





Objective. To compare the survival outcomes between myositis-associated usual interstitial pneumonia (MA-UIP) and idiopathic pulmonary fibrosis (IPF-UIP).
Methods. Adult MA-UIP and IPF-UIP patients were identified using CTD and IPF registries. The MA-UIP cohort included myositis or anti-synthetase syndrome patients with interstitial lung disease while manifesting UIP on high-resolution CT chest and/or a lung biopsy revealing UIP histology. IPF subjects met American Thoracic Society criteria and similarly had UIP histopathology. Kaplan–Meier survival curves compared cumulative and pulmonary event-free survival (event = transplant or death) between (i) all MA-UIP and IPF-UIP subjects, (ii) MA-UIP with biopsy proven UIP (n = 25) vs IPF-UIP subjects matched for age, gender and baseline forced vital capacity (±10%). Cox proportional hazards ratios compared the survival controlling for co-variates.
Results. Eighty-one IPF-UIP and 43 MA-UIP subjects were identified. The median cumulative and event-free survival time in IPF vs MA-UIP was 5.25/1.8 years vs 16.2/10.8 years, respectively. Cumulative and event-free survival was significantly worse in IPF-UIP vs MA-UIP [hazards ratio of IPF-UIP was 2.9 (95% CI: 1.5, 5.6) and 5.0 (95% CI: 2.8, 8.7) (P < 0.001), respectively]. IPF-UIP event-free survival (but not cumulative) remained significantly worse than MA-UIP with a hazards ratio of 6.4 (95% CI: 3.0, 13.8) after controlling for age at interstitial lung disease diagnosis, gender, ethnicity and baseline forced vital capacity%. Respiratory failure was the most common cause of death in both groups. A sub-analysis of 25 biopsy-proven MA-UIP subjects showed similar results.
Conclusion. MA-UIP patients demonstrated a significant survival advantage over a matched IPF cohort, suggesting that despite similar histological and radiographic findings at presentation, the prognosis of MA-UIP is superior to that of IPF-UIP.
Rheumatology key messages
Myositis associated interstitial lung disease has better prognosis than idiopathic pulmonary fibrosis, despite similar pathology.
Myositis associated interstitial lung disease should be recognized and treated early with immunosuppressive medication.
Introduction
The lung is the most commonly affected extramuscular organ in myositis, and the frequency of interstitial lung disease (ILD) among patients with myositis varies between 5 and 65% depending on the method of ascertainment [1]. This is especially true in myositis patients possessing anti-synthetase autoantibodies as 86% (77/90) of anti-Jo-1 antibody positive patients in one study met criteria for ILD using clinical, radiographic and pulmonary function data [2]. Similarly, many patients with non-Jo-1 anti-synthetase autoantibodies have no muscle weakness yet they manifest features of ILD [3]. This subset of myositis patients presenting with dyspnoea must be recognized as they may not have classic PM or DM, but rather more subtle features of autoimmune disease or the anti-synthetase syndrome such as fever, arthritis, skin rashes and RP. In fact, ILD preceded the diagnosis of myositis in 19% of patients in one study [4], while another report showed that ILD was diagnosed before myositis in 33% of patients [5].
Much like the idiopathic interstitial pneumonias (IIPs), the lung pathology in myositis includes non-specific interstitial pneumonia, organizing pneumonia, acute interstitial pneumonia and usual interstitial pneumonia (UIP) [2, 6–8]. Thus, the clinical, radiographic and histopathological features of lung involvement in myositis resemble, and can be indistinguishable from, that seen in IIP patients, specifically those with idiopathic pulmonary fibrosis (IPF) where UIP predominates. In UIP, there is both immunological and fibrotic targeting of the airway and interstitial structures leading to the same histology in both myositis-associated UIP (MA-UIP) and IPF-associated UIP (IPF-UIP). The prognosis of IPF-UIP is poor but patients with CTD-ILD, even with UIP pathology, may have a better response to immunosuppressive treatment if recognized earlier in their disease course even though they share a similar histopathological appearance. However, there is paucity of literature on outcome of MA-UIP or ILD associated with anti-synthetase syndrome in comparison with IPF-UIP. The aim of this study was to compare the survival and pulmonary outcomes in subjects with MA-UIP having either a UIP histopathology on lung biopsy or radiographic UIP features vs classic IPF patients (IPF-UIP) sharing similar histological and radiographic UIP findings.
Methods
Cohorts
The University of Pittsburgh CTD Registry encompasses more than three decades of prospective data and a serum sample repository collected on consecutive outpatients and inpatients with various autoimmune diseases evaluated at the University of Pittsburgh. Clinical, laboratory, serological, radiographic and histopathological data have been entered into a computer, and organ system definitions have been created for this registry. Similarly, the Simmons Center for ILD at the University of Pittsburgh Medical Center has been collecting clinical, laboratory, radiographic and pulmonary function data on ILD subjects since 2001. Adult myositis patients with ILD (DM, PM, myositis in overlap with another CTD) or patients with the anti-synthetase syndrome and ILD enrolled in the CTD database between 1985 and 2014 were identified. PM and DM were defined as patients meeting probable or definite Bohan and Peter classification criteria and anti-synthetase (+) patients were defined as having one of the eight recognized anti-synthetase autoantibodies (i.e. anti-Jo-1, PL-7, PL-12, EJ, OJ, KS, Zo and Tyr) regardless of their CTD diagnosis [3, 9, 10]. ILD was defined as pulmonary findings on high-resolution computed tomography (HRCT) as interpreted by a thoracic radiologist (C.F.). The MA-ILD cohort was further selected only for PM, DM and anti-synthetase syndrome patients with radiographic features of UIP on HRCT such that the MA-UIP cohort was established. A thoracic radiologist (C.F.) independently reviewed all HRCT scans on the MA-ILD cohort in a systematic fashion determining the diagnosis of radiographic UIP pattern independent of any previous reporting. UIP pattern was defined as per the American Thoracic Society definition [11] meeting all three criteria of honeycombing, reticulations and basilar predominance with absence of features that argue against UIP. A comparator group of IPF diagnosis was established from a Simmons Center for ILD cohort such that they had UIP pattern on HRCT as well as histopathological diagnosis of UIP as per American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the IIPs [11]. This comparator group was designated as IPF-UIP in this study. The biopsy (+) MA-UIP subset was matched 1:1 with a comparator subset of IPF-UIP cohort for age at diagnosis (±10 years), gender and baseline percentage of predicted forced vital capacity (FVC%) (±10%). Clinical (i.e. demographic, presenting and follow-up symptoms, O2 requirements, etc.), pulmonary function test (PFT; FVC%, percentage of predicted forced expiratory volume in 1 s (FEV1%), percentage of predicted diffusing capacity for carbon monoxide (DLCO%)), radiographic (HRCT) and outcome variables (e.g. transplant, death, cause of death and survival time) were retrieved from both CTD and ILD databases on both cohorts. The study was approved by the University of Pittsburgh Institutional Review Board and all patients provided written consent.
Serological data
Autoantibody identification in the CTD Registry was completed using a combination of protein and RNA immunoprecipitation in our University of Pittsburgh research lab as previously described [3, 10].
Outcomes
The CTD database and/or EMR provided outcome information on PFTs, HRCT chest, lung transplant and mortality. Patients with an unknown status or indeterminate cause of death were submitted to the National Death Index and the resultant cause of death codes, along with independent chart review (R.A., C.M., C.V.O.), were used to determine the primary cause of death. All patients with unknown clinical status and no National Death Index match were submitted to the Social Security Death Index to determine status and date of death. Cumulative survival and pulmonary event-free survival were evaluated as outcome variables with event defined as lung transplant or death. PFT worsening was defined as time to ⩾20% FVC% decline from baseline. Comparisons of outcome variables were made between MA-UIP and IPF and biopsy-proven MA-UIP vs biopsy proven IPF-UIP.
Statistics
Baseline clinical characteristics including PFT, radiographic and transplant or death data were compared between MA-UIP and IPF using t test, Mann–Whitney test or chi-square test based on the distribution of the data. Kaplan–Meier survival curves and the log rank test compared cumulative, pulmonary event-free survival and PFT worsening between MA-UIP and IPF and biopsy-proven MA-UIP (n = 25) vs biopsy proven IPF-UIP (matched as outlined above). Cox proportional hazards ratios (HRs) compared cumulative and pulmonary event-free survival controlling for co-variates including gender, ethnicity, age at ILD diagnosis and baseline FVC% as applicable.
Results
Overall
Eighty-one IPF patients with UIP histopathology were identified from the ILD database. Forty-three patients were identified with MA-UIP based on radiographic HRCT findings including 13 PM, 9 DM, 14 anti-synthetase syndrome (without a PM or DM diagnosis) and 7 myositis in overlap with another CTD. The autoantibody findings included 22 with anti-Jo-1, 10 anti-PL-12, 4 anti-PL-7, 3 anti-EJ, 3 anti-KS and 1 with only the anti-SSA autoantibody. Thus, all but one MA-UIP subject possessed anti-synthetase autoantibodies. For 25 patients of the MA-UIP cohort, surgical lung biopsies were available confirming histopathologically UIP that formed the biopsy + MA-UIP subset. Table 1 compares the demographic features and baseline PFT findings between the MA-UIP and IPF-UIP subjects. IPF-UIP patients were significantly older than the MA-ILD subjects and were more commonly male and Caucasian but the baseline FVC% and DLCO% were comparable between the two cohorts. There were more smokers (current or past) in the IPF-UIP cohort. There were 16 deaths (37%) in the MA-UIP cohort as compared with 36 (44%) in the IPF-UIP cohort (non-significant), but patients with MA-UIP died at a younger age [54 (11.7) years] compared with those in the IPF-UIP [68 (7.5) years] group (P < 0.001). Twenty-three per cent (10/43) of the MA-UIP patients received a lung transplant while 56% (45/81) went to lung transplant in the IPF-UIP cohort (P = 0.001) and MA-UIP patients were younger at the time of lung transplantation [54 (6.4) vs 64 (8.8) years of age, P < 0.001]. The pulmonary end point of death or transplant was reached significantly less often [53.5% (23/43) vs 77.8% (63/81), P = 0.005] in the MA-UIP subjects than those with IPF-UIP.
Baseline clinical and demographic features of the two usual interstitial pneumonia cohorts
| Clinical features | MA-UIP (n = 43) | IPF-UIP (n = 81) | P-value |
|---|---|---|---|
| Age at ILD diagnosis, mean (s.d.), years | 46 (11.0) | 63 (8.4) | <0.001 |
| Gender, male, % | 35 | 73 | <0.001 |
| Caucasian, % | 83 | 98 | 0.004 |
| Baseline FVC%, mean (s.d.) | 60 (19.6) | 65 (15.3) | 0.11 |
| Baseline DLCO%, mean (s.d.) | 47 (18.3) | 47 (17.3) | 1.0 |
| Death, n (%) | 16 (37) | 36 (44) | 0.43 |
| Age at death, mean (s.d.), years | 54 (11.7) | 68 (7.5) | <0.001 |
| Transplant, n (%) | 10 (23) | 45 (56) | 0.001 |
| Age of transplant, mean (s.d.), years | 54 (6.4) | 64 (8.8) | <0.001 |
| Pulmonary event (death or transplant), n (%) | 23 (53.5) | 63 (77.8) | 0.005 |
| Tobacco use (current or past), n (%) | 16 (40)a | 56 (69.1) | 0.002 |
| Clinical features | MA-UIP (n = 43) | IPF-UIP (n = 81) | P-value |
|---|---|---|---|
| Age at ILD diagnosis, mean (s.d.), years | 46 (11.0) | 63 (8.4) | <0.001 |
| Gender, male, % | 35 | 73 | <0.001 |
| Caucasian, % | 83 | 98 | 0.004 |
| Baseline FVC%, mean (s.d.) | 60 (19.6) | 65 (15.3) | 0.11 |
| Baseline DLCO%, mean (s.d.) | 47 (18.3) | 47 (17.3) | 1.0 |
| Death, n (%) | 16 (37) | 36 (44) | 0.43 |
| Age at death, mean (s.d.), years | 54 (11.7) | 68 (7.5) | <0.001 |
| Transplant, n (%) | 10 (23) | 45 (56) | 0.001 |
| Age of transplant, mean (s.d.), years | 54 (6.4) | 64 (8.8) | <0.001 |
| Pulmonary event (death or transplant), n (%) | 23 (53.5) | 63 (77.8) | 0.005 |
| Tobacco use (current or past), n (%) | 16 (40)a | 56 (69.1) | 0.002 |
Smoking data were not available for two patients. DLCO%: percentage of predicted diffusing capacity of the lung for carbon monoxide; FVC%: percentage of predicted forced vital capacity; IPF-UIP: idiopathic pulmonary fibrosis; MA-UIP: myositis-associated usual interstitial pneumonia.
Baseline clinical and demographic features of the two usual interstitial pneumonia cohorts
| Clinical features | MA-UIP (n = 43) | IPF-UIP (n = 81) | P-value |
|---|---|---|---|
| Age at ILD diagnosis, mean (s.d.), years | 46 (11.0) | 63 (8.4) | <0.001 |
| Gender, male, % | 35 | 73 | <0.001 |
| Caucasian, % | 83 | 98 | 0.004 |
| Baseline FVC%, mean (s.d.) | 60 (19.6) | 65 (15.3) | 0.11 |
| Baseline DLCO%, mean (s.d.) | 47 (18.3) | 47 (17.3) | 1.0 |
| Death, n (%) | 16 (37) | 36 (44) | 0.43 |
| Age at death, mean (s.d.), years | 54 (11.7) | 68 (7.5) | <0.001 |
| Transplant, n (%) | 10 (23) | 45 (56) | 0.001 |
| Age of transplant, mean (s.d.), years | 54 (6.4) | 64 (8.8) | <0.001 |
| Pulmonary event (death or transplant), n (%) | 23 (53.5) | 63 (77.8) | 0.005 |
| Tobacco use (current or past), n (%) | 16 (40)a | 56 (69.1) | 0.002 |
| Clinical features | MA-UIP (n = 43) | IPF-UIP (n = 81) | P-value |
|---|---|---|---|
| Age at ILD diagnosis, mean (s.d.), years | 46 (11.0) | 63 (8.4) | <0.001 |
| Gender, male, % | 35 | 73 | <0.001 |
| Caucasian, % | 83 | 98 | 0.004 |
| Baseline FVC%, mean (s.d.) | 60 (19.6) | 65 (15.3) | 0.11 |
| Baseline DLCO%, mean (s.d.) | 47 (18.3) | 47 (17.3) | 1.0 |
| Death, n (%) | 16 (37) | 36 (44) | 0.43 |
| Age at death, mean (s.d.), years | 54 (11.7) | 68 (7.5) | <0.001 |
| Transplant, n (%) | 10 (23) | 45 (56) | 0.001 |
| Age of transplant, mean (s.d.), years | 54 (6.4) | 64 (8.8) | <0.001 |
| Pulmonary event (death or transplant), n (%) | 23 (53.5) | 63 (77.8) | 0.005 |
| Tobacco use (current or past), n (%) | 16 (40)a | 56 (69.1) | 0.002 |
Smoking data were not available for two patients. DLCO%: percentage of predicted diffusing capacity of the lung for carbon monoxide; FVC%: percentage of predicted forced vital capacity; IPF-UIP: idiopathic pulmonary fibrosis; MA-UIP: myositis-associated usual interstitial pneumonia.
Survival comparison between MA-UIP vs IPF-UIP
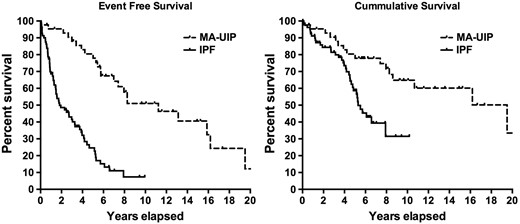
Kaplan–Meier survival curves between the two usual interstitial pneumonia cohorts
IPF: idiopathic pulmonary fibrosis; MA-UIP: myositis-associated usual interstitial pneumonia; Event: Death or Lung Transplant.
Cumulative and event-free survival in the two usual interstitial pneumonia cohorts
| MA-UIP (n = 43) | IPF-UIP (n = 81) | |
|---|---|---|
| Median cumulative survival (years) | 16.1 | 5.2 |
| Median event-free survival (years) | 10.8 | 1.8 |
| Unadjusted cumulative survival, % | ||
| 5 years | 80 | 59 |
| 10 years | 65 | 32 |
| Unadjusted event-free survival, % | ||
| 5 years | 80 | 25 |
| 10 years | 50 | 0 |
| MA-UIP (n = 43) | IPF-UIP (n = 81) | |
|---|---|---|
| Median cumulative survival (years) | 16.1 | 5.2 |
| Median event-free survival (years) | 10.8 | 1.8 |
| Unadjusted cumulative survival, % | ||
| 5 years | 80 | 59 |
| 10 years | 65 | 32 |
| Unadjusted event-free survival, % | ||
| 5 years | 80 | 25 |
| 10 years | 50 | 0 |
IPF-UIP: idiopathic pulmonary fibrosis; MA-UIP: myositis-associated usual interstitial pneumonia; Event: Death or Lung Transplant.
Cumulative and event-free survival in the two usual interstitial pneumonia cohorts
| MA-UIP (n = 43) | IPF-UIP (n = 81) | |
|---|---|---|
| Median cumulative survival (years) | 16.1 | 5.2 |
| Median event-free survival (years) | 10.8 | 1.8 |
| Unadjusted cumulative survival, % | ||
| 5 years | 80 | 59 |
| 10 years | 65 | 32 |
| Unadjusted event-free survival, % | ||
| 5 years | 80 | 25 |
| 10 years | 50 | 0 |
| MA-UIP (n = 43) | IPF-UIP (n = 81) | |
|---|---|---|
| Median cumulative survival (years) | 16.1 | 5.2 |
| Median event-free survival (years) | 10.8 | 1.8 |
| Unadjusted cumulative survival, % | ||
| 5 years | 80 | 59 |
| 10 years | 65 | 32 |
| Unadjusted event-free survival, % | ||
| 5 years | 80 | 25 |
| 10 years | 50 | 0 |
IPF-UIP: idiopathic pulmonary fibrosis; MA-UIP: myositis-associated usual interstitial pneumonia; Event: Death or Lung Transplant.
Pulmonary function results
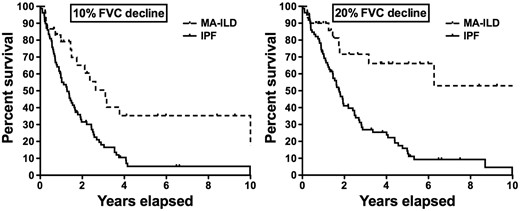
Time to decline in FVC% between the two usual interstitial pneumonia cohorts
FVC%: percentage of predicted forced vital capacity; IPF: idiopathic pulmonary fibrosis; MA-UIP: myositis-associated usual interstitial pneumonia.
Survival and pulmonary function comparison between biopsy-proven MA-UIP vs IPF-UIP
MA-UIP with biopsy-proven UIP had a significantly better event-free survival compared with the matched IPF-UIP cohort after adjusting for age, gender, ethnicity and baseline FVC [HR = 4.0 (95% CI: 1.9, 8.5), P < 0.01]. However, cumulative survival was not significantly different, likely due to the high frequency of lung transplantation in the IPF-UIP patients. Similarly, pulmonary function decline was significantly better in biopsy-matched MA-UIP (20% FVC decline: HR = 2.9, P = 0.03; 10% FVC decline: HR = 2.7, P = 0.026), after controlling for age, gender, ethnicity and baseline FVC. Smoking status had no influence on these results.
Cause of death
A pulmonary cause for death or lung transplantation were prevalent in both the MA-UIP and IPF-UIP cohorts, but IPF-UIP subjects fared worse than the MA-UIP patients despite controlling for age, gender and baseline FVC%. Respiratory failure was the most common cause of death in both groups followed by infection. Specifically, among 36 IPF-UIP deaths, 14 were from respiratory failure due to worsening pulmonary fibrosis, while other causes included infection (n = 6), malignancy (n = 5), multi-organ failure (n = 2), renal failure (n = 2), liver failure (n = 1), CNS haemorrhage (n = 1), transplant graft failure (n = 1) and unknown causes (n = 4). Among MA-UIP patients, there were 16 deaths with eight due to respiratory failure from pulmonary fibrosis while three died from infection, one from lung cancer, one from pulmonary artery hypertension and three from an unknown cause.
Discussion
This study represents the first report comparing survival and pulmonary outcomes in two prospectively collected patient cohorts of myositis associated UIP and idiopathic UIP (IPF). Adult myositis patients or those possessing any one of several anti-synthetase autoantibodies with clinical, radiographic and/or histological features of UIP had improved survival and pulmonary outcomes compared with a matched cohort of IPF patients with UIP features. This was observed despite similar histopathological findings in a subset of the MA-UIP subjects compared with those with IPF-UIP. Nevertheless, the survival was poor in both groups of patients and respiratory failure or other complications of pulmonary fibrosis were the predominant cause of death in both the MA-UIP and IPF cohorts. Similarly, the functional pulmonary status as reflected by pulmonary function tests also worsened less dramatically in the MA-UIP group compared with the IPF patients.
These are important observation for several reasons. First, in the patient presenting with dyspnoea and features of ILD it is essential to carefully elucidate any clinical features of autoimmune disease to identify those individuals with possible myositis or anti-synthetase syndrome as the aetiology for their ILD. As reported by other investigators, these features may be subtle, requiring a careful rheumatological history and physical examination [3, 10, 12, 13] as well as the identification of anti-synthetase antibodies. Secondly, IPF-UIP patients may receive newer therapies for IPF or be considered for lung transplantation, whereas patients with MA-ILD require aggressive immunosuppression. Although lung transplantation may indeed improve survival in the patient with IPF-UIP, our study still demonstrated worse survival and pulmonary outcomes in the IPF-UIP subset despite more frequent lung transplantation compared with that observed in the matched cohort of MA-UIP patients. Thirdly, the demonstration of the other, non-Jo-1 anti-synthetase autoantibodies in the MA-UIP cohort highlights the importance of measuring these autoantibodies since many of these patients have no muscle weakness and less overt features of myositis compared with the classic myositis population [3, 10]. Autoantibody screening for patients presenting with ILD should include myositis specific autoantibodies specifically anti-synthetase autoantibodies as these patients may have a negative ANA.
This study has some limitations. We recognize that patients with MA-UIP and IPF-UIP were identified retrospectively. However, the nature of the myositis and ILD databases at the University of Pittsburgh is that consecutive patients with myositis and other connective tissue diseases (including the anti-synthetase syndrome) as well as ILD are prospectively entered into a database. In addition, all CTD subjects undergo the same testing including the detection of autoantibodies by immunoprecipitation and the completion of routine and pulmonary-directed diagnostic studies with longitudinal follow-up. Second, the IPF-UIP patients did not undergo the immunoprecipitation screening of myositis autoantibodies in our research labs as MA-UIP patients did.
However, they were screened with a comprehensive commercial autoantibody panel by pulmonologists as part of their usual care, which included a myositis panel. Nevertheless, none of the IPF-UIP patients had a clinical suspicion of autoimmunity and all met American Thoracic Society criteria for IPF and were seen and enrolled in an IPF cohort by expert pulmonologists working in an ILD clinical and research centre. Third, given that the cohort of IPF-UIP were all biopsy proven whereas MA-UIP were a mix of biopsy proven and CT chest documented UIP, the two groups were not identical in the ascertainment. Therefore, we performed a sub-analysis of only biopsy proven MA-UIP and compared it with the IPF-UIP cohort and found similar results. Finally, we did not include the variety of immunosuppressive treatment regimens that patients with MA-UIP received as this was not the intent of this paper. The point is that it is important to recognize the autoimmune aetiology of ILD in order that patients receive potentially life-saving immunosuppressive therapy to at least improve survival or mitigate the need for lung transplantation.
In summary, despite similar histopathological and radiographic findings at presentation, the prognosis of MA-UIP was superior to that of IPF-UIP. Thus, in patients presenting with histological and imaging features of UIP, it is critical to distinguish those with underlying myositis or the anti-synthetase syndrome from those with true IPF. Further studies exploring the baseline predictive histopathological differences for improved survival as well as the optimal treatment for ILD in myositis are necessary.
Funding: No specific funding was received from any funding bodies in the public, commercial or not-for-profit sectors to carry out the work described in this manuscript.
Disclosure statement: C.O. received grants/research support from Genentech for a clinical trial. K.G. has received consultancy fees from Gilead Pharmaceuticals and Phillips Respironics. All other authors have declared no conflicts of interest.

{kind=link}
{kind=link}
Comments