap syndromes with features of other connective tissue disorders occur more commonly [6–9].
Despite considerable advances in the management of childhood IIMs, the conditions are still associated with significant morbidity and mortality, representing a major long-term medical, social and economic burden on patients, their families and health care systems [10]. Evidence suggests that early aggressive management of JDM improves outcome, whereas a long duration of untreated disease is associated with longer time to reach remission and higher rates of complications such as ongoing skin disease or functional impairment [11–13]. Emerging data suggest an increased risk of morbidities, such as cardiovascular disease, and osteoporosis in adulthood although few long-term outcome data are yet available.
Improving both long- and short-term outcomes requires early recognition, prompt referral to specialist care and appropriate treatment of patients with JDM and other forms of myositis. These goals have been hindered by the lack of an evidence base in paediatric myositis. Little is known about the basic mechanisms underlying the pathogenesis of childhood myositis, and there is almost no level 1 evidence base from trials in JDM for any specific therapeutic interventions that are in current use. There is, therefore, an urgent need for robust basic, translational and clinical research in this field and the generation of standardized protocols of management. Until recently, it was difficult to carry out detailed investigations of genetics, disease mechanisms or biomarker studies, let alone clinical trials, in the paediatric IIMs due to their rarity, the lack of validated tools for the assessment of disease activity or outcome and the absence of any standardized treatment protocols.
Here, we present the 10-year experience of the Juvenile Dermatomyositis National (UK and Ireland) Cohort Biomarker Study and Repository for Idiopathic Inflammatory Myopathies, formerly known as the Juvenile Dermatomyositis National (UK and Ireland) Registry and Repository for Idiopathic Inflammatory Myopathies. The aims of this registry are to define the clinical characteristics, biomarkers and response to treatments of childhood IIMs, while at the same time developing the specific infrastructure and tools needed to enable participation in interventional trials (Table 1). Through the generation of standardized protocols for investigation and management, this study aims to make significant and important contributions to the recognition, understanding and management of paediatric myositis, and so to address the needs of children with these severe, life-threatening diseases.
Aims of the UK JDM cohort biomarker study and repository
| Determine the demographics of JDM |
| Define disease presentation, activity, damage, current management protocols and response to current medication use |
| Determine prognostic biomarkers (genetic, serological, gene expression or others) |
| Assess the validity and reliability of disease activity scores that can be used in the clinical assessment of response to therapy as well as future clinical trials |
| Develop a cohort of patients suitable for recruitment to clinical interventional trials |
| Facilitate national and international collaborative research studies and trials in JDM |
| Create a consented repository of data and sample collection to investigate the immunological and genetic abnormalities of JDM with a view to identifying future therapeutic targets |
| Determine the demographics of JDM |
| Define disease presentation, activity, damage, current management protocols and response to current medication use |
| Determine prognostic biomarkers (genetic, serological, gene expression or others) |
| Assess the validity and reliability of disease activity scores that can be used in the clinical assessment of response to therapy as well as future clinical trials |
| Develop a cohort of patients suitable for recruitment to clinical interventional trials |
| Facilitate national and international collaborative research studies and trials in JDM |
| Create a consented repository of data and sample collection to investigate the immunological and genetic abnormalities of JDM with a view to identifying future therapeutic targets |
Aims of the UK JDM cohort biomarker study and repository
| Determine the demographics of JDM |
| Define disease presentation, activity, damage, current management protocols and response to current medication use |
| Determine prognostic biomarkers (genetic, serological, gene expression or others) |
| Assess the validity and reliability of disease activity scores that can be used in the clinical assessment of response to therapy as well as future clinical trials |
| Develop a cohort of patients suitable for recruitment to clinical interventional trials |
| Facilitate national and international collaborative research studies and trials in JDM |
| Create a consented repository of data and sample collection to investigate the immunological and genetic abnormalities of JDM with a view to identifying future therapeutic targets |
| Determine the demographics of JDM |
| Define disease presentation, activity, damage, current management protocols and response to current medication use |
| Determine prognostic biomarkers (genetic, serological, gene expression or others) |
| Assess the validity and reliability of disease activity scores that can be used in the clinical assessment of response to therapy as well as future clinical trials |
| Develop a cohort of patients suitable for recruitment to clinical interventional trials |
| Facilitate national and international collaborative research studies and trials in JDM |
| Create a consented repository of data and sample collection to investigate the immunological and genetic abnormalities of JDM with a view to identifying future therapeutic targets |
Methods
The establishment of a national registry and repository for childhood IIMs [the Juvenile Dermatomyositis National (UK and Ireland) Cohort Biomarker Study and Repository for Idiopathic Inflammatory Myopathies] was made possible following an initial generous grant from the C. Hayes Research Trust. The study was launched in 2000 and to date has recruited 285 children.
Establishing the network of centres and research group
The coordinating centre, based at UCL Institute of Child Health, London, initiated discussions with the British Society of Paediatric and Adolescent Rheumatology requesting expressions of interest from units wishing to contribute to the registry. Units deemed able to recruit adequate numbers of patients (regional tertiary paediatric rheumatology referral centres) were offered funding for allied health professional/research nursing sessions to recruit patients, and undertake data entry and sample collection. Central to the success of the registry was a series of initiating visits to participating centres offering discussion about practicalities of recruitment, form completion, use of validated assessment tools for muscle function and disease activity [including the childhood myositis assessment scale (CMAS) and manual muscle testing (MMT)] [14–17] and protocols for sample collection.
Ethical approval was obtained for collection of data and biological samples within the Biobank, and the study was carried out in accordance with the Declaration of Helsinki. Patients and families are fully aware that consent includes donation of the contributed biological material. A separate, broad ethical approval was obtained, for the use of the Biobank samples in the investigation of the pathogenesis of childhood myositis. The group of committed researchers was formally established as the Juvenile Dermatomyositis Research Group (JDRG), ensuring ownership of, and commitment to, the project. An independent steering committee was established, including external clinical, ethical and scientific advisors, and consumer representation, to oversee the project and scrutinize applications to use the clinical data and/or biological samples from the registry/repository. Authorship and membership guidelines were agreed, and a standardized application process and agreement forms for access to data or samples developed, for use by external collaborators.
Recruitment, data entry and sample Biobank
Data collection forms were developed through a series of consensus meetings using the nominal group technique with the principle investigators (PIs) and members of the research teams. Agreement was reached regarding key typical and less common clinical features to be recorded. A standardized set of assessments, including those for muscle function strength and stamina, and laboratory investigations were agreed. Baseline and follow-up forms were developed, recording history, clinical features, muscle assessment, laboratory results and details of treatment. Annual incidence figures provided an estimate of 20–30 new cases a year within the UK. Given this rarity of both JDM and other paediatric IIMs, it was agreed to recruit both established cases (i.e. including data obtained retrospectively) and new cases, recruited at diagnosis. Contributing centres were all referral centres for specialist paediatric rheumatology care but were encouraged to facilitate recruitment from, and shared care with, other clinicians and hospitals locally, using established networks of care. This both increased recruitment of patients to the registry and provided a mechanism for dissemination of the best practice. All patients included have given written informed parent/patient consent/assent.
A central database was established, with full technical support, and maintained by a dedicated data manager, who also provides support to local centres. All data are anonymized. During the first 10 years of the registry, data have been collected on paper forms and entered onto the database at the coordinating centre. Work is ongoing to develop web-based data entry, which should minimize the need for data entry centrally saving both time and potential transcription errors. Paper data collection will remain an option for any unit unable to enter online. The Biobank of clinical samples has been integral to the study throughout, meaning that this is now a unique resource, with samples stored from the time of presentation and during the disease course. In accordance with ethical approval for paediatric studies, venous blood is collected only when venepuncture is clinically indicated, and sent to the coordinating laboratory to be processed. Peripheral blood mononuclear cells (PBMCs), serum and genomic DNA are prepared and stored following agreed standard operating procedures using standard methods. One centre on site with the coordinating laboratory can complete sample processing within 4 h of venepuncture and also, therefore, stores plasma samples for proteomics. Biopsy material is stored (after diagnostic work has been completed) in cases on whom muscle or other biopsy is performed.
Results
Membership of the JDRG network
Nine centres originally agreed to participate in the JDRG. All PIs have had specialist training in paediatric rheumatology and work in specialist referral centres for paediatric rheumatology. The full contributor list is provided in the acknowledgements. An indication of the success of this registry is that all original centres recruiting and contributing data to the registry continue to do so, and several new centres wishing to join have recently approached the coordinating centre. New centres are provided with specific training before starting to recruit patients.
Data and sample collection
Fundamental to the establishment of the registry was agreement by consensus on a core set of data and the development of standardized forms for this collection (see supplementary data Forms 1 and 2, available at Rheumatology Online). Data are collected at recruitment (Form 1) including information on disease up to the date of recruitment and then serially (Form 2), with 3–4 monthly data for the first 2 years and then at least once a year. For the latter, annual data summing 1 year are now collected onto a separate form (see supplementary data Form 3, available at Rheumatology Online). Together with common and rarer symptoms and examination features, data collection includes assessment of disease activity using the following tools: CMAS, MMT of strength, Childhood HAQ (CHAQ), physician’s global assessment by visual analogue scale (VAS), parent/patient assessment by VAS and laboratory tests (Forms 1 and 2). These measures are all part of the recent proposed core sets of measures to measure disease activity in JDM and the CMAS, MMT and CHAQ are all validated tools in JDM [14–17]. Clinicians familiar with the data collection forms are able to complete Forms 1 and 2 in ∼10 min. Some contributors use Form 2 as their clinical record, reducing the burden of additional time for form filling. Biobank samples are stored, as a minimum, at recruitment (serum, DNA and PBMCs), and then serum (and DNA where required) on an annual basis, whereas PBMC samples are stored at baseline, 1 and 5 years. Biopsy material has been successfully sent from many centres around the country, either a frozen block of tissue or as fixed dried slides ([18] and J. Holton, V. Varsani and L.R. Wedderburn, submitted).
Recruitment and patient demographics
To be eligible for the study, a child must have definite or suspected myositis with symptoms starting before the 16th birthday. This cut-off age reflects current UK clinical practice for defining paediatric myositis. Conditions presenting primarily with myositis were considered eligible, resulting in the inclusion of a number of children with unusual diagnoses. Children were approached to enrol in the study sequentially from all sites, and were included if they had a diagnosis of definite, probable or possible JDM or PM as defined by the contributing physician [2, 3]. Since there are no agreed international criteria for the definition of overlap syndromes in children, and features of overlap vary between cases, children with DM or PM, who also had significant overlap with scleroderma, polyarthritis lupus or MCTD (provided myositis was the predominant clinical feature), were recorded as entered by the contributing physician. Other forms of myositis such as focal, eosinophilic, orbital, inclusion body or sarcoid myositis, or other IIMs were included. The only exclusion criterion was entry onto another registry.
In 10 years of the study, 296 patients from nine centres were approached as eligible for the study, of which 285 were recruited. Four children were subsequently shown not to have myositis as a primary diagnosis, five patients had insufficient data for analysis or were lost to follow-up and one family requested to be removed from the registry. Therefore, analysis is possible on 275 patients, of which 175 (63.6%) were recruited prospectively, within 12 months of onset. There have been three deaths of patients within the registry. From the 275 cases recruited, 86% have contributed blood samples and 81 (29%) have contributed muscle biopsy material to the Biobank. Our estimate of the prevalence of JDM in the UK, based upon incidence and national population data in 2000, was ∼450 children: it is, therefore, apparent that a percentage of cases have not yet been reached through our network. It was hoped that each regional centre would facilitate recruitment from smaller centres and colleagues in other specialties (dermatology and neurology), to ensure a truly representative spectrum of patients. In practice, recruitment has largely remained within major centres and the less severely affected children may, therefore, be under-represented in the cohort.
The diagnoses assigned to all 275 cases are shown in Table 2. Demographic and baseline clinical data for the 258 patients with a diagnosis of definite or probable JDM, and definite or probable JPM are shown separately. JPM was diagnosed in only seven (2.5%) children in this cohort. Of the cases assigned with a diagnosis of JDM, 35 had features of overlap with either scleroderma (19), lupus (7), MCTD (5) or severe arthritis (4). As these overlap cases were noted at the discretion of the recruiting physician they may not reflect the overall prevalence of overlap features within the registry cohort. We have previously published data outlining the frequency of clinical features such as arthritis (36%) or features suggestive of scleroderma overlap (24%), for patients within the registry [6, 19]. Of the 258 children with a diagnosis of probable or definite JDM or JPM, 182 (70.5%) were girls, giving a female to male ratio of 2.39 : 1. The median age of onset was 6.3 years, similar to other large cohorts [10, 20–22]. At disease onset, 37% patients were ≤4 years old. The distribution of age of onset is illustrated in Fig. 1. Median time between onset of symptoms and diagnosis was 4 months. It is noteworthy that 117 (42%) children had muscle biopsy but only 21 (7.6%) had an EMG during diagnostic work-up. This is in keeping with our previous study, which surveyed 92 centres from 32 countries caring for children with JDM, and showed that 70% of centres had access to muscle MRI, but only 61 and 55%, respectively, used muscle biopsy and EMG in the investigation of children with suspected myositis [23]. This study and our data confirm the need for developing improved diagnostic criteria based on modern methods of diagnosis.
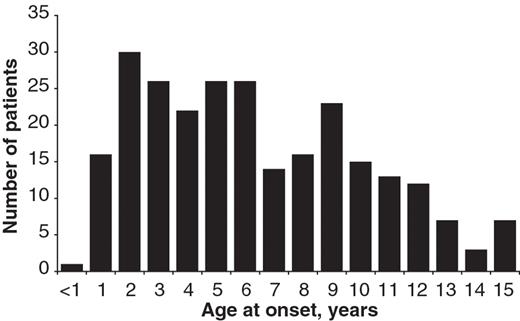
Age distribution at onset of myositis symptoms in the 258 children recruited to the Juvenile Dermatomyositis National (UK and Ireland) Cohort Biomarker Study and Repository for Idiopathic Inflammatory Myopathies, with definite or probable JDM or definite or probable JPM.
Demographics of patients in the registry (analysis of 275 patients)a
| Diagnosis | n (%) |
|---|---|
| Definite JDM | 203 (73.8) |
| Probable JDM | 48 (17.5) |
| Definite JPM | 4 (1.5) |
| Probable JPM | 3 (1.1) |
| Other IIMsb | 4 (1.5) |
| Focal myositis | 5 (1.8) |
| MCTD | 8 (2.9) |
| Diagnosis | n (%) |
|---|---|
| Definite JDM | 203 (73.8) |
| Probable JDM | 48 (17.5) |
| Definite JPM | 4 (1.5) |
| Probable JPM | 3 (1.1) |
| Other IIMsb | 4 (1.5) |
| Focal myositis | 5 (1.8) |
| MCTD | 8 (2.9) |
| Demographic data, n (%) | In cases of definite or probable JDM or JPM (n = 258) |
|---|---|
| Sex | |
| Girls | 182 (70.5) |
| Boys | 76 (29.5) |
| Ethnicity | |
| White | 207 (80.2) |
| Black—Caribbean | 9 (3.5) |
| Black—African | 7 (2.7) |
| Black—Other | 4 (1.6) |
| Indian | 6 (2.3) |
| Pakistani | 10 (3.9) |
| Bangladeshi | 1 (0.4) |
| Chinese | 0 |
| Other ethnicity | 14 (5.4) |
| Demographic data, n (%) | In cases of definite or probable JDM or JPM (n = 258) |
|---|---|
| Sex | |
| Girls | 182 (70.5) |
| Boys | 76 (29.5) |
| Ethnicity | |
| White | 207 (80.2) |
| Black—Caribbean | 9 (3.5) |
| Black—African | 7 (2.7) |
| Black—Other | 4 (1.6) |
| Indian | 6 (2.3) |
| Pakistani | 10 (3.9) |
| Bangladeshi | 1 (0.4) |
| Chinese | 0 |
| Other ethnicity | 14 (5.4) |
| Baseline characteristics, cases of definite or probable JDM or JPM | |
|---|---|
| Age at onset, median (IQR), years | 6.3 (3.8–9.6) |
| Time between symptom onset and diagnosis, median (IQR), months | 4 (2–10) |
| Physician’s global assessment at the time of diagnosis, median (IQR)c | 4.8 (2.4–7.0) |
| CHAQ at the time of diagnosis, median (IQR)c | 1.375 (0.625–2.375) |
| CMAS at time of diagnosis, median (IQR)c | 31 (17–47) |
| Baseline characteristics, cases of definite or probable JDM or JPM | |
|---|---|
| Age at onset, median (IQR), years | 6.3 (3.8–9.6) |
| Time between symptom onset and diagnosis, median (IQR), months | 4 (2–10) |
| Physician’s global assessment at the time of diagnosis, median (IQR)c | 4.8 (2.4–7.0) |
| CHAQ at the time of diagnosis, median (IQR)c | 1.375 (0.625–2.375) |
| CMAS at time of diagnosis, median (IQR)c | 31 (17–47) |
aMedian length of completed follow-up data = 3.0 years, IQR = 1.1–5.0 years. bThese four cases were: two brothers with neonatal-onset multi-system inflammatory disorder that included prominent inflammatory myositis, one post-streptococcal myositis and one viral myositis. cFor those recruited prospectively where baseline measure at the time of diagnosis is available. IQR: interquartile range.
Demographics of patients in the registry (analysis of 275 patients)a
| Diagnosis | n (%) |
|---|---|
| Definite JDM | 203 (73.8) |
| Probable JDM | 48 (17.5) |
| Definite JPM | 4 (1.5) |
| Probable JPM | 3 (1.1) |
| Other IIMsb | 4 (1.5) |
| Focal myositis | 5 (1.8) |
| MCTD | 8 (2.9) |
| Diagnosis | n (%) |
|---|---|
| Definite JDM | 203 (73.8) |
| Probable JDM | 48 (17.5) |
| Definite JPM | 4 (1.5) |
| Probable JPM | 3 (1.1) |
| Other IIMsb | 4 (1.5) |
| Focal myositis | 5 (1.8) |
| MCTD | 8 (2.9) |
| Demographic data, n (%) | In cases of definite or probable JDM or JPM (n = 258) |
|---|---|
| Sex | |
| Girls | 182 (70.5) |
| Boys | 76 (29.5) |
| Ethnicity | |
| White | 207 (80.2) |
| Black—Caribbean | 9 (3.5) |
| Black—African | 7 (2.7) |
| Black—Other | 4 (1.6) |
| Indian | 6 (2.3) |
| Pakistani | 10 (3.9) |
| Bangladeshi | 1 (0.4) |
| Chinese | 0 |
| Other ethnicity | 14 (5.4) |
| Demographic data, n (%) | In cases of definite or probable JDM or JPM (n = 258) |
|---|---|
| Sex | |
| Girls | 182 (70.5) |
| Boys | 76 (29.5) |
| Ethnicity | |
| White | 207 (80.2) |
| Black—Caribbean | 9 (3.5) |
| Black—African | 7 (2.7) |
| Black—Other | 4 (1.6) |
| Indian | 6 (2.3) |
| Pakistani | 10 (3.9) |
| Bangladeshi | 1 (0.4) |
| Chinese | 0 |
| Other ethnicity | 14 (5.4) |
| Baseline characteristics, cases of definite or probable JDM or JPM | |
|---|---|
| Age at onset, median (IQR), years | 6.3 (3.8–9.6) |
| Time between symptom onset and diagnosis, median (IQR), months | 4 (2–10) |
| Physician’s global assessment at the time of diagnosis, median (IQR)c | 4.8 (2.4–7.0) |
| CHAQ at the time of diagnosis, median (IQR)c | 1.375 (0.625–2.375) |
| CMAS at time of diagnosis, median (IQR)c | 31 (17–47) |
| Baseline characteristics, cases of definite or probable JDM or JPM | |
|---|---|
| Age at onset, median (IQR), years | 6.3 (3.8–9.6) |
| Time between symptom onset and diagnosis, median (IQR), months | 4 (2–10) |
| Physician’s global assessment at the time of diagnosis, median (IQR)c | 4.8 (2.4–7.0) |
| CHAQ at the time of diagnosis, median (IQR)c | 1.375 (0.625–2.375) |
| CMAS at time of diagnosis, median (IQR)c | 31 (17–47) |
aMedian length of completed follow-up data = 3.0 years, IQR = 1.1–5.0 years. bThese four cases were: two brothers with neonatal-onset multi-system inflammatory disorder that included prominent inflammatory myositis, one post-streptococcal myositis and one viral myositis. cFor those recruited prospectively where baseline measure at the time of diagnosis is available. IQR: interquartile range.
Funding and research studies resulting from the Juvenile Dermatomyositis National (UK and Ireland) Cohort Biomarker Study and Repository for Idiopathic Inflammatory Myopathies
The initial establishment of this study was made possible by a grant from the C. Hayes Research Trust. Since that time the study has attracted considerable research funding, some for specific projects using the Cohort study as a platform and others for infrastructure support. These include grants from the Wellcome Trust, Myositis Support Group, UK, Raynaud’s and Scleroderma Association, Arthritis Research, UK (formerly Arthritis Research Campaign) and Action Medical Research. Recently, the study has been adopted onto the UK Government-funded Medicines for Children Research Network portfolio, making it eligible for UK government-sourced funding for support for health service research costs. A dedicated website for public and researcher information about the study and the JDRG has been launched (www.juveniledermatomyositis.org.uk).
In the 10 years since inception, the JDM cohort biomarker study and repository has generated 20 specific projects (Table 3), led both by members of the JDRG and by outside investigators, and agreed by the Steering Committee. Many of these have generated new knowledge, including pathological studies [24–30], generation of a standardized assessment of muscle biopsy [18], imaging studies [31], a study on the effects of exercise on MRI features in muscle [32], characterization of novel auto-antibodies in JDM [6, 33, 34], serological/genetic correlative studies and candidate gene association studies [6, 35, 36]. In addition, the study has generated analysis of our initial large cohort of children with myositis [19] as well as specific studies focusing on complex cases such as children with orophayrngeal involvement [37] and the use of novel therapies [38, 39]. Further clinical and basic studies are ongoing.
Projects approved for study within the registry, 2000–10
| Year approved | Project title | References |
|---|---|---|
| 2002 | Immunological characterization of the infiltrate in JDM muscle | [22, 25] |
| 2002 | Tracking of lymphocyte changes before or after flare of active disease | |
| 2002 | Analysis of MHC Class 1 expression in an animal model of myositis | [27, 28] |
| 2002 | Recognition of MHC Class 1 molecules on muscle cells by lymphocytes (CD8 T cells or NK cells) | [22, 25, 29] |
| 2002 | The role of muscle ultrasound and MRI in juvenile dermatomyositis | [30, 31] |
| 2004 | Investigation of alterations of MRP8/14 proteins in juvenile dermatomyositis | Ongoing study |
| 2004 | Autoantibodies and HLA haplotypes in juvenile dermatomyositis, juvenile scleroderma and overlap syndromes | [8, 32, 33] |
| 2004 | Do functional gene variants in anti-inflammatory pathways alter the course of disease and allow greater inflammatory response? | [34, 35] |
| 2005 | Design and testing/validation of a score tool for assessing severity in JDM | [29] |
| 2005 | Fetuin-A and calcinosis in juvenile dermatomyositis | [26] |
| 2005 | Genotyping analysis in JDM for phenotype–genotype correlation | [8, 34, 35] |
| 2006 | Is there evidence of maternal microchimerism in muscle biopsies from children with juvenile dermatomyositis? | Ongoing study |
| 2006 | Extension for genotyping analysis in JDM for phenotype: genotype correlation | [35] |
| 2008 | Genome-wide association study of the idiopathic inflammatory myopathies (collaboration with MYOGEN consortium) | Ongoing study |
| 2008 | Vasculopathy of JDM | Ongoing study |
| 2009 | Fatigue in children with juvenile dermatomyositis | Ongoing study |
| 2009 | A survey of current practice in treatment of juvenile dermatomyositis in the UK and Ireland | Ongoing study |
| 2009 | Development of classification criteria for the idiopathic inflammatory myopathies and their major subgroups (collaboration with IMACS) | Ongoing study |
| 2009 | Five year single blind, phase III effectiveness randomized actively controlled clinical trial in new onset juvenile dermatomyositis: prednisolone versus prednisolone plus cyclosporin A versus prednisolone plus methotrexate (collaboration through PRINTO) | Ongoing study |
| 2010 | IMACS Outcomes Repository (collaboration with IMACS) | Ongoing study |
| Year approved | Project title | References |
|---|---|---|
| 2002 | Immunological characterization of the infiltrate in JDM muscle | [22, 25] |
| 2002 | Tracking of lymphocyte changes before or after flare of active disease | |
| 2002 | Analysis of MHC Class 1 expression in an animal model of myositis | [27, 28] |
| 2002 | Recognition of MHC Class 1 molecules on muscle cells by lymphocytes (CD8 T cells or NK cells) | [22, 25, 29] |
| 2002 | The role of muscle ultrasound and MRI in juvenile dermatomyositis | [30, 31] |
| 2004 | Investigation of alterations of MRP8/14 proteins in juvenile dermatomyositis | Ongoing study |
| 2004 | Autoantibodies and HLA haplotypes in juvenile dermatomyositis, juvenile scleroderma and overlap syndromes | [8, 32, 33] |
| 2004 | Do functional gene variants in anti-inflammatory pathways alter the course of disease and allow greater inflammatory response? | [34, 35] |
| 2005 | Design and testing/validation of a score tool for assessing severity in JDM | [29] |
| 2005 | Fetuin-A and calcinosis in juvenile dermatomyositis | [26] |
| 2005 | Genotyping analysis in JDM for phenotype–genotype correlation | [8, 34, 35] |
| 2006 | Is there evidence of maternal microchimerism in muscle biopsies from children with juvenile dermatomyositis? | Ongoing study |
| 2006 | Extension for genotyping analysis in JDM for phenotype: genotype correlation | [35] |
| 2008 | Genome-wide association study of the idiopathic inflammatory myopathies (collaboration with MYOGEN consortium) | Ongoing study |
| 2008 | Vasculopathy of JDM | Ongoing study |
| 2009 | Fatigue in children with juvenile dermatomyositis | Ongoing study |
| 2009 | A survey of current practice in treatment of juvenile dermatomyositis in the UK and Ireland | Ongoing study |
| 2009 | Development of classification criteria for the idiopathic inflammatory myopathies and their major subgroups (collaboration with IMACS) | Ongoing study |
| 2009 | Five year single blind, phase III effectiveness randomized actively controlled clinical trial in new onset juvenile dermatomyositis: prednisolone versus prednisolone plus cyclosporin A versus prednisolone plus methotrexate (collaboration through PRINTO) | Ongoing study |
| 2010 | IMACS Outcomes Repository (collaboration with IMACS) | Ongoing study |
Projects approved for study within the registry, 2000–10
| Year approved | Project title | References |
|---|---|---|
| 2002 | Immunological characterization of the infiltrate in JDM muscle | [22, 25] |
| 2002 | Tracking of lymphocyte changes before or after flare of active disease | |
| 2002 | Analysis of MHC Class 1 expression in an animal model of myositis | [27, 28] |
| 2002 | Recognition of MHC Class 1 molecules on muscle cells by lymphocytes (CD8 T cells or NK cells) | [22, 25, 29] |
| 2002 | The role of muscle ultrasound and MRI in juvenile dermatomyositis | [30, 31] |
| 2004 | Investigation of alterations of MRP8/14 proteins in juvenile dermatomyositis | Ongoing study |
| 2004 | Autoantibodies and HLA haplotypes in juvenile dermatomyositis, juvenile scleroderma and overlap syndromes | [8, 32, 33] |
| 2004 | Do functional gene variants in anti-inflammatory pathways alter the course of disease and allow greater inflammatory response? | [34, 35] |
| 2005 | Design and testing/validation of a score tool for assessing severity in JDM | [29] |
| 2005 | Fetuin-A and calcinosis in juvenile dermatomyositis | [26] |
| 2005 | Genotyping analysis in JDM for phenotype–genotype correlation | [8, 34, 35] |
| 2006 | Is there evidence of maternal microchimerism in muscle biopsies from children with juvenile dermatomyositis? | Ongoing study |
| 2006 | Extension for genotyping analysis in JDM for phenotype: genotype correlation | [35] |
| 2008 | Genome-wide association study of the idiopathic inflammatory myopathies (collaboration with MYOGEN consortium) | Ongoing study |
| 2008 | Vasculopathy of JDM | Ongoing study |
| 2009 | Fatigue in children with juvenile dermatomyositis | Ongoing study |
| 2009 | A survey of current practice in treatment of juvenile dermatomyositis in the UK and Ireland | Ongoing study |
| 2009 | Development of classification criteria for the idiopathic inflammatory myopathies and their major subgroups (collaboration with IMACS) | Ongoing study |
| 2009 | Five year single blind, phase III effectiveness randomized actively controlled clinical trial in new onset juvenile dermatomyositis: prednisolone versus prednisolone plus cyclosporin A versus prednisolone plus methotrexate (collaboration through PRINTO) | Ongoing study |
| 2010 | IMACS Outcomes Repository (collaboration with IMACS) | Ongoing study |
| Year approved | Project title | References |
|---|---|---|
| 2002 | Immunological characterization of the infiltrate in JDM muscle | [22, 25] |
| 2002 | Tracking of lymphocyte changes before or after flare of active disease | |
| 2002 | Analysis of MHC Class 1 expression in an animal model of myositis | [27, 28] |
| 2002 | Recognition of MHC Class 1 molecules on muscle cells by lymphocytes (CD8 T cells or NK cells) | [22, 25, 29] |
| 2002 | The role of muscle ultrasound and MRI in juvenile dermatomyositis | [30, 31] |
| 2004 | Investigation of alterations of MRP8/14 proteins in juvenile dermatomyositis | Ongoing study |
| 2004 | Autoantibodies and HLA haplotypes in juvenile dermatomyositis, juvenile scleroderma and overlap syndromes | [8, 32, 33] |
| 2004 | Do functional gene variants in anti-inflammatory pathways alter the course of disease and allow greater inflammatory response? | [34, 35] |
| 2005 | Design and testing/validation of a score tool for assessing severity in JDM | [29] |
| 2005 | Fetuin-A and calcinosis in juvenile dermatomyositis | [26] |
| 2005 | Genotyping analysis in JDM for phenotype–genotype correlation | [8, 34, 35] |
| 2006 | Is there evidence of maternal microchimerism in muscle biopsies from children with juvenile dermatomyositis? | Ongoing study |
| 2006 | Extension for genotyping analysis in JDM for phenotype: genotype correlation | [35] |
| 2008 | Genome-wide association study of the idiopathic inflammatory myopathies (collaboration with MYOGEN consortium) | Ongoing study |
| 2008 | Vasculopathy of JDM | Ongoing study |
| 2009 | Fatigue in children with juvenile dermatomyositis | Ongoing study |
| 2009 | A survey of current practice in treatment of juvenile dermatomyositis in the UK and Ireland | Ongoing study |
| 2009 | Development of classification criteria for the idiopathic inflammatory myopathies and their major subgroups (collaboration with IMACS) | Ongoing study |
| 2009 | Five year single blind, phase III effectiveness randomized actively controlled clinical trial in new onset juvenile dermatomyositis: prednisolone versus prednisolone plus cyclosporin A versus prednisolone plus methotrexate (collaboration through PRINTO) | Ongoing study |
| 2010 | IMACS Outcomes Repository (collaboration with IMACS) | Ongoing study |
In line with our goals, the resource and expertise facilitated through this Cohort study and the JDRG have led to numerous collaborative efforts. These include an international survey on diagnostic methods in JDM [23], contributions to studies to generate disease activity and damage measures [40, 41], long-term outcome studies in JDM [10], consensus-driven protocols [42] and ongoing involvement in other national and international projects. The JDRG and its members are closely involved in the JDM strategy of the UK Clinical Studies Group for paediatric rheumatology (www.mcrn.org.uk), contribution to ongoing clinical trials, and in the International Myositis Assessment and Clinical Studies Group (IMACS), European Myositis Network (EUMYONET) and International Myositis Genetics Consortium (MYOGEN). Genetic studies have included a large contribution from this cohort to a world-wide collaborative effort to carry out a genome-wide association study of myositis.
Effects of the Juvenile Dermatomyositis Cohort Biomarker Study and Repository for Idiopathic Inflammatory Myopathies on clinical practice
In addition to the ongoing research studies on juvenile myositis, there is evidence that the use of our study data forms has led to a more standardized approach for the assessment of affected children (N.M., unpublished data). The decision to include the CMAS as part of the standard assessment and the training provided during setting up of the registry have led to its widespread adoption as a reliable clinical tool for the assessment of muscle strength in units around the UK. The establishment of the JDRG has provided a forum for regular communication between members, and facilitated discussion regarding treatment approaches. There is emerging evidence that this has led to a gradual shift in practice from the initial cohort [19] to the present day. Treatment of JDM is not yet evidence based but the group has facilitated emerging consensus. Standardized treatment protocols are currently under discussion and will hopefully be adopted by the group, providing a more secure basis for assessing outcome and the basis of future therapeutic studies.
Discussion
The establishment of this registry and repository of biological samples from children with juvenile IIMs has provided a unique opportunity and resource with which to improve our knowledge and understanding of these rare childhood disorders. The cohort is now of a size to allow studies of long-term morbidity and outcomes. Several other registries and large cohort studies of JDM have been described [7, 10, 20–22, 43], but to our knowledge this is one of the very few multicentre studies to include serial clinical data, the majority collected prospectively, directly linked to serial biological sampling. This will allow testing of biomarkers and their predictive potential: such studies are underway. Our cohort has similar demographics to previously described cohorts. The majority of children were diagnosed within a relatively short time of disease onset (median time to diagnosis 4 months). This could reflect a bias in that children on the registry represent those who are cared for in regional paediatric rheumatology centres. Three deaths within this cohort give a mortality rate of 1%; however, we are aware that this may be an underestimate if some children who presented with very severe disease died before being recruited to the study.
The development of this project posed many challenges, some foreseen, others not. There have also been unanticipated opportunities and benefits. Generous start-up funding was pivotal to the initial successful establishment of the registry. Funding to enable dedicated staff time in both the coordinating and local recruiting centres was important. Perhaps the most crucial factor in the success of this project was the early establishment of ownership and inclusive membership within the group. Ensuring that all centres benefited from the initial funding, visits from the coordinating centre team to each unit and a significant amount of time spent on a consultation process to define the data collection has engendered a sense of collective responsibility for the project. The inclusion of all centres into an inclusive research group, the JDRG, and ensuring that the Chair of the Steering Committee was not from the coordinating centre were helpful. The success of this approach is perhaps best reflected in the fact that centres have subsequently continued to contribute data and samples without dedicated funding.
In keeping with many similar registry projects and non-hypothesis-driven research, defining the correct data set was a significant challenge. It may only be once the projects are underway that the superfluous or missed data items become apparent. In this project it was only once the work had begun to study specific antibodies that differed between children with overlap conditions and ‘pure’ JDM, that it was realized that the initial data set failed to capture the presence or absence of Raynaud’s symptoms, crucial for clinical characterization for this sub-study [6]. This necessitated going back to the recruiting units for further information and the data set was subsequently amended. Similarly, it is clear that there are items of redundant data within the registry and simplifying the collection with a single annual follow-up form has subsequently been agreed by the group. Although it is desirable to maintain a constant data set throughout, it is also important to be able to retain some flexibility to revise this if necessary. A major challenge for this registry has been the inclusion of a large amount of data collected retrospectively on patients with JDM. In such an uncommon condition it was felt important to include all the cases and not only new ones on whom data can be gathered prospectively. It was recognized that this would inevitably result in the inclusion of many cases with incomplete data and that some projects or analyses would only be possible with the prospectively recruited cases. All cases can contribute valuable numbers to large genetic studies to identify susceptibility loci for myositis, even if recruited retrospectively.
This registry has been highly successful in generating projects and publications contributing to our body of knowledge about these rare disorders. It has also drawn together a group of dedicated researchers, facilitating their collaboration with larger international projects. Since its inception, the group, and the data it has collected, have contributed to various large international projects aiming to define and validate core set measures of disease activity and damage in JDM and outcome studies [40, 41, 44]. Although our data set does not formally document these core set measures, the individual components of the tools were already collected as part of the data set, and therefore available for study. The inclusion of biological samples from all cases has made possible genetic and biological studies, in collaboration with investigators successfully obtaining funding and steering committee permission for their project. An unexpected benefit from the establishment of the registry has been the effect of standardizing data collection and the use of the CMAS to assess muscle function as routine clinical practice within the group. The CMAS has been validated for JDM but was not previously in widespread use in UK centres. The collaboration has facilitated discussion about treatment and a move to the more widespread use of earlier more aggressive therapy. It is hoped to build on this experience and establish standardized treatment protocols, which would enable further outcome studies.
Lessons learned during the establishment of this registry and Biobank are applicable to other similar projects. Key to the success of any such project is the establishment of collective ownership by all contributors. The initial time taken to establish the data collection should not be underestimated. Researchers and funding agencies should be aware that it may take several years and a significant investment of both time and money before such a project will become productive.

Acknowledgements
The JDRG would like to thank all of the patients and their families who contributed to the study. We would like to thank E. and J. Hayes for the long-standing support for this project. We thank all local research coordinators and PIs who have made this research possible. The members of the JDRG who contributed were as follows: Dr Liza McCann and Ian Roberts (The Royal Liverpool Children’s Hospital, Alder Hey, Liverpool); Dr Phil Riley and Dr Eileen Baildam (Royal Manchester Children’s Hospital, Manchester); Dr Clive Ryder and Mrs Janis Scott (Birmingham Children’s Hospital, Birmingham); Dr Sue Wyatt and Mrs Gillian Jackson (Leeds General Infirmary, Leeds); Dr Joyce Davidson, Dr Janet Gardner-Medwin and Ms Sue Ferguson (The Royal Hospital for Sick Children, Yorkhill, Glasgow); Dr Mark Friswell, Dr Helen Foster and Mrs Alison Swift (The Royal Victoria Infirmary, Newcastle); Dr Helen Venning and Mrs Elizabeth Hutchinson (Queens Medical Centre, Nottingham); Dr Lucy R. Wedderburn, Dr Clarissa A. Pilkington, Dr N. Hasson, Ms Sue Maillard, Ms Elizabeth Halkon, Ms Virginia Brown, Ms Audrey Juggins, Dr Sally Smith, Ms Sian Evans, Ms Hemlata Varsani, Ms Laura Beard and Dr Elli Enayat (Great Ormond Street Hospital, London); and Dr Kevin Murray (Princess Margaret Hospital, Perth, Western Australia).
Funding: The JDM cohort study has been supported by generous grants from the Cathal Hayes Research Trust, the Wellcome Trust UK (085860), Action Medical Research UK (SP4252), Henry smith Charity, Arthritis Research UK (formerly the Arthritis Research Campaign) (14518 and 18796), Raynaud’s and Scleroderma Association and the UK Myositis Support Group. The study has been adopted onto the Comprehensive Research Network through the Medicines for Children Research Network (MCRN, www.mcrn.org.uk). Funding to pay the Open Access publication charges for this article was provided by the Wellcome Trust.
Disclosure statement: The authors have declared no conflicts of interest.

{kind=link}
Comments